

ご利用について
医療専門家向けの本PDQがん情報要約では、ウィルムス腫瘍とその他の小児腎腫瘍の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児腎腫瘍に関する一般情報
-
小児および青年のがん患者の生存において、劇的な改善が達成されている。1975年から2010年の間に、小児がんの死亡率は50%以上低下した。[ 1 ]15歳未満のウィルムス腫瘍の患児については、5年生存率が同じ期間で74%から88%に改善した。[ 1 ]小児および青年がん生存者には、治療から数ヵ月または数年経過後もがん療法の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。(小児および青年がん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)に関するPDQ要約を参照のこと。)
小児腎腫瘍は、全小児がん症例の約7%を占める。小児腎腫瘍の大半はウィルムス腫瘍であるが、15~19歳の年齢群だけでみると、大半の腫瘍が腎細胞がんである。ウィルムス腫瘍は、片側の腎臓(片側性)のみを侵すこともあれば、両側の腎臓(両側性)を侵すこともある。それほど多くない種類の小児腎腫瘍としては、ラブドイド腫瘍、明細胞肉腫、先天性間葉芽腎腫、腎臓のユーイング肉腫、原発性腎筋上皮がん、嚢胞性部分的分化型腎芽腫、多房性嚢胞性腎腫、原発性腎滑膜肉腫、腎未熟型肉腫などがある。腎芽腫症は非悪性腫瘍の一種である。[ 2 ][ 3 ]
参考文献- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Ahmed HU, Arya M, Levitt G, et al.: Part I: Primary malignant non-Wilms' renal tumours in children. Lancet Oncol 8 (8): 730-7, 2007.[PUBMED Abstract]
- Ahmed HU, Arya M, Levitt G, et al.: Part II: Treatment of primary malignant non-Wilms' renal tumours in children. Lancet Oncol 8 (9): 842-8, 2007.[PUBMED Abstract]
- ウィルムス腫瘍
-
ウィルムス腫瘍の発生率
ウィルムス腫瘍は乳児および小児において最も頻度の高い腎腫瘍である。ウィルムス腫瘍の発生率は15歳未満の小児100万人当たり8.2例、または乳児1万人当たり1例である。[ 1 ]米国では、毎年約650例のウィルムス腫瘍が診断されている。アジア人の発生率ははるかに低い。
片側性のウィルムス腫瘍の男女比は0.92/1.00であるが、両側性は0.60/1.00である。診断時平均年齢は、ウィルムス腫瘍の片側性症例で生後44ヵ月、両側性症例で生後31ヵ月である。[ 2 ][ 3 ]ウィルムス腫瘍患児の約10%に、関連する先天性奇形症候群がみられる。[ 4 ]
ウィルムス腫瘍に関連する症候群およびその他の病態
ウィルムス腫瘍は、その他の点ではがん発症の素因がない健康な小児で典型的に発生するが、ウィルムス腫瘍の小児の約10%は先天異常を有することが報告されている。[ 4 ][ 5 ]先天異常およびウィルムス腫瘍を有する患者において、症例の60%でnephrogenic rest(造腎組織遺残)が報告されている。[ 6 ]パリのInstitut Curieが検討した一連のウィルムス腫瘍患者295人のうち、52人(17.6%)に異常または症候群が認められ、そのうちの43人は重大な問題を有すると判定されたほか、14人には遺伝学的に腫瘍素因症候群が証明された。[ 7 ]
ウィルムス腫瘍の患児には、片側過形成や、停留精巣および尿道下裂などの尿路奇形がみられることがある。患児には、過成長、無虹彩症、遺伝的奇形、およびその他の異常など、識別可能な表現型の症候群がみられることがある。これらの症候群によって、この疾患が有する遺伝子異常に関する情報への手がかりが得られる。これらの表現型の症候群および他の病態は、過成長および非過成長のカテゴリーに分類されている(表1を参照のこと)。過成長の症候群および病態は、出生前および出生後に体細胞が過度に発育することが原因である。[ 8 ][ 9 ]
ウィルムス腫瘍の絶対リスクは、基礎にある疾患または異常で異なることを認識することが重要である。例えば、片側過形成のほとんどの患者はウィルムス腫瘍を発症しない。
表1.ウィルムス腫瘍に関連する症候群および病態a 症候群/病態 遺伝子 過成長の表現型 非過成長の表現型 高リスクのウィルムス腫瘍(20%超) CLOVES = 先天的脂肪腫性過成長、血管奇形、表皮母斑、および骨格/脊椎奇形;MULIBREY = 筋肉:(MU)scle、肝臓:(LI)ver、脳:(BR)ain、および眼球:(EY)eの特徴的な異常;WAGR = ウィルムス腫瘍、無虹彩症、泌尿生殖器奇形、および精神遅滞。 a出典:Treger et al.[ 10 ] WAGR症候群 WT1欠失 X Denys-Drash症候群 WT1ミスセンス変異 X パールマン症候群 DIS3L2変異 X BRCA2(FANCD1)またはPALB2(FANCN)の両アレル性変異を伴うファンコニー貧血 BRCA2、PALB2 X 染色分体早期解離/多彩異数性モザイク症候群 BUB1BまたはTRIP13の両アレル性変異 X 中リスクのウィルムス腫瘍(5~20%) Frasier症候群 WT1イントロン9におけるスプライス部位変異 X ベックウィズ-ヴィーデマン症候群 片親性ダイソミーまたはH19エピ変異 X Simpson-Golabi-Behmel症候群 GPC3変異 X 低リスクのウィルムス腫瘍(5%未満) ブルーム症候群 BLMの両アレル性変異 X DICER1症候群 DICER1変異 X リー-フラウメニ症候群 TP53、CHEK2 X 孤立性片側過形成症 X 副甲状腺機能亢進症顎腫瘍症候群 CDC73(HRPT2としても知られる)変異 X MULIBREY低身長症 TRIM37変異 X CLOVES症候群を含むPIK3CA関連分節性過成長 PIK3CA変異 X 9q22.3微小欠失症候群 9q22.3 X ソトス症候群 NSD1 X 家族性ウィルムス腫瘍 FWT1 X FWT2 外性器異常 WT1 X 散発性無虹彩症 WT1 X 18トリソミー X WT1およびWT2などのウィルムス腫瘍に関連する遺伝子に関する情報については、本要約のウィルムス腫瘍のゲノム情報のセクションを参照のこと。
ウィルムス腫瘍の症候性原因
WT1に関連する症候群には以下のものがある:
WT2に関連する症候群には以下のものがある:
その他のウィルムス腫瘍の症候性原因には以下のものがある:
ウィルムス腫瘍の非症候性原因
ウィルムス腫瘍の非症候性原因には以下のものがある:
ウィルムス腫瘍のゲノム情報
小児の他の胎児性新生物に類似したウィルムス腫瘍は、少数の遺伝子異常の後に典型的に発生する。1件の研究では、ウィルムス腫瘍117例に対してゲノムワイドシークエンシング、mRNAおよびmiRNA発現、DNAコピー数、およびメチル化解析が実施され、その後ウィルムス腫瘍651例に対して標的シークエンシングが実施された。[ 59 ]腫瘍は、予後良好な組織型(FH)で再燃したウィルムス腫瘍またはびまん性退形成が認められるウィルムス腫瘍のいずれかで選択された。研究により、以下が示された:[ 59 ]
ウィルムス腫瘍症例の約3分の1には、WT1、CTNNB1、またはWTXの変異が関わっている。[ 60 ][ 61 ]ウィルムス腫瘍症例の別のサブセットは、DROSHA、DGCR8、DICER1、およびXPO5などのmiRNAプロセシング遺伝子(miRNAPG)の変異により生じる。[ 62 ][ 63 ][ 64 ][ 65 ]ウィルムス腫瘍で反復的に変異を生じ、初期の腎発生にきわめて重要な他の遺伝子には、SIX1およびSIX2(初期の腎発生に重要な役割を果たす転写因子)[ 62 ][ 63 ]、EP300、CREBBP、MYCNなどがある。[ 59 ]ウィルムス腫瘍における変異のうち、30~50%は腎発生における転写伸長過程で収束するようであり、MLLT1、BCOR、MAP3K4、BRD7、およびHDAC4といった遺伝子が含まれる。[ 59 ]退形成型ウィルムス腫瘍はTP53変異の存在を特徴とする。
WAGR(ウィルムス腫瘍、無虹彩症、泌尿生殖器奇形、および精神遅滞)症候群、ベックウィズ-ヴィーデマン症候群、片側肥大症、Denys-Drash症候群、パールマン症候群などの多くの遺伝性疾患の患者でウィルムス腫瘍の発生率の増加が観察されている。[ 66 ]家族性ウィルムス腫瘍症例で認められている他の遺伝的原因にはRESTおよびCTR9の生殖細胞変異などがある。[ 50 ][ 67 ]
ウィルムス腫瘍のゲノム情報および遺伝的特徴を以下に要約する。
WT1遺伝子
WT1遺伝子は11番染色体短腕(11p13)に位置する。WT1は泌尿生殖器の正常な発達に必要な転写因子であり、腎芽体の分化にとって重要である。[ 68 ]WT1の変異は散発性ウィルムス腫瘍の10~20%に認められる。[ 60 ][ 68 ][ 69 ]
WT1の変異を有するウィルムス腫瘍の特徴は以下の通りである:
WT1の生殖細胞変異は、ウィルムス腫瘍に加えて、以下の疾患の1つがある患児により多くみられる:
WT1の生殖細胞変異による症候性疾患には、WAGR症候群、Denys-Drash症候群[ 17 ]、およびFrasier症候群などがある。[ 14 ]
生殖細胞系のWT1点変異は、腎症、46,XY性分化異常症、およびウィルムス腫瘍のさまざまなリスクを特徴とする遺伝的症候群を引き起こす。[ 79 ][ 80 ]
WT1の変異の遺伝子型/表現型の相関を評価した諸研究から、ウィルムス腫瘍のリスクは切断型変異で最も高く(17例中14例、82%)、ミスセンス変異でそれより低い(67例中27例、42%)ことが示されている。リスクが最も低かったのはKTSスプライス部位の変異である(27例中1例、4%)。[ 79 ][ 80 ]両側性ウィルムス腫瘍は、WT1切断型変異の症例(14例中9例)の方がWT1ミスセンス変異の症例(27例中3例)より多かった。[ 79 ][ 80 ]これらのゲノム研究から、Denys-Drash症候群の患児でウィルムス腫瘍のリスクが高く、Frasier症候群の患児でウィルムス腫瘍のリスクが低いという以前の推定が確認される。
WAGR症候群およびウィルムス腫瘍に伴う晩期合併症(晩期障害)には以下のものがある:
(ウィルムス腫瘍に伴う晩期合併症(晩期障害)に関する詳しい情報については、ウィルムス腫瘍とその他の小児腎腫瘍の治療に関するPDQ要約のウィルムス腫瘍治療後の晩期合併症(晩期障害)のセクションを参照のこと。)
CTNNB1遺伝子
CTNNB1はウィルムス腫瘍において最も一般的に変異が見られる遺伝子であり、ウィルムス腫瘍患者の15%に発生すると報告されている。[ 59 ][ 61 ][ 69 ][ 71 ][ 83 ]このようなCTNNB1変異により、腎発生に大きな役割を果たすWNT経路の活性化が生じる。[ 84 ]CTNNB1の変異は、WT1の変異を伴って発生することが多く、WT1に変異を認めるウィルムス腫瘍のほとんどの症例は、同時にCTNNB1の変異を有している。[ 69 ][ 71 ][ 83 ]CTNNB1の変異は、MLLT1の変異を伴う場合を除き、WT1またはWTXの変異がみられない状況で発見されるのはまれなため、WT1蛋白に損傷がない状況での腫瘍発生には、β-カテニンの活性化のみでは十分ではないと考えられる。[ 61 ][ 85 ]CTNNB1変異は、腫瘍には認められるが、nephrogenic restには認められないため、ウィルムス腫瘍発生における晩期事象であると考えられる。[ 74 ]
X染色体上のWTX遺伝子
AMER1とも呼ばれるWTXはX染色体のXq11.1に存在する。この遺伝子はウィルムス腫瘍症例の15~20%で変異している。[ 60 ][ 61 ][ 69 ][ 86 ][ 87 ]WTXの生殖細胞変異は、X連鎖性の硬化性骨異形成症である頭部硬化症を伴う先天性線状骨障害を引き起こす(MIM300373)。[ 88 ]WTXの生殖細胞変異を有しているにもかかわらず、先天性線状骨障害患者には腫瘍を発症する素因はない。[ 88 ]WTX蛋白はβ-カテニンの分解およびAPC蛋白の細胞内分布の両方に関わっていると考えられる。[ 85 ][ 89 ]WTXはWTX遺伝子の一部または全部に関わる欠失により変異することが最も多く、有害な点変異が生じることはそれほど多くない。[ 60 ][ 69 ][ 86 ]WTX変異を有するウィルムス腫瘍症例のほとんどは後成的な11p15異常を有する。[ 69 ]
WTX変異は男女に等しく分布しており、WTXの不活性化は、臨床像または予後に対して明確な影響を及ぼさない。[ 60 ]
染色体11p15(WT2)上のインプリンティングクラスター領域(ICR)とベックウィズ-ヴィーデマン症候群
ウィルムス腫瘍の第2の遺伝子座であるWT2は、染色体11p15.5のインプリンティング領域にマップされており、生殖細胞変異であればベックウィズ-ヴィーデマン症候群を引き起こす。ウィルムス腫瘍患児の約3%は、増殖調節遺伝子座11p15.5に過成長の臨床症状を伴わない後成的または遺伝的な生殖細胞の変化を有する。ベックウィズ-ヴィーデマン症候群の患児と同様に、これらの患児でも両側性ウィルムス腫瘍または家族性ウィルムス腫瘍の発生率は高い。[ 51 ]
ベックウィズ-ヴィーデマン症候群の患者でウィルムス腫瘍を有する患者の約1/5が両側性腫瘍を呈するが、異時性の両側性腫瘍も認められる。[ 27 ][ 28 ][ 29 ]ベックウィズ-ヴィーデマン症候群の有病率は、National Wilms Tumor Study(NWTS)に報告されたウィルムス腫瘍の小児で約1%である。[ 2 ][ 29 ]
ベックウィズ-ヴィーデマン症候群患者の約80%は11p15ドメインの分子欠損を有する。[ 90 ]ベックウィズ-ヴィーデマン症候群の基礎をなすさまざまな分子機構が同定されている。これらの異常の一部は遺伝的であるが(CDKN1Cの母方アレルの生殖細胞変異、11p15の父方片親性イソダイソミー、または11p15ドメイン部の重複)、より多くは後成的である(母方ICR2/KvDMR1のメチル化喪失または母方ICR1のメチル化)。[ 51 ][ 91 ]
WT2遺伝子座に存在するいくつかの候補遺伝子は、独立した2つのインプリンティングドメイン、IGF2/H19およびKIP2/LIT1を構成している。[ 91 ]LOHは母方染色体にのみ影響を与え、父方の遺伝子を活性化し、母方の活性遺伝子を不活化する働きがある。この領域における遺伝子のインプリントの消失またはスイッチ(メチル化状態の変化)も頻繁に観察されており、同様の機能異常をもたらす。[ 51 ][ 90 ][ 91 ]
エピジェノタイプと表現型との関係はベックウィズ-ヴィーデマン症候群において明らかにされており、この症候群では11p15領域の変異の種類に応じてがんの発生率が異なっている。[ 92 ]
特定の遺伝子型-表現型の相関を特徴とするベックウィズ-ヴィーデマン症候群には、以下の4つの主要な分子的サブタイプがある:
- ICR1のメチル化(ICR1-GoM)。症例の5~10%は、テロメアICR1-GoMにより引き起こされ、IGF2遺伝子(正常であれば父方アレルのみによって発現)の両アレル性発現に加え、腫瘍抑制性のH19遺伝子の発現減少が生じる。ウィルムス腫瘍の発生率は22.8%である。[ 93 ]
- ICR2のメチル化喪失(ICR2-LoM)。ベックウィズ-ヴィーデマン症候群症例の50%は、ICR2-LoMにより引き起こされ、正常であれば母系染色体のみによって発現するCDKN1C遺伝子の発現減少を来す。腫瘍の発生率は、非常に低い(2.5%)。[ 93 ]
- 片親性ダイソミー(UPD)。染色体11p15.5に位置するモザイクUPDでは、両方のインプリンティング遺伝子クラスターでの発現変化が観察され、本症例の20~25%を占めている。ウィルムス腫瘍の発生率は6.2%で、次に肝芽腫(4.7%)および副腎がん(1.5%)が多い。[ 93 ]ベックウィズ-ヴィーデマン症候群症例の1%未満は、11p15領域を巻き込んだ染色体再構成によって発生する。
- CDKN1C変異。母系遺伝性のCDKN1C機能喪失変異は、本症例の約5%を占めている。この種類は、神経芽腫の4.3%の発生率と関係している。[ 93 ]
父方の11p15イソダイソミーを有する患者では神経芽腫または肝芽腫などの他の腫瘍が報告された。[ 21 ][ 25 ][ 94 ]ベックウィズ-ヴィーデマン症候群の患者が肝芽腫を発症する相対リスクは一般集団におけるリスクの2,280倍である。[ 29 ]
インプリンティングの消失または遺伝子のメチル化は他の遺伝子座ではまれにしか認められず、11p15.5におけるインプリンティングの消失の特異性を支持している。[ 95 ]興味深いことに、ヨーロッパの小児におけるよりも発生率が低いアジアの小児におけるウィルムス腫瘍は、nephrogenic restまたはIGF2のインプリンティングの消失と関連していない。[ 96 ]
その他の遺伝子および染色体の変異
ウィルムス腫瘍の発生機序および生物学に関与しているその他の遺伝子および染色体異常には以下のものがある:
図2は、予後良好な組織型を示したにもかかわらず再燃を来したことから選択されたウィルムス腫瘍症例の選択コホートのゲノムの全体像を要約したものである。[ 75 ]予後良好な組織型のウィルムス腫瘍75症例が遺伝子発現データの教師なし解析によりクラスター化され、6つのクラスターが得られた。利用可能な遺伝子発現データのあるMLLT1変異腫瘍6例中5例はクラスター3であり、2例はCTNNB1変異を伴っていた。このクラスターはWT1の変異または小セグメントの欠失を来した腫瘍4例も含んでおり、そのすべてがCTNNB1の変異、またはWTXの小セグメントの欠失もしくは変異のいずれかも来していた。これには11p15のインプリンティングを保持する相当数の腫瘍も含まれていた(すべてのMLLT1変異腫瘍を含む)。miRNAPG変異症例はまとめてクラスター化され、MLLT1およびWT1/WTX/CTNNB1変異症例の両方と相互排他的であった。
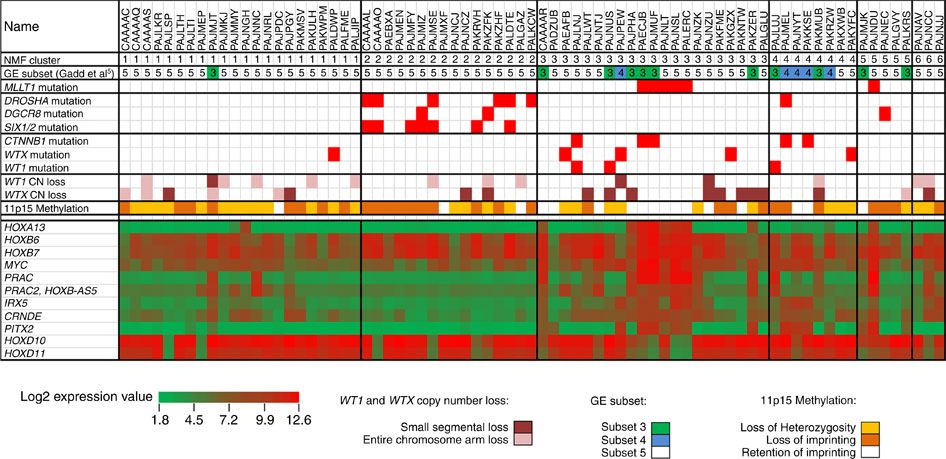
図2.遺伝子発現データの教師なし解析。予後良好な組織型のウィルムス腫瘍75例についての非負値行列因子分解(Non-negative Matrix Factorization、NMF)解析では、6つのクラスターが得られた。遺伝子発現データが利用可能なMLLT1変異腫瘍6例のうち、5例はクラスター3で、2例はCTNNB1変異を伴っていた。このクラスターには11p15のインプリンティングを保持する相当数の腫瘍も含まれており(すべてのMLLT1変異腫瘍を含む)、他のクラスターとは対照的に、ほとんどの症例で11p15のヘテロ接合性の消失またはインプリンティングの保持がみられた。miRNAPG変異のほぼすべての症例は、NMFクラスター2に含まれ、WT1、WTX、およびCTNNB1のほとんどの変異は、NMFクラスター3および4に含まれていた。Copyright © 2015 Perlman, E. J. et al.MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology wilms tumours. Nat. Commun. 6:10013 doi: 10.1038/ncomms10013 (2015).この論文は、http://creativecommons.org/licenses/by/4.0/に記載されている通り、Creative Commons Attribution 4.0 International Licenseの条件により、Macmillan Publishers Limitedの1部門であるNature Publishing Groupにより配布されている。 両側性ウィルムス腫瘍
ウィルムス腫瘍の個人の約5~10%は両側性または多中心性の腫瘍を有する。両側性病変の有病率は、遺伝的素因症候群の個人の方が素因症候群を認めない個人よりも高い。例えば、両側性ウィルムス腫瘍の545例において、真(bona fide)の生殖細胞病原性多様体は22%の患者で認められた。[ 120 ]最も一般的な素因多様体は、WT1変異と11p15のインプリンティングの消失である。[ 20 ][ 68 ]
WT1変異を伴う両側性ウィルムス腫瘍は小児患者の早期発症に関連があり(月齢10ヵ月での発症に対し、変異を伴わない患児では月齢39ヵ月)、エクソン8のWT1ナンセンス変異が高頻度で認められる。両側性ウィルムス腫瘍患者の3%に罹患した家族がいる。[ 121 ]
ウィルムス腫瘍の素因がある小児のスクリーニング
通常、ウィルムス腫瘍を発症する素因が有意に高い小児(例えば、ベックウィズ-ヴィーデマン症候群または他の過成長症候群、WAGR症候群、Denys-Drash症候群、散発性無虹彩症、または孤立性片側過形成のほとんどの小児)では、少なくとも8歳になるまで3ヵ月ごとに超音波検査によるスクリーニングを実施する。[ 78 ][ 122 ]早期で無症状の小型のウィルムス腫瘍が発見されることがあり、腎温存手術で切除できる可能性がある。[ 122 ]
それぞれの過成長症候群に対する腫瘍スクリーニングプログラムが提案されている。これらのプログラムは、発表されている年齢、腫瘍型の発生率、および2016 American Association for Cancer Research (AACR) Childhood Cancer Predisposition Workshopでの推奨に基づいていた。特定の症候群に対して遺伝的またはエピジェネティックなサブグループに基づく異なるがんリスクのデータが得られており、ヨーロッパではサブグループ別の推奨が発表されているが、こうした実践は米国で採用されていない。AACRワークショップ委員会により、ウィルムス腫瘍のリスクが1%を超えるすべての症候群に対して統一されたスクリーニングアプローチが提案された。ベックウィズ-ヴィーデマン症候群、18トリソミー、およびSimpson-Golabi-Behmel症候群の患者にはまた、血清αフェトプロテイン(AFP)測定および超音波検査による肝芽腫の追加スクリーニングも推奨されている。[ 123 ]
遺伝カウンセリング
ウィルムス腫瘍患者にみられる奇形の発現頻度から、遺伝カウンセリング、分子的および遺伝的探索ならびにフォローアップの必要性が強調される。
フランスの研究では、以下のいずれかに該当する患者は遺伝カウンセリングに紹介する必要があると結論された:[ 7 ]
奇形が認められないか小奇形が1つのみ存在する場合は、単純な腫瘍学的フォローアップの適応となる。[ 7 ]
以下に該当する患者には、遺伝カウンセリングの実施後にWT1変異の検査を考慮すべきである:
ベックウィズ-ヴィーデマン症候群の何らかの症状、片側過形成、または両側性もしくは家族性ウィルムス腫瘍を呈する患者の場合は、11p15の異常の検査を考慮すべきである。
ウィルムス腫瘍の臨床的特徴
ウィルムス腫瘍のほとんどの患者は無症状の腹部腫瘤を呈し、小児検診で親または小児科医により発見される。素因となる臨床症候群が確認されている小児では、ルーチンのスクリーニングで腎腫瘍が発見されることがある。他の所見には、以下のものがある:
ウィルムス腫瘍または他の腎悪性腫瘍を有する小児は、以下を契機に診療を受ける場合もある:
ウィルムス腫瘍の診断的評価および病期評価
ウィルムス腫瘍および他の小児腎腫瘍の診断と病期決定には、以下の検査および手技が用いられる:
- 身体診察と病歴聴取。腎腫瘤のある小児には、無虹彩症、発達遅滞、尿道下裂、停留精巣、仮性半陰陽、過成長、片側過形成など、関連する症候群の徴候を入念に評価する。
- 全血球算定(CBC)。
- 肝機能検査。
- 腎機能検査。
- 尿検査。
- 腹部画像検査。
- 胸部CTを最初に実施している場合は、胸部X線は不要である。
- 胸部CT。ウィルムス腫瘍で多くみられる転移部位は、肺および肝である。約15%の患者が肺転移を呈する。CTは、転移した肺結節の検出で最も高感度の方法である。
- フッ素18-フルオロデオキシグルコース(18F-FDG)-ポジトロン放射断層撮影(PET)-CT。ウィルムス腫瘍には18F-FDGが集積するため、18F-FDG PET-CT画像検査では従来のCTスキャン画像検査よりも多くの臨床適用可能な情報が得られる。PET-CTは、特に両側性腫瘍の患者または術前化学療法を受けた患者に有用な場合がある。18F-FDG PET-CTでは腫瘍および転移巣のFDG集積領域が強調されるが、この所見は組織学的に確認された活動性病変に対応する。[ 138 ]
- フォンウィルブランド病の精密検査。ウィルムス腫瘍を発症した患者の約1~8%には後天性のフォンウィルブランド病が認められるが、その多くは無症状である。フォンウィルブランド因子多量体はウィルムス腫瘍に結合するため、その血漿中濃度は低値となる。[ 139 ]一部の臨床医は、手術前にフォンウィルブランド病の評価を実施することを推奨している。
- 生検または切除および両側性ウィルムス腫瘍の問題。臨床的に切除可能なウィルムス腫瘍と思われる腎腫瘤のある小児では、生検中に腫瘍細胞が拡散する恐れがあるため、生検は行われていない。生検によって、そうした患者の病期がIII期に進行する場合がある。北米では、ほとんどの症例で最初の治療は一次的腎摘出術である。一次的腎摘出術を実施できない場合は、開腹または複数コアによる生検が必要となる。一次的腎摘出術に対する禁忌には、以下のものがある:
手術不能なウィルムス腫瘍から化学療法前に採取した生検組織は、組織学的検討と初期治療の決定に用いることができる。しかしながら、手術不能な腫瘍の組織型を判定するための生検の実施については、生検により局所の腫瘍が拡散する恐れがあり、ウィルムス腫瘍の組織学的分類は生検では判定できないため、依然として議論の余地がある。[ 140 ]
小児が最初の手技として生検を受ける場合、小児は肉眼的残存腫瘍を有するため、III期と考えられる。
臨床的にI期またはII期のウィルムス腫瘍とされる腎腫瘤のある小児では、生検中に腫瘍細胞が拡散する恐れがあるため、生検は行われていない。生検によって、そうした患者の病期がIII期に進行する場合がある。代わりに腎摘出術(北米)または化学療法(欧州)を実施する。そのため、病理学的診断は腎摘出標本を検査して初めて得られる。
両側性ウィルムス腫瘍を有する小児はしばしば、生検なしで治療される。[ 141 ]
腎腫瘤のX線像がウィルムス腫瘍の典型像と一致しない場合は、腎腫瘤の生検が適応となり、即時腎摘除術は施行されない。手術不能なウィルムス腫瘍から化学療法前に採取した生検組織は、組織学的検討と初期治療の決定に用いることができる。[ 140 ]
腫瘍の不均一性のために、いずれの生検標本においても退形成の組織型の検出は困難な場合がある。NWTS-4およびNWTS-5(COG-Q9401/NCT00002611)から得られたデータによると、ウィルムス腫瘍にみられる組織学的不均一性のために、かなりの数の患者が退形成の組織型でありながら、化学療法後の根治的手術の時点までそれと判明せず、事前の生検(針生検または切開生検のいずれでも)では見落とされること[ 142 ]が示されている点を認識しておくことが重要である。
ウィルムス腫瘍患児における対側腎病変の検出により、病期および患者の初期管理が変化する可能性があることから、先行して手術を行わない腎温存アプローチの役割が示されている。NWTS-4試験の結果を基にルーチンで行われる対側腎の術中検索はもはや推奨されないため、ベースライン画像での対側腎病変の検出は重要である。[ 133 ][ 136 ]最初の画像検査により、両側性の経過が示唆される場合、著者らは両側性ウィルムス腫瘍としての治療を推奨している。他の病変の起源が確定していない場合、著者らは、腎摘出術を進める前にその病変の病理学的評価を推奨している。[ 133 ][ 136 ]
両側性ウィルムス腫瘍の小児では、その小児が典型的な年齢でX線像で示されている場合は、生検を回避できる。このことは、189人中187人が生検なしで最初に治療されたCOG AREN0534(NCT00945009)研究で評価された。全員がウィルムス腫瘍を有した。6週間の治療後にRECIST1.1基準による奏効が30%未満の場合は、退形成、間質分化型、および横紋筋腫分化を評価するために両側生検が実施された。退形成が発見された場合は、化学療法の治療が変更された。他の2つが発見された場合は、さらなる化学療法により腫瘍の退縮が得られる可能性は低く、著者らは根治的手術を推奨した。[ 141 ]
- すべてのウィルムス腫瘍患者の病期を局所的に判定するには、リンパ節サンプリングが必要である。リンパ節は、短期と長期の両方の生存について予後的価値が大きいことが示されている。肉眼による検査は不正確なことで悪名高く、偽陰性率は31.3%および偽陽性率は18.1%である。[ 143 ]
臨床および画像所見に基づきウィルムス腫瘍と疑われた腎腫瘤のうち、約5%は他の病態と診断される。[ 144 ]
ウィルムス腫瘍が疑われる患者では、その他の術前病期診断検査を実施して、ウィルムス腫瘍の血管内進展または破裂について評価する。[ 135 ]
ウィルムス腫瘍の予後および予後因子
ウィルムス腫瘍は、患児の大半において治癒可能な腫瘍である。1980年代以降、予後良好な組織型のウィルムス腫瘍の5年生存率は常に90%を超えている。[ 147 ]治療期間、放射線量、照射範囲、放射線療法を受ける患者の割合はすべて減少しているにもかかわらず、このような良好な治療成績が得られている。[ 148 ]
ウィルムス腫瘍患者の予後は以下の因子に依存する:[ 149 ][ 150 ][ 151 ][ 152 ]
ウィルムス腫瘍の比較的年齢の高い青年および成人
16歳以上の患者のウィルムス腫瘍はまれであり、100万人当たりの年間発生率は0.2例未満である。[ 154 ]ヨーロッパでは、ウィルムス腫瘍の成人患者(15歳以上と定義される)の診断時の年齢中央値は34歳である;しかしながら、60歳以上の患者の報告がある。[ 154 ]ウィルムス腫瘍の3%は成人に発生する。ウィルムス腫瘍が成人の全腎腫瘍に占める割合は1%未満であり、最も多い成人の腎がんである腎細胞がんと考えられていたものに対する腎摘出術後の予想外の所見である場合がある。
青年および若年成人(AYA)患者(15~39歳)の転帰は小児の転帰より不良である。Surveillance, Epidemiology, and End Results(SEER)データベースのウィルムス腫瘍患者の解析において、AYA患者(n = 104)は小児患者(n = 2,574)よりも5年OS率が統計的に不良であった(69% vs 94%;P < 0.001)。[ 155 ][証拠レベル:3iA]小児試験で治療を受けた成人についてはより良好な結果が報告されている。National Wilms Tumor Study(NWTS)がNWTS-1、2、および3試験からのウィルムス腫瘍の成人患者の転帰について報告している。NWTS-1試験での成人の3年OS率は24%(対して小児では74%)であり、NWTS-3試験では5年OS率が82.6%に改善したが、各試験で治療を受けた成人患者数は31人以下であった。[ 156 ][ 157 ][ 158 ]これらのデータは、ウィルムス腫瘍の成人患者の多くが、適切な治療を受けた場合は治癒が期待できること、特に腫瘍が拡がっていない場合および/または完全に切除された場合はその可能性が高いことを示唆している。成人患者の転帰が劣っていることは、小児と成人の腫瘍の生物学的特性の相違、不正確な診断、不適切な病期決定(例えば、限局性疾患として病期分類されやすい、またはリンパ節サンプリングが行われないことが多い)、過小治療/遵守率の低さ(例えば、放射線療法の非実施)、内科腫瘍医および病理医が成人のウィルムス腫瘍に不慣れであること(誤診や診断の遅れにつながる可能性がある)、適切なリスク調整治療開始の遅れ、および成人用に特化された治療プロトコルの不在の結果である可能性がある。難治性または再発疾患の成人に対しては、腫瘍における潜在的な治療標的に対するスクリーニングを考慮すべきである。[ 159 ]
International Society of Pediatric Oncology(SIOP)およびCOGの腎腫瘍委員会による以下の推奨は、成人のウィルムス腫瘍患者の転帰を改善するための統一的アプローチを奨励している。[ 160 ]
ウィルムス腫瘍の組織学的所見
ウィルムス腫瘍が組織学的に診断された患者のほとんどは、現行の治療法で良好な経過を示すが、約10%の患者は予後不良と関連する病理組織学的特徴を有し、組織型によっては再燃率および死亡率が高い。ウィルムス腫瘍は腫瘍と腎臓の病理組織学的基準により、次の2つの予後グループに分類できる:
予後良好な組織型(FH)
ウィルムス腫瘍は、正常腎と組織学的に類似した三相型の発育を呈し、芽体細胞、上皮(小管)細胞、および間質細胞で構成される。すべての腫瘍が三相型であるわけではなく、一相型パターンがあると診断が困難になる場合もある。
組織学的特徴と予後または治療への反応性との関連が示唆されている一方で、これらの特徴のうち、退形成を除いて、北米の治療アルゴリズムで統計的有意性に達しているものはなく、そのため初期治療を指示するものはない。[ 161 ]
退形成の組織型
退形成の組織型は、ウィルムス腫瘍症例の約10%を占める。退形成の組織型は、ウィルムス腫瘍患者における反応と生存に関する単独で最も重要な組織学的予測因子である。比較的年齢の高い患者(10~16歳)に起こる腫瘍では、退形成の組織型の発生率が高い。[ 162 ]両側性腫瘍では、12~14%の割合で片側の腎臓に退形成の組織型がみられることが報告されている。[ 163 ][ 164 ]
退形成の診断を確定するには、以下の2つの組織学的基準を満たさなければならない:
TP53遺伝子の変異と一致する17p上の変化では、退形成組織型の病巣との関係が認められている。[ 109 ]巣状性退形成は、原発腫瘍内に退形成の明瞭な限局性領域が1ヵ所以上存在することと定義されている。これらの因子はすべて、別の遺伝子病変を獲得したウィルムス腫瘍細胞の亜集団から退形成が晩期イベントとして進展するという仮説を支持している。[ 165 ]巣状性退形成は、びまん性退形成ほど予後不良ではない。[ 151 ][ 166 ][ 167 ]
退形成は腫瘍の侵攻性よりもむしろ治療への反応性と最もよく相関する。退形成は、びまん性に分布し、進行期に同定される場合に不良な予後と最も一貫して関連する。これらの腫瘍は、予後良好な組織型のウィルムス腫瘍の小児に以前から用いられている化学療法に対する抵抗性が高い。[ 151 ]
nephrogenic rest(造腎組織遺残)
nephrogenic restは、異常に残留している胎生期の腎前駆細胞の細胞群である。nephrogenic restは非選択の小児剖検例の約1%、片側性ウィルムス腫瘍の腎の35%、および両側性ウィルムス腫瘍の腎のほぼ100%で検出される。[ 168 ][ 169 ]術前化学療法は、nephrogenic restの全有病率に影響しないようである。先天異常は、片側性ウィルムス腫瘍患者の9%および両側性ウィルムス腫瘍患者の33%を含めて、nephrogenic restを有する患者の12%で報告されている。[ 6 ]
腎芽腫症という用語は、びまん性または多巣性のnephrogenic restが存在することと定義されている。nephrogenic restは、遺残のカテゴリー(腎葉内または腎葉周囲のnephrogenic rest)および増殖期(初発または休眠状態のnephrogenic rest、過形成性のnephrogenic rest、退縮または硬化しているnephrogenic rest)によって下位分類できる。びまん性過形成腎葉周囲腎芽腫症は、一側または両側の腎臓周囲に厚い皮質を形成する腎芽腫症の固有の1カテゴリーであり、前腫瘍状態と考えられる。ウィルムス腫瘍とびまん性過形成腎葉周囲のnephrogenic restの鑑別は困難な場合があり、病変と周囲の腎実質との接合部を調べることが不可欠である。切開生検では、病変部と正常な腎実質との辺縁を含めない限り、診断価値がない。[ 170 ]
nephrogenic restの種類と割合は、片側性または両側性腫瘍の患者により異なる。両側性ウィルムス腫瘍の患者では、高い割合(52%)で腎葉周囲の遺残がみられ、腎葉内または両方の遺残がみられた割合(32%)を上回っていたほか、片側性腫瘍の患者(腎葉周囲が18%、腎葉内または両方が20%)よりも遺残の相対的割合が高かった。[ 81 ]腎葉内のnephrogenic restは、間質細胞型ウィルムス腫瘍および診断時年齢の低さに関連している。[ 6 ]
腎芽腫に対して切除された腎臓内にnephrogenic restを認めた患者は、nephrogenic restの種類に関係なく、残りの腎臓においても腫瘍形成リスクが高いとみなされる。このリスクは患者の年齢とともに低下する。[ 47 ]
両側性びまん性過形成腎葉周囲腎芽腫症では、一般にウィルムス腫瘍の発生リスクを低下させるために化学療法による治療が行われるが、それでもウィルムス腫瘍の発生リスクは高いままで、1件のシリーズでは55%とされている。[ 170 ]長期間にわたって化学療法による治療を受けている患者は、依然としてウィルムス腫瘍の発生リスクが高い状態にある。これらの患者にウィルムス腫瘍が発生すると、おそらくはこれらの症例では退形成の発生率が高く(症例の1/3を超える)、生き残った異常な腎細胞で退形成の発生と選択が起きた結果として、他の両側性ウィルムス腫瘍患者よりも予後が悪くなる。[ 170 ][ 171 ]
腎臓外のnephrogenic restはまれであるが、腎外性ウィルムス腫瘍に進行する恐れがある。[ 172 ]
ウィルムス腫瘍の病期情報
腫瘍の病期は、画像検査の結果と腎摘出時の手術および病理所見の両方により決定する。予後良好な組織型の腫瘍にも退形成の組織型の腫瘍にも同じ病期が適用される。したがって、病期情報は2つの基準によって分類される(例えば、予後良好な組織型のII期、または退形成の組織型のII期)。[ 161 ][ 173 ]
病期分類システムは、NWTSが最初に開発し、COGにより現在も使用されている。北米で用いられる病期分類システムおよび病期ごとの発生率の概要を以下に示す。[ 161 ]
I期
I期ウィルムス腫瘍(患者の43%)は、以下に挙げる基準をすべて満たしていなければならない:
非常にリスクの低いI期など、特定の治療プロトコルに適格な腫瘍では、所属リンパ節を顕微鏡で検査しなければならない。最も正確な病期を得るには、臨床的に異常なリンパ節がない場合でも、すべての患者に対してリンパ節サンプリングが強く推奨される。
II期
II期ウィルムス腫瘍(患者の20%)では、腫瘍が完全に切除され、切除辺縁またはその外側に腫瘍の証拠が認められない。腫瘍が腎臓を越えて進展していることは、以下の基準のいずれかにより証明される:
リンパ節サンプリングの結果がすべて陰性である。
腫瘍の生検など、側腹に限局した破裂または漏出は、現在、COG Renal Tumor Committee(COG RTC)によりIII期に含められる;ただし、このアプローチを裏付けるデータには異論がある。[ 140 ][ 174 ]
III期
III期ウィルムス腫瘍(患者の21%)では、術後、腹部に限局している残存性の非血液性腫瘍が認められる。以下のいずれか1つが生じる:
リンパ節浸潤および顕微鏡的残存腫瘍は、予後良好な組織型のIII期ウィルムス腫瘍患者の転帰を強く予測すると報告されている。[ 175 ]
IV期
IV期ウィルムス腫瘍(患者の11%)では、以下のいずれかに該当する:
副腎内の腫瘍の存在は転移とは判定されず、病期分類は他にあるすべての病期分類パラメータにより決定される。原発腫瘍には上記基準に従い局所的病期を割り当て、その病期で局所療法が決定される。例えば、ある患者はIV期で、局所はIII期の場合もある。
V期
V期ウィルムス腫瘍(患者の5%)は、診断時に腫瘍の両側性浸潤を呈する。現在のパラダイムでは、両側性ウィルムス腫瘍のすべての患者が最初の6週間または12週間に同じ治療を受ける。根治的手術後、治療は残りの腎臓の最も高い病期および治療後の病理学に基づいて実施される。[ 141 ]
ウィルムス腫瘍の治療
ウィルムス腫瘍に対する治療法選択肢の概要
ウィルムス腫瘍は比較的まれな腫瘍であるため、この腫瘍の患者はすべて登録して臨床試験に組み入れることを考えるべきである。至適治療法を決定し実施するには、ウィルムス腫瘍患児の治療経験を有するがん専門医(小児外科医および/または小児泌尿器科専門医、小児放射線腫瘍医、および小児腫瘍医)からなる集学的チームによる治療計画が必要である。
ウィルムス腫瘍の小児を治療するためのランダム化臨床研究の大半は、2つの大規模臨床グループ(COG RTCおよびSIOP)により実施されている。この2グループ間の違いは、病期判定および分類に影響を与える。ウィルムス腫瘍の治療に対する標準アプローチは2つある:治療の第一段階として、COG RTCはすべての片側性腫瘍に対して即時手術を、SIOPは術前化学療法を使用する。いずれのグループも術後化学療法を使用するが、例外として、化学療法を受けていない選択された症例、および進行期の選択された症例ではリスク調整アプローチにおいて放射線療法が使用される。
本要約ではNWTS(現在のCOG RTC)の結果および研究に焦点を当てている。
NWTS-1、NWTS-2、NWTS-3、NWTS-4、およびNWTS-5の主要な治療および研究に関する結論は次の通りである:
- 腫瘍がI期またはII期の予後良好な組織型の患児では、腎摘出術後にビンクリスチンおよびダクチノマイシンからなる併用化学療法を施行した場合、ルーチンの側腹への術後放射線療法は必要ない。[ 178 ]
- III期の予後良好な組織型の患者の予後が最も良好であるのは、(a)ダクチノマイシン、ビンクリスチン、ドキソルビシン、および10.8Gyの側腹への放射線療法、または(b)ダクチノマイシン、ビンクリスチン、および20Gyの側腹への放射線療法による治療を行った場合である。肉眼的残存病変を増加させる可能性のある広範な腹腔内病変または広範な腹腔内腫瘍漏出には、腹部全体への照射が適応となる。[ 178 ]
- IV期の予後良好な組織型の腫瘍を有する患者では、ビンクリスチン、ダクチノマイシン、およびドキソルビシンの併用に、プロトコル投与量のシクロホスファミド(10mg/kg/日、6週間ごとに3日間)を追加しても予後の改善はみられない。[ 178 ]
- ダクチノマイシンのコース当たりの単回投与量(I期~II期の予後良好な組織型、I期の退形成組織型、III期の予後良好な組織型、III期~IV期、またはI期~IV期の腎明細胞肉腫)は分割投与コースと同等であり、EFSは同じ結果で、より大きな用量強度が達成されるとともに、毒性と経費の軽減に関連している。[ 182 ]
- I期およびII期の予後良好な組織型の患者に対する治療は18週間で十分であり、III期およびIV期の患者に対しては、15ヵ月の治療の代わりに、6ヵ月の治療が行われることがある。[ 148 ][ 176 ][ 182 ][ 183 ][ 184 ]
- 1q増加は、予後良好な組織型の片側ウィルムス腫瘍における不良な生存に関係している。1q増加は最も強力な転帰の予測因子であり、1q増加が認められる状況では、1pまたは16qのいずれの欠失も重要ではない。予後良好な組織型の片側ウィルムス腫瘍で1q増幅がみられない状況における1pおよび/または16qの欠失は、ある程度の予後的意義を保持しており、高い再発リスクに関係している。[ 97 ][ 99 ]
手術
以下の手術の原則はNWTS試験から導き出されたものである:
- 外科医の最も重要な役割は、絶対に腫瘍を破裂させることなく完全に摘出し、腫瘍の進度を評価することである。経腹壁的手技または胸腹部切開術による根治的腎摘出術およびリンパ節サンプリングが選択手技である。[
185
]側腹切開は、腎臓の露出が限定的であるため実施しない。
腫瘍が切除可能な患者で、術前または術中の生検は、いずれも現在のCOG病期分類体系で腫瘍の病期を進行させる可能性があるため、実施されない。[ 185 ]
- 対側腎のルーチンの検索は、技術的に適切な画像検査により両側性の経過が示唆されない場合には必要でない。最初の画像検査により、両側腎の腫瘍が示唆される場合は、腎温存手術を促す治療法を採用すべきである。[ 136 ]
- ウィルムス腫瘍の約2%の症例は尿管浸潤を伴う。肉眼的血尿、無機能腎、または水腎症が認められるときは、腫瘍が尿管に達している可能性があるので膀胱鏡検査が推奨される。 腫瘍漏出を避けるために一塊切除術が望ましい。[ 186 ]
- 外科医は術中の漏出リスクに注意する必要があり、COG研究AREN03B2(NCT00898365)に登録された患者1,131人における術中漏出例のレビューに記載されているように、特に右側の大型腫瘍を有する患者では慎重を期すべきである。[ 187 ]
- 画像評価で明らかなIV期(例、肺転移)であっても、腎腫瘍の切除を考慮すべきである。遠隔転移の設定で、局所のI期またはII期のウィルムス腫瘍の治療に局所放射線療法は必要ない。
腎温存手術については議論の余地があり、以下に該当する小児を除いて推奨されない:[ 188 ][ 189 ];[ 190 ][証拠レベル:3iiB]
腎温存手術は、非常にリスクの低い腫瘍を有する患者であっても腎臓内に腫瘍があることから、診断時のほとんどの患者で実施可能ではないと考えられる。[ 192 ]北米では、片側性ウィルムス腫瘍に対して化学療法を施行して腫瘍容積を減量した後に行う腎温存手術(腎部分切除術)は、研究段階にあると考えられている。[ 193 ][ 194 ]
腎門および大動脈周囲のリンパ節サンプリングは、そのリンパ節が正常にみえても、実施が適切である。[ 185 ][ 195 ]さらに、浸潤が疑われるすべてのリンパ節を採取し精査すべきである。切除した断端部、残存した腫瘍、および転移浸潤が疑われるすべてのリンパ節に、チタン製クリップで標識を付ける。
ウィルムス腫瘍が隣接臓器に浸潤することはまれである;したがって、近隣臓器の切除が適応となることはめったにない。横隔膜を越えた他臓器と副腎の切除を含む、より広範な切除を実施した場合には、合併症の発生率が高くなる。この知見から、現在のCOGプロトコルでは、腎摘出術で追加臓器の切除が必要となる患者では初回生検、術前補助化学療法、その後に二次切除を検討すべきであると推奨されている。[ 196 ]肝転移の一次切除は推奨されない。[ 197 ]
化学療法
腎摘出術前の術前化学療法は、以下の状況で適応とされ、こうした状況は生検が必要な状況下で前述されている(詳しい情報については、本要約のウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと):[ 185 ][ 196 ][ 198 ][ 199 ][ 200 ][ 201 ]
術前化学療法は生検後に行う。生検は側腹部から実施してもよい。[ 145 ][ 202 ][ 203 ][ 204 ][ 205 ][ 206 ]正確な組織型評価および分子検査には十分な組織が不可欠である。退形成組織型でない限り、術前化学療法には、ビンクリスチンおよびダクチノマイシンに加えてドキソルビシンを含める;このような場合は、化学療法にレジメンIによる治療を含める(表2を参照のこと)。化学療法により、腫瘍のサイズおよび血管の供給が減少するため、一般に腫瘍の除去が容易になる:また、外科的合併症の発現頻度が減少する可能性もある。[ 140 ][ 145 ][ 196 ][ 198 ][ 207 ][ 208 ]
北米では、術前にcontained ruptureの所見を有する患者に対する術前化学療法の施行が術中の漏出を防止することが示唆されているが、これについては議論の余地がある。[ 209 ][ 210 ]CTによる後腹膜のcontained ruptureの術前診断は、経験を積んだ小児放射線科医でも困難である。[ 134 ]
化学療法を受ける新生児および生後12ヵ月未満の乳児では、より高い年齢の小児に比べて、化学療法の用量を50%に減量する必要がある。[ 211 ]乳児(生後12ヵ月未満)に対する用量は、体重キログラム当たりで計算し、体表面積当たりでは計算しない。この減量により、NWTS研究の対象となった乳児の同年齢群で報告されている毒性作用を軽減し、良好な全転帰を維持することができる。[ 212 ]
ウィルムス腫瘍の患児では肝毒性作用(過去に静脈閉塞疾患と呼ばれた洞様毛細血管閉塞症候群)が報告されているため、初期治療コース中はこれらの患者の肝機能検査値を綿密に監視すべきである。[ 213 ][ 214 ]ダクチノマイシンまたはドキソルビシンは、放射線療法を実施している間は投与してはならない。治療中に腎不全を発症する患者は、ビンクリスチン、ダクチノマイシン、およびドキソルビシンによる化学療法を継続して受けることができる。ビンクリスチンおよびドキソルビシンは全用量で投与可能である;しかしながら、ダクチノマイシンは重度の好中球減少症と関連している。これらの薬物の減量は不要な可能性があるが、患者が治療を受けている間は正確な薬理的および薬物動態学的検査が必要である。[ 215 ][ 216 ]
予後良好な組織型のウィルムス腫瘍で、1p/16qのヘテロ接合性の消失が認められる患者に対する治療の強化によりEFSが改善する。AREN0532(NCT00352534)およびAREN0533(NCT00379340)試験において、I期およびII期の予後良好な組織型のウィルムス腫瘍で、DD-4Aレジメン(ダクチノマイシン、ビンクリスチン、およびドキソルビシン)で治療された患者が87.3%の4年EFS率を示したのに対し、NWTS-5試験で治療されたI期およびII期患者に対する4年EFS率は68.8%(P = 0.042)であった。III期およびIV期疾患の患者は、レジメンMで治療された場合の4年EFS率が90.2%であったのに対し、NWTS-5試験で治療されたIII期およびIV期患者に対する4年EFS率は61.3%(P = 0.001)であった。4年生存率の改善傾向は、I期およびII期患者とIII期およびIV期患者でみられた。[ 217 ][証拠レベル:3iiiDi]
生検を実施する際、または局所腫瘍がIII期の場合には、腫瘍床への術後放射線療法が必要である。手術と放射線療法を受けたウィルムス腫瘍患者1,488人を対象にした研究において、手術後の放射線療法の開始を15日以上遅らせると、非転移性ウィルムス腫瘍患者に対する死亡リスクが増加した。[ 218 ][証拠レベル:3iiiA]
表2では、ウィルムス腫瘍を治療するための使用が受け入れられている化学療法レジメンを記述する。
表2.ウィルムス腫瘍に対して受け入れられている化学療法レジメン レジメン名 レジメンの記述 レジメンEE-4A[ ] 腎摘出術後18週間のビンクリスチン、ダクチノマイシン レジメンDD-4A[ ] 24週間のビンクリスチン、ダクチノマイシン、ドキソルビシン;ベースラインの腎摘出術、または生検とその後の腎摘出術 レジメンI[ ] 腎摘出術後24週間のビンクリスチン、ドキソルビシン、シクロホスファミド、エトポシド レジメンM[ ] ビンクリスチン、ダクチノマイシン、ドキソルビシン、シクロホスファミド、およびエトポシドとその後の放射線療法 レジメンUH1[ ] ビンクリスチン、ドキソルビシン、シクロホスファミド、カルボプラチン、およびエトポシド × 30週間 + 放射線療法 レジメンUH2[ ] ビンクリスチン、ドキソルビシン、シクロホスファミド、カルボプラチン、エトポシド、ビンクリスチン、およびイリノテカン × 36週間 + 放射線療法 放射線療法
放射線療法は局所制御を改善し、転移病変部位を治療するために用いられる。放射線療法は歴史的に病期と組織型に依存しているが、最近では腫瘍の分子署名も指針となっている。[ 221 ]
-
COGのアプローチ:
先行手術では組織型の確認と腫瘍の範囲が提供され、放射線療法を含む補助療法の理論的根拠が得られる。組織型に加えて、不良な局所制御に対する手術後の危険因子には:(1)不完全切除、(2)切除断端陽性、および(3)リンパ節転移がある。I期またはII期の予後良好な組織型のウィルムス腫瘍患者には放射線療法は使用されない。予後良好な組織型のウィルムス腫瘍患者について、III期腫瘍では治療に側腹または腹部放射線療法が用いられる。予後不良な組織型(巣状性退形成またはびまん性退形成)の症例では、すべての患者に対して側腹または腹部放射線療法が適応とされる。(詳しい情報については表3を参照のこと。)
表3.小児腫瘍学グループのAREN0532、AREN0533、およびAREN0321プロトコルにおける放射線療法のガイドライン 局所/局所領域病変 XRT = 放射線療法。 a1日1.5Gyの分割での全腹部放射線療法を必要とする。切除不能なびまん性腹腔内播種を有する患者は21Gyの照射を受ける。 b全肺照射は、1日1.5Gyの分割で実施される。 cすべての患者が放射線療法を受けるわけではない。 dブースト照射は肉眼的病変に実施される。 I期 II期 III期 III期(びまん性の漏出、腹膜転移、手術前の破裂)a 予後良好である組織型 放射線療法なし 放射線療法なし 10.8Gy 10.5Gy 巣状性退形成 10.8Gy 10.8Gy 10.8Gy 10.5Gy びまん性退形成 10.8Gy 10.8Gy 19.8Gy 10.5Gy + 9Gyのブースト照射 転移病変 IV期肺転移 IV期肝転移 IV期脳転移 IV期骨転移 予後良好である組織型 生後12ヵ月未満に対する10.5Gyb、c;生後12ヵ月超に対する12Gyb、c 19.8Gy +/- 5.4~10.8Gyのブースト照射d 16歳未満に対する21.6Gy + 10.8Gyのブースト照射;16歳超に対する30.6Gy 16歳未満に対する25.2Gy;16歳超に対する30.6Gy 巣状性またはびまん性の退形成 生後12ヵ月未満に対する10.5Gyb;生後12ヵ月超に対する12Gyb 19.8Gy +/- 5.4~10.8Gyのブースト照射d 16歳未満に対する21.6Gy + 10.8Gyのブースト照射;16歳超に対する30.6Gy 16歳未満に対する25.2Gy;16歳超に対する30.6Gy -
SIOPのアプローチ:
放射線療法が必要な小児は、以前のSIOP試験の経験に基づいて側腹および/または転移部位への術後治療を受ける。SIOP 1~9試験で、術前放射線療法または術前化学療法により、腫瘍漏出を起こす患者の割合が20%超から5%に低下したことが実証された。SIOP 5試験における術前放射線療法に対する術前化学療法の非劣性、および術前放射線療法に伴う二次悪性腫瘍の懸念から、SIOPでは標準の初回治療として術前化学療法を推奨するに至った。[ 179 ]時間の経過とともに、術後放射線療法で治療される小児の割合は、90%超からSIOP6~9試験、SIOP 93-01、およびSIOP-2001のそれぞれ15%および25%に低下した。[ 177 ]
I期ウィルムス腫瘍の治療
表4に、公表されている結果に基づいて、I期ウィルムス腫瘍患者に対する標準治療法の選択肢および生存データの概要を示す。
表4.I期ウィルムス腫瘍に対する標準治療法選択肢の概要a 組織型 4年RFSまたはEFS 4年OS 治療(化学療法レジメンの定義については DA = びまん性の退形成型;EFS = イベントフリー生存率;FA = 巣状性退形成型;FH = 予後良好な組織型;LOH = ヘテロ接合性の消失;OS = 全生存率;RFS = 無再燃生存率;XRT = 放射線療法。 a出典:Grundy et al.[ 99 ]、Shamberger et al.[ 152 ]、Fernandez et al.[ 221 ]、Dix et al.[ 217 ]、およびDaw et al.[ 226 ] b診断後4.12年経過時に1人の患者が肺に再燃を来した。 FHで、2歳未満/腫瘍重量550g未満 90% 100% リンパ節生検のみを含む手術 FHで、2歳以上/腫瘍重量550g以上 94%のRFS率 98% 腎摘出術 + リンパ節検体採取に続いてレジメンEE-4A 1p/16qのLOHを伴うFH(n = 8) 100%のEFS率 100% 腎摘出術 + リンパ節検体採取に続いてレジメンDD-4A FA 100% 100%(n = 8) 腎摘出術 + リンパ節検体採取に続いてレジメンDD-4AおよびXRT DA 100%b 100%(n = 10) 腎摘出術 + リンパ節検体採取に続いてレジメンDD-4AおよびXRT AREN0532(NCT00352534)試験において、診断時に2歳未満で腫瘍組織が550g未満であった予後良好な組織型のI期ウィルムス腫瘍の患者には、腎摘出術単独が適切な治療であるという仮説の妥当性が、COGにより確認された。NWTS-5試験では、診断時に2歳未満で腫瘍組織が550g未満であった予後良好な組織型のI期ウィルムス腫瘍の小児を対象として、この治療法が検討された。
証拠(診断時に2歳未満でI期の予後良好な組織型の腫瘍が550g未満の小児に対する手術単独):
- AREN0532(NCT00352534)試験は、NWTS-5の診断時年齢が2歳未満で、腫瘍重量が550g未満のI期予後良好である組織型(FH)のウィルムス腫瘍を有する小児について補助化学療法を省略できるという知見を確認するためにデザインされた。計116人の患者が非常にリスクの低いウィルムス腫瘍の基準を満たして、研究に登録された。[ 152 ][ 221 ][ 227 ]
- COGにより、すべての年齢の予後良好である組織型(上皮優勢型を示す)のI期ウィルムス腫瘍患者の転帰が報告された。AREN03B2に登録された予後良好である組織型のI期ウィルムス腫瘍の約20%が上皮優勢型であった。予後良好組織型である上皮優勢型のI期ウィルムス腫瘍患者177人のこの集団において、117人の患者がEE4Aで治療され、57人の患者は非常に低リスクのウィルムス腫瘍であると分類され、観察のみで治療された。[ 228 ][証拠レベル:3iiiA]
- AREN0321(NCT00335556)研究により、I期の退形成型ウィルムス腫瘍患者の治療成績はビンクリスチン/ダクチノマイシン療法へのドキソルビシンおよび側腹放射線療法の追加で改善されることが実証された。[ 226 ]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
II期ウィルムス腫瘍の治療
表5に、公表されている結果に基づいて、II期ウィルムス腫瘍患者に対する標準治療法の選択肢および生存データの概要を示す。
表5.II期ウィルムス腫瘍に対する標準治療法選択肢の概要a 組織型 4年RFSまたはEFS 4年OS 治療(化学療法レジメンの定義については DA = びまん性の退形成型;EFS = イベントフリー生存率;FA = 巣状性退形成型;FH = 予後良好な組織型;LOH = ヘテロ接合性の消失;OS = 全生存率;RFS = 無再燃生存率;XRT = 放射線療法。 a出典:Grundy et al.[ 99 ]、Dome et al.[ 151 ]、Dix et al.[ 217 ]、およびDaw et al.[ 220 ] FH 86%のRFS率 98% 腎摘出術 + リンパ節検体採取に続いてレジメンEE-4A 1p/16qのLOHを伴うFH(n = 24) 83%のEFS率 100% 腎摘出術 + リンパ節検体採取に続いてレジメンDD-4A FA 80%のEFS率 80%(n = 5) 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンDD-4A DA 84%のEFS 84%(n = 19) 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンUH1 NWTS-3、NWTS-4、およびNWTS-5では、術中に漏出が生じた患者は次の2つのグループに分類される:(1)腹腔全体に及ぶびまん性の漏出が生じた患者;(2)側腹に限局される局所漏出が生じた患者。びまん性の漏出が生じている患者には、腹部全体に対する放射線療法および3剤併用化学療法(ビンクリスチン、ダクチノマイシン、およびドキソルビシン)による治療が行われ、局所漏出がみられる患者にはビンクリスチンおよびダクチノマイシンのみによる治療が行われる。NWTS-3およびNWTS-4での治療を受けた患者の解析で、局所漏出が生じたII期の患者は、局所漏出が生じなかったII期の患者に比べてOSが低いことが指摘されたが、これに基づきCOG研究では、局所漏出の患者に対してドキソルビシンおよび側腹への放射線照射による治療が施されている。[ 229 ]このアプローチについては見解が分かれており、検証は行われていない;したがって、標準治療とするべきではない。
NWTS-4の予後良好な組織型のII期ウィルムス腫瘍患者499人を対象としたレビューでは、患者95人で腫瘍漏出が生じた。手術中に腫瘍漏出が生じ、ビンクリスチンおよびダクチノマイシンによる治療を受け、側腹への放射線療法は受けていない患者の8年RFS率およびOS率はそれぞれ75.7%および90.3%であり、腫瘍漏出が生じなかった患者における85%および95.6%より低かった。これらの相違はいずれも統計的に有意ではなかった。[ 174 ]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
III期ウィルムス腫瘍の治療
表6に、公表されている結果に基づいて、III期ウィルムス腫瘍患者に対する標準治療法の選択肢および生存データの概要を示す。
表6.III期ウィルムス腫瘍に対する標準治療法選択肢の概要a 組織型 4年RFSまたはEFS 4年OS 治療(化学療法レジメンの定義については DA = びまん性の退形成型;EFS = イベントフリー生存率;FA = 巣状性退形成型;FH = 予後良好な組織型;LOH = ヘテロ接合性の消失;OS = 全生存率;RFS = 無再燃生存率;XRT = 放射線療法。 a出典:Grundy et al.[ 99 ]、Dome et al.[ 151 ]、Fernandez et al.[ 230 ]、Dix et al.[ 217 ]、およびDaw et al.[ 220 ] FH(すべての患者) 88%のEFS率 97% 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンDD-4A FH(1pおよび/または16qのLOHを伴わない)およびリンパ節転移陽性 85%のEFS率 97% 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンDD-4A FH(1pおよび/または16qのLOHを伴わない)およびリンパ節転移陰性 97%のEFS率 99% 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンDD-4A FH(1pおよび16qのLOHを伴う)(n = 31) 87%のEFS率 94% 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンM FA 88%のRFS率 100%(n = 8) 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンDD-4A FA(術前治療) 71%のRFS率 71%(n = 7) レジメンDD-4Aによる術前治療に続いて腎摘出術 + リンパ節検体採取および腹部へのXRT DA 46%のEFS率 53%(n = 16) レジメンIによる術前治療に続いて腎摘出術 + リンパ節検体採取および腹部へのXRT DA 82%のEFS 91%(n = 23) 即時腎摘出術 + リンパ節検体採取に続いて腹部へのXRTおよびレジメンUH1 III期FHウィルムス腫瘍でCOG AREN0532プロトコルで治療された患者588人において、1pまたは16qにおけるヘテロ接合性の消失はEFSに影響するが、OSには影響しないことが示された。リンパ節転移の状態とヘテロ接合性の消失の状態を組み合わせると、両方が認められない場合に非常に良好なEFSおよびOSの強力な予測因子となり、4年EFS率は97%およびOS率は99%であった。[ 230 ][証拠レベル:2Di]リンパ節転移陽性と1pまたは16qにおけるヘテロ接合性の消失の両方が認められる患者に対する治療成績は不良となり、4年EFS率は74%であった。しかしながら、4年OS率は影響を受けず、92%であった。[ 230 ]これらの結果に基づいて、AREN0533試験に登録された患者について、1p/16qにおけるヘテロ接合性の消失を有する患者では治療が強化された。III期およびIV期ウィルムス腫瘍でヘテロ接合性の消失を有する患者は、レジメンMで治療された。4年EFS率は90.2%およびOS率は96.1%であったのに対し、NWTS-5試験の患者に対する4年EFS率は61.3%(P = 0.001)および4年OS率は86.0%(P = 0.087)であった。生存率の改善が示唆された;しかしながら、この研究は生存率の差を判定する検出力を有していなかった。[ 217 ][証拠レベル:3iiiDi]
非転移性ウィルムス腫瘍患者に対する放射線療法の早期開始は、集学的治療法のきわめて重要な要素である。手術と放射線療法を受けたウィルムス腫瘍患者1,488人を対象にしたレビューにおいて、手術と放射線療法の間隔が14日を超えると、死亡リスクが増加した(ハザード比、2.13;P = 0.013)。このことは、手術から14日以内に放射線療法を開始することの重要性を強調しており、ウィルムス腫瘍の治療プロトコルで規定されている。[ 218 ][証拠レベル:3iiiA]
純粋に局所的漏出に基づいてIII期に分類される患者については、本要約のII期ウィルムス腫瘍の治療のセクションを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
IV期ウィルムス腫瘍の治療
表7に、公表されている結果に基づいて、IV期ウィルムス腫瘍患者に対する標準治療法の選択肢および生存データの概要を示す。
表7.IV期ウィルムス腫瘍に対する標準治療法選択肢の概要a 組織型 4年RFSまたはEFS 4年OS 治療(化学療法レジメンの定義については CR = 完全奏効;DA = びまん性の退形成;EFS = イベントフリー生存率;FA = 巣状性退形成;FH = 予後良好な組織型;LOH = ヘテロ接合性の消失;OS = 全生存率;RFS = 無再燃生存率;XRT = 放射線療法。 a出典:Grundy et al.[ 99 ]、Dome et al.[ 151 ]、Dix et al.[ 219 ]、Dix et al.[ 217 ]、およびDaw et al.[ 220 ] b腹部へのXRTは腎腫瘍の局所的病期に基づいて計画される。 c肺へのXRTは、胸部X線/胸部コンピュータ断層撮影で肺転移の証拠を認める患者にのみ実施する。 d詳しい情報については、AREN0533(NCT00379340)研究を参照のこと。 FH 76%のRFS率 86% 腎摘出術 + リンパ節検体採取の後、腹部へのXRTb、転移部位への放射線照射、両側肺へのXRTc、およびレジメンDD-4A FH(孤立性の肺結節を伴う) 85%のEFS率 96% 腎摘出術 + リンパ節検体採取の後、腹部へのXRTb、+/-両側肺へのXRTc、およびレジメンDD-4AまたはレジメンMd FH(孤立性の肺結節を伴い、DD-4Aに対してCRを達成) 83%のEFS率 94% 腎摘出術 + リンパ節検体採取に続いて腹部へのXRTbおよびレジメンDD-4A FH(孤立性の肺結節を伴い、DD-4Aに対して完全奏効を未達成) 92%のEFS率 96% 腎摘出術 + リンパ節検体採取の後、腹部へのXRTbと両側肺へのXRTc、およびレジメンM FH(1pおよび/または16qのLOHを伴う)(n = 20) 95%のEFS率 100% 腎摘出術 + リンパ節検体採取の後、腹部へのXRTb、転移部位への放射線照射b、およびレジメンM FA 61%のEFS率 72%(n = 11) 腎摘出術 + リンパ節検体採取の後、腹部へのXRTb、転移部位への放射線照射、両側肺へのXRTc、およびレジメンDD-4A DA 33%のEFS率 33%(n = 10) 即時腎摘出術 + リンパ節検体採取の後、腹部へのXRTb、転移部位への放射線照射、肺全体へのXRTc、およびレジメンI DA(術前治療) 60%のEFS 70%(n = 10) レジメンUH2による術前治療に続いて腎摘出術 + リンパ節検体採取の後、腹部へのXRTb、転移部位への放射線照射、および肺全体へのXRTc IV期の腫瘍は、肺、肝、骨、または脳への血行性転移の存在によって定義され、最も多い転移部位は肺である。過去には肺転移の検出に胸部X線が使用されていた。CTが導入されたことで、胸部X線では認められなかった肺結節が胸部CTにより多くの患者に検出されたため論争が生じた。CT検査のみで肺結節が検出され(胸部X線では陰性)、新たに予後良好な組織型のウィルムス腫瘍と診断された患者の管理では、急性および晩期毒性を伴う強化療法を追加する必要があるかどうかについての議論が生じた。
証拠(胸部CTでのみ検出される肺結節の治療):
- NWTS-4およびNWTS-5に参加し、CTのみで肺結節が検出された患者186人を対象としたレトロスペクティブなレビューで、ドキソルビシン、ビンクリスチン、ダクチノマイシンを投与する治療と、2薬のみを投与する治療との比較結果が報告された。[ 231 ]
欧州の複数のレトロスペクティブ研究で、胸部X線により肺転移が診断された患者に対し、肺への放射線療法を省略した場合の影響が調査された。欧州の研究者らは、SIOP-93-01(NCT00003804)試験で治療を受け、胸部X線で肺転移が同定されたウィルムス腫瘍患者のほとんどに対する治療において、放射線療法を省略した。欧州での腎腫瘍に対するアプローチは北米でとられる手法とは異なっている。画像検査で腎腫瘍の存在が判明している患者全員に対し、腎摘出術に先立ってビンクリスチン、ダクチノマイシン、ドキソルビシンによる9週間の化学療法が実施された。
証拠(肺への放射線照射の省略):
- レトロスペクティブSIOP研究では、新たにウィルムス腫瘍と診断され肺転移が認められた患者234人に対し、腎摘出術前の化学療法に対する肺転移巣の反応に応じた治療が行われた。[
232
]
- 治療の6週間後に完全寛解を示した患者(67%)には同じ化学療法を継続し、肺への放射線療法は必要とされなかった。
- 肺転移巣が残存する患者は、転移巣切除について評価された。
- 切除が不完全または手術不能で肺転移巣が残存していた患者には、9週間にわたってイホスファミド/アントラサイクリンとカルボプラチン/エトポシドを交互に繰り返す、より積極的な化学療法が実施された。
- COG AREN0533(NCT00379340)研究では、予後良好な組織型のウィルムス腫瘍と孤立性の肺転移がみられる患者に対して、ヨーロッパでの経験に基づいて肺への放射線照射に対する曝露を低下させながらEFSを改善するための新たな戦略が適用された。治療法は、肺結節の反応および1pおよび16qにおける腫瘍特異的なヘテロ接合性の消失に基づいて調整された。[ 219 ][証拠レベル:3iiiDi]
COGの試験では、治療で肺への放射線が保留された患者がヨーロッパの試験よりも少なかったものの、これらの研究間にはいくつかの違いがあるために、直接比較できない点に留意することが重要である。[ 219 ][ 232 ]ヨーロッパの患者は、北米の患者よりも肺転移の再評価前にダクチノマイシンおよびドキソルビシンの投与間隔を狭めた(dose-dense)レジメンを受ける(ヨーロッパではダクチノマイシン、135ug/kgおよびドキソルビシン、100mg/m2であったのに対し、北米ではダクチノマイシン、45ug/kgおよびドキソルビシン、45mg/m2)。ヨーロッパの研究では、化学療法または肺の転移巣切除により完全寛解が達成された患者に対して肺への放射線療法の省略が可能である一方で、米国では化学療法単独で完全寛解が達成された患者でのみ放射線療法が省略された。ヨーロッパの研究では、画像検査が中央で評価されなかった一方、米国では、中央で評価され、AREN0533(NCT00379340)試験では、完全寛解の定義がより厳格となっている可能性がある。
IV期ウィルムス腫瘍の患者にとって、診断時における肝転移の存在は独立した予後不良因子ではない。[ 197 ]
AREN0321(NCT00335556)研究において、びまん性退形成型ウィルムス腫瘍で測定可能な病変を有する患者に対してビンクリスチンおよびイリノテカンの併用(VI)が初期治療段階で検証された。びまん性退形成型IV期ウィルムス腫瘍で測定可能な病変を有する患者14人が治療域療法(window therapy)を受けた;1人の患者が完全奏効(CR)を達成し、10人の患者が部分奏効(PR)を達成し、疾患の安定が得られた患者はいなかった。これにより、CRおよびPR率は79%となった。治療域療法でVIに反応した患者は、受けたレジメン(UH2)にVIが組み込まれていた。この試験で心/肺毒性が観察されたため、研究は妨げられ、ドキソルビシン、シクロホスファミド、およびエトポシド(カルボプラチンと併用される場合)の用量を低下するように修正された。びまん性退形成型ウィルムス腫瘍を新たに診断された患者において、修正されたレジメンのさらなる研究が計画されている。[ 220 ][証拠レベル:3iiiDii]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
V期のウィルムス腫瘍および両側性ウィルムス腫瘍の発症素因がある患者の治療
現在、V期ウィルムス腫瘍(診断時の両側性ウィルムス腫瘍)の治療およびウィルムス腫瘍を発症する素因を有する患者に対する標準のアプローチは存在しない;しかしながら、両側性ウィルムス腫瘍を有する患者の治療に関する1件のプロスペクティブ研究が初めて完了しており、このアプローチのガイダンスが提供されている。[ 141 ]
両側性ウィルムス腫瘍の患児の管理は、きわめて困難である。治療の目的は、すべての腫瘍を根治すること、および可能な限り多くの正常腎組織を温存することであり、これらを満たした患児では慢性腎不全のリスクが減少することが期待される。[ 233 ]
歴史的に、NWTS-4およびNWTS-5試験とヨーロッパで実施された試験によると、両側性ウィルムス腫瘍患者は限局性ウィルムス腫瘍の患者と比べてEFSおよびOSが低かった。NWTS-4研究の報告によると、予後良好な組織型の両側性ウィルムス腫瘍患者では、8年EFS率が74%、OS率が89%であった;退形成の組織型の患者では、EFS率が40%、OS率が45%であった。[ 164 ]NWTS-5研究の報告によると、すべての両側性ウィルムス腫瘍患者に対する4年EFS率が56%、OS率が81%であった;4年EFS率は予後良好な組織型の患者(65%)、巣状性退形成組織型の患者(76%)、およびびまん性退形成組織型の患者(25%)についても報告された。[ 99 ][ 151 ]欧州でも、両側性ウィルムス腫瘍患者に関する同様の転帰が報告されている。[ 163 ][ 234 ]オランダの単一施設の研究(N = 41)では、重大な合併症として腎不全(32%)および二次腫瘍(20%)が認められた。[ 234 ]オランダの研究における末期腎不全の高い発生率は、追跡調査期間が他より長いことを反映している可能性がある。
V期のウィルムス腫瘍に対する治療法の選択肢には、以下のものある:
両側性ウィルムス腫瘍に対する術前化学療法および切除
両側性ウィルムス腫瘍の患者に対する治療の目標は全体的な転帰を損なうことなく可能な限り多くの腎組織を保存することである。このアプローチは、基礎にある生殖細胞系の遺伝子異常のほか、腎組織の治療関連の機能喪失が原因の可能性がある末期の腎疾患の晩期合併症(晩期障害)を回避するために用いられる。末期の腎疾患は、片側性ウィルムス腫瘍の患者(1%未満)よりも両側性ウィルムス腫瘍の患者(非症候性の小児で12%)においての方が頻繁に発生する。腎機能の転帰は両側腎温存手術後は他の種類の手術後よりもかなり良好である。[ 141 ]
従来、患者に対して両側の腎生検と各腎の病期判定が実施され、続いて術前化学療法が行われてきた。最初のプロスペクティブ多施設治療試験(COG AREN0534[NCT00945009])では、画像検査の結果がウィルムス腫瘍と一致する場合、治療前生検は必要とされなかった。[ 141 ]ウィルムス腫瘍以外の腎腫瘍の両側での発生率は非常に低いため、このアプローチが採用された。また、針生検およびくさび状生検は、ウィルムス腫瘍の退形成を同定する上で成功率が高いとはいえない。[ 142 ]10歳以上の年齢や非定型的な画像的特徴などのまれな臨床状況の設定において、ウィルムス腫瘍以外の診断を考慮すべき場合は、組織診断が実施される。[ 141 ]
術前化学療法で治療する患者には、4~8週間後に腫瘍の病状を評価することが必要である。臨床試験で治療を受けていない患者では、微小な縮小が化学療法に誘発された分化を反映していることもあれば、退形成組織型を反映していることもあるため、生検または切除を行う理想の時期は明らかでない。明らかに切除不能な腫瘍に対して計画された切除または生検は、診断の12週間後までに試行する。両側性ウィルムス腫瘍患者において腫瘍の病状を評価せずに治療を継続すると、退形成組織型または化学療法に誘発された分化(横紋筋腫分化など)を見落とすことがあり、腫瘍制御のための追加の有益性が得られずに患者への毒性作用が増加する可能性がある。退形成組織型の腫瘍は両側性ウィルムス腫瘍患者の10%に生じ、化学療法への反応が不良である。[ 164 ]
退形成組織型の診断を確定した後、完全切除を実施する。この診断を組織学的に確定することは容易ではない。NWTS-4の患者27人のシリーズでは、20例(74%)で病理の不一致(片側退形成腫瘍)が認められ、両側腎から組織を得る必要が強調されている。後にびまん性の退形成型腫瘍を有することが判明した7人の患者は、診断を確定するためにコア生検を受けたが、退形成は明らかにならなかった。退形成は、開腹くさび状生検を実施した場合、患者9人中わずか3人で同定され、腎部分摘除術または腎全摘術を受けた患者では9人中7人で同定された。[ 164 ]
生検またはセカンドルック手術後の化学療法および/または放射線療法の実施は、初期治療に対する腫瘍の反応に応じて決定する。初期治療に対する反応が不十分であることが二次治療で観察された患者、または退形成が認められる場合は、より積極的な治療が必要である。[ 173 ][ 235 ][ 236 ]
末期の腎疾患は両側性ウィルムス腫瘍患者において臨床的に最も重篤な罹病であり、基礎にある生殖細胞系の遺伝子異常のほか、腎組織の治療関連の機能喪失が原因の可能性がある。両側性ウィルムス腫瘍に対する治療後は、腎機能の長期モニタリングが必要である。
証拠(両側性ウィルムス腫瘍に対する術前化学療法および切除):
- 両側性ウィルムス腫瘍における最初のプロスペクティブ研究(AREN0534[NCT00945009])は、腎組織を温存しながら術前化学療法を強化し(ビンクリスチン、ダクチノマイシン、およびドキソルビシンの3剤を利用する)、診断から12週間以内に根治的手術を完了し、組織学的奏効に基づいて術後化学療法を修正することでEFSおよびOSの改善を目指した。[ 141 ]
- Associazione Italiana di Ematologia e Oncologia Pediatrica(AIEOP)の施設に登録された両側性ウィルムス腫瘍の小児93人を対象にした21年間にわたるレトロスペクティブ・レビューにおいて、43人の患者が術前にビンクリスチンおよびダクチノマイシンで治療され、37人の患者がビンクリスチン、ダクチノマイシン、およびドキソルビシンで治療された。術前化学療法の期間は1~40週間であった(中央値、12週間)。[ 163 ]
- SIOP-93-01(NCT00003804)ガイドラインに従って術前療法を受けたウィルムス腫瘍の患者49人を対象にしたレトロスペクティブ・レビューにおいて、手術の時期は腫瘍退縮の証拠が画像検査でもはや確認されない場合と決定された。腎温存手術前の平均治療期間は80日であった。[ 237 ]
- St. Jude Children's Research Hospitalのレトロスペクティブ・レビューでは、研究者らが、予後良好な組織型の両側性ウィルムス腫瘍の小児に対する術前化学療法とその後の腎温存手技の経験について記述している。[ 238 ]
腎移植
V期ウィルムス腫瘍の患児では、ほとんどの再燃が診断後2年以内に発生するため、通常は悪性腫瘍の証拠がみられずに1~2年が経過するまで腎移植は延期される。[ 240 ]同様に、全例で両側腎摘出術が必要となるDenys-Drash症候群とウィルムス腫瘍を併発する患児に対する腎移植も、一般に初期治療の完了後1~2年は延期される。[ 240 ]
臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
(再発疾患に関する情報については、本要約の再発小児腎腫瘍の治療のセクションを参照のこと。)
治療後のフォローアップ
ウィルムス腫瘍の治療が完了し、両側性ウィルムス腫瘍などの遺伝的素因に一致する特徴を呈している患者には、スクリーニングとして、特定の症候群のリスクが高い期間(WT1関連症候群の場合は5年;ベックウィズ-ヴィーデマン症候群の場合は8年)にわたり、3ヵ月ごとに異時性腫瘍に対する腎臓超音波検査を実施する。
ウィルムス腫瘍の治療後の晩期合併症(晩期障害)
ウィルムス腫瘍の治療を受けた患児では、以下の問題の発生リスクが高い:
(小児および青年におけるがん治療の晩期合併症(晩期障害)に関する詳しい考察については、小児がん治療の晩期合併症(晩期障害)のPDQ要約を参照のこと。)
参考文献- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2016. Bethesda, Md: National Cancer Institute, 2019. Available online. Last accessed February 27, 2020.[PUBMED Abstract]
- Breslow N, Olshan A, Beckwith JB, et al.: Epidemiology of Wilms tumor. Med Pediatr Oncol 21 (3): 172-81, 1993.[PUBMED Abstract]
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. Bethesda, Md: National Cancer Institute, 2009. Also available online. Last accessed January 31, 2020.[PUBMED Abstract]
- Scott RH, Stiller CA, Walker L, et al.: Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J Med Genet 43 (9): 705-15, 2006.[PUBMED Abstract]
- Narod SA, Hawkins MM, Robertson CM, et al.: Congenital anomalies and childhood cancer in Great Britain. Am J Hum Genet 60 (3): 474-85, 1997.[PUBMED Abstract]
- Vujanić GM, Apps JR, Moroz V, et al.: Nephrogenic rests in Wilms tumors treated with preoperative chemotherapy: The UK SIOP Wilms Tumor 2001 Trial experience. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- Dumoucel S, Gauthier-Villars M, Stoppa-Lyonnet D, et al.: Malformations, genetic abnormalities, and Wilms tumor. Pediatr Blood Cancer 61 (1): 140-4, 2014.[PUBMED Abstract]
- Gracia Bouthelier R, Lapunzina P: Follow-up and risk of tumors in overgrowth syndromes. J Pediatr Endocrinol Metab 18 (Suppl 1): 1227-35, 2005.[PUBMED Abstract]
- Lapunzina P: Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 137 (1): 53-71, 2005.[PUBMED Abstract]
- Treger TD, Chowdhury T, Pritchard-Jones K, et al.: The genetic changes of Wilms tumour. Nat Rev Nephrol 15 (4): 240-251, 2019.[PUBMED Abstract]
- Clericuzio CL: Clinical phenotypes and Wilms tumor. Med Pediatr Oncol 21 (3): 182-7, 1993.[PUBMED Abstract]
- Fischbach BV, Trout KL, Lewis J, et al.: WAGR syndrome: a clinical review of 54 cases. Pediatrics 116 (4): 984-8, 2005.[PUBMED Abstract]
- Breslow NE, Norris R, Norkool PA, et al.: Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol 21 (24): 4579-85, 2003.[PUBMED Abstract]
- Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al.: The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms' tumor. Hum Mutat 13 (2): 146-53, 1999.[PUBMED Abstract]
- Koziell AB, Grundy R, Barratt TM, et al.: Evidence for the genetic heterogeneity of nephropathic phenotypes associated with Denys-Drash and Frasier syndromes. Am J Hum Genet 64 (6): 1778-81, 1999.[PUBMED Abstract]
- Royer-Pokora B, Beier M, Henzler M, et al.: Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A 127 (3): 249-57, 2004.[PUBMED Abstract]
- Pelletier J, Bruening W, Kashtan CE, et al.: Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67 (2): 437-47, 1991.[PUBMED Abstract]
- Mueller RF: The Denys-Drash syndrome. J Med Genet 31 (6): 471-7, 1994.[PUBMED Abstract]
- Barbaux S, Niaudet P, Gubler MC, et al.: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17 (4): 467-70, 1997.[PUBMED Abstract]
- Porteus MH, Narkool P, Neuberg D, et al.: Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms' tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 18 (10): 2026-31, 2000.[PUBMED Abstract]
- Rump P, Zeegers MP, van Essen AJ: Tumor risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am J Med Genet A 136 (1): 95-104, 2005.[PUBMED Abstract]
- Choufani S, Shuman C, Weksberg R: Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 163C (2): 131-40, 2013.[PUBMED Abstract]
- Eggermann T, Algar E, Lapunzina P, et al.: Clinical utility gene card for: Beckwith-Wiedemann Syndrome. Eur J Hum Genet 22 (3): , 2014.[PUBMED Abstract]
- Ibrahim A, Kirby G, Hardy C, et al.: Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics 6 (1): 11, 2014.[PUBMED Abstract]
- Brioude F, Lacoste A, Netchine I, et al.: Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr 80 (6): 457-65, 2013.[PUBMED Abstract]
- Mussa A, Russo S, Larizza L, et al.: (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet 89 (4): 403-415, 2016.[PUBMED Abstract]
- Green DM, Breslow NE, Beckwith JB, et al.: Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol 21 (3): 188-92, 1993.[PUBMED Abstract]
- DeBaun MR, Siegel MJ, Choyke PL: Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. J Pediatr 132 (3 Pt 1): 401-4, 1998.[PUBMED Abstract]
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.[PUBMED Abstract]
- Milani D, Pezzani L, Tabano S, et al.: Beckwith-Wiedemann and IMAGe syndromes: two very different diseases caused by mutations on the same gene. Appl Clin Genet 7: 169-75, 2014.[PUBMED Abstract]
- Morris MR, Astuti D, Maher ER: Perlman syndrome: overgrowth, Wilms tumor predisposition and DIS3L2. Am J Med Genet C Semin Med Genet 163C (2): 106-13, 2013.[PUBMED Abstract]
- Astuti D, Morris MR, Cooper WN, et al.: Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44 (3): 277-84, 2012.[PUBMED Abstract]
- Golabi M, Leung A, Lopez C: Simpson-Golabi-Behmel Syndrome Type 1. In: Pagon RA, Adam MP, Bird TD, et al., eds.: GeneReviews. Seattle, Wash: University of Washington, 1993-2018, pp. Available online. Last accessed June 08, 2020.[PUBMED Abstract]
- Peterman CM, Fevurly RD, Alomari AI, et al.: Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr Blood Cancer 64 (12): , 2017.[PUBMED Abstract]
- Fagali C, Kok F, Nicola P, et al.: MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur J Med Genet 52 (5): 333-6, 2009 Sep-Oct.[PUBMED Abstract]
- Isidor B, Bourdeaut F, Lafon D, et al.: Wilms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet 21 (7): 784-7, 2013.[PUBMED Abstract]
- Cairney AE, Andrews M, Greenberg M, et al.: Wilms tumor in three patients with Bloom syndrome. J Pediatr 111 (3): 414-6, 1987.[PUBMED Abstract]
- Hartley AL, Birch JM, Tricker K, et al.: Wilms' tumor in the Li-Fraumeni cancer family syndrome. Cancer Genet Cytogenet 67 (2): 133-5, 1993.[PUBMED Abstract]
- Bourdeaut F, Guiochon-Mantel A, Fabre M, et al.: Alagille syndrome and nephroblastoma: Unusual coincidence of two rare disorders. Pediatr Blood Cancer 50 (4): 908-11, 2008.[PUBMED Abstract]
- Russell B, Johnston JJ, Biesecker LG, et al.: Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet A 167A (9): 2122-31, 2015.[PUBMED Abstract]
- Bonaïti-Pellié C, Chompret A, Tournade MF, et al.: Genetics and epidemiology of Wilms' tumor: the French Wilms' tumor study. Med Pediatr Oncol 20 (4): 284-91, 1992.[PUBMED Abstract]
- Winther JF, Sankila R, Boice JD, et al.: Cancer in siblings of children with cancer in the Nordic countries: a population-based cohort study. Lancet 358 (9283): 711-7, 2001.[PUBMED Abstract]
- Breslow NE, Olson J, Moksness J, et al.: Familial Wilms' tumor: a descriptive study. Med Pediatr Oncol 27 (5): 398-403, 1996.[PUBMED Abstract]
- Li FP, Williams WR, Gimbrere K, et al.: Heritable fraction of unilateral Wilms tumor. Pediatrics 81 (1): 147-9, 1988.[PUBMED Abstract]
- Ruteshouser EC, Huff V: Familial Wilms tumor. Am J Med Genet C Semin Med Genet 129 (1): 29-34, 2004.[PUBMED Abstract]
- Paulino AC, Thakkar B, Henderson WG: Metachronous bilateral Wilms' tumor: the importance of time interval to the development of a second tumor. Cancer 82 (2): 415-20, 1998.[PUBMED Abstract]
- Coppes MJ, Arnold M, Beckwith JB, et al.: Factors affecting the risk of contralateral Wilms tumor development: a report from the National Wilms Tumor Study Group. Cancer 85 (7): 1616-25, 1999.[PUBMED Abstract]
- Grundy P, Koufos A, Morgan K, et al.: Familial predisposition to Wilms' tumour does not map to the short arm of chromosome 11. Nature 336 (6197): 374-6, 1988.[PUBMED Abstract]
- Little SE, Hanks SP, King-Underwood L, et al.: Frequency and heritability of WT1 mutations in nonsyndromic Wilms' tumor patients: a UK Children's Cancer Study Group Study. J Clin Oncol 22 (20): 4140-6, 2004.[PUBMED Abstract]
- Hanks S, Perdeaux ER, Seal S, et al.: Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 5: 4398, 2014.[PUBMED Abstract]
- Scott RH, Douglas J, Baskcomb L, et al.: Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 40 (11): 1329-34, 2008.[PUBMED Abstract]
- Grønskov K, Olsen JH, Sand A, et al.: Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet 109 (1): 11-8, 2001.[PUBMED Abstract]
- Clericuzio C, Hingorani M, Crolla JA, et al.: Clinical utility gene card for: WAGR syndrome. Eur J Hum Genet 19 (4): , 2011.[PUBMED Abstract]
- Hoyme HE, Seaver LH, Jones KL, et al.: Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79 (4): 274-8, 1998.[PUBMED Abstract]
- Shanske AL: Trisomy 18 in a second 20-year-old woman. Am J Med Genet A 140 (9): 966-7, 2006.[PUBMED Abstract]
- Reid S, Renwick A, Seal S, et al.: Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 42 (2): 147-51, 2005.[PUBMED Abstract]
- Hirsch B, Shimamura A, Moreau L, et al.: Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 103 (7): 2554-9, 2004.[PUBMED Abstract]
- Reid S, Schindler D, Hanenberg H, et al.: Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39 (2): 162-4, 2007.[PUBMED Abstract]
- Gadd S, Huff V, Walz AL, et al.: A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49 (10): 1487-1494, 2017.[PUBMED Abstract]
- Wegert J, Wittmann S, Leuschner I, et al.: WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48 (12): 1102-11, 2009.[PUBMED Abstract]
- Ruteshouser EC, Robinson SM, Huff V: Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47 (6): 461-70, 2008.[PUBMED Abstract]
- Walz AL, Ooms A, Gadd S, et al.: Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27 (2): 286-97, 2015.[PUBMED Abstract]
- Wegert J, Ishaque N, Vardapour R, et al.: Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27 (2): 298-311, 2015.[PUBMED Abstract]
- Rakheja D, Chen KS, Liu Y, et al.: Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802, 2014.[PUBMED Abstract]
- Torrezan GT, Ferreira EN, Nakahata AM, et al.: Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039, 2014.[PUBMED Abstract]
- Dome JS, Huff V: Wilms Tumor Predisposition. In: Pagon RA, Adam MP, Bird TD, et al., eds.: GeneReviews. Seattle, Wash: University of Washington, 1993-2018, pp. Available online. Last accessed June 08, 2020.[PUBMED Abstract]
- Mahamdallie SS, Hanks S, Karlin KL, et al.: Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet 47 (12): 1471-4, 2015.[PUBMED Abstract]
- Huff V: Wilms tumor genetics. Am J Med Genet 79 (4): 260-7, 1998.[PUBMED Abstract]
- Scott RH, Murray A, Baskcomb L, et al.: Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 3 (3): 327-35, 2012.[PUBMED Abstract]
- Corbin M, de Reyniès A, Rickman DS, et al.: WNT/beta-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosomes Cancer 48 (9): 816-27, 2009.[PUBMED Abstract]
- Maiti S, Alam R, Amos CI, et al.: Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 60 (22): 6288-92, 2000.[PUBMED Abstract]
- Gadd S, Huff V, Huang CC, et al.: Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia 14 (8): 742-56, 2012.[PUBMED Abstract]
- Breslow NE, Beckwith JB, Perlman EJ, et al.: Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer 47 (3): 260-7, 2006.[PUBMED Abstract]
- Fukuzawa R, Heathcott RW, More HE, et al.: Sequential WT1 and CTNNB1 mutations and alterations of beta-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J Clin Pathol 60 (9): 1013-6, 2007.[PUBMED Abstract]
- Perlman EJ, Gadd S, Arold ST, et al.: MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6: 10013, 2015.[PUBMED Abstract]
- Diller L, Ghahremani M, Morgan J, et al.: Constitutional WT1 mutations in Wilms' tumor patients. J Clin Oncol 16 (11): 3634-40, 1998.[PUBMED Abstract]
- Perlman EJ, Grundy PE, Anderson JR, et al.: WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children's oncology group study. J Clin Oncol 29 (6): 698-703, 2011.[PUBMED Abstract]
- Scott RH, Walker L, Olsen ØE, et al.: Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 91 (12): 995-9, 2006.[PUBMED Abstract]
- Lipska BS, Ranchin B, Iatropoulos P, et al.: Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85 (5): 1169-78, 2014.[PUBMED Abstract]
- Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, et al.: Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol 10 (5): 825-31, 2015.[PUBMED Abstract]
- Lange J, Peterson SM, Takashima JR, et al.: Risk factors for end stage renal disease in non-WT1-syndromic Wilms tumor. J Urol 186 (2): 378-86, 2011.[PUBMED Abstract]
- Breslow NE, Takashima JR, Ritchey ML, et al.: Renal failure in the Denys-Drash and Wilms' tumor-aniridia syndromes. Cancer Res 60 (15): 4030-2, 2000.[PUBMED Abstract]
- Koesters R, Ridder R, Kopp-Schneider A, et al.: Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms' tumors. Cancer Res 59 (16): 3880-2, 1999.[PUBMED Abstract]
- Koesters R, Niggli F, von Knebel Doeberitz M, et al.: Nuclear accumulation of beta-catenin protein in Wilms' tumours. J Pathol 199 (1): 68-76, 2003.[PUBMED Abstract]
- Major MB, Camp ND, Berndt JD, et al.: Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 316 (5827): 1043-6, 2007.[PUBMED Abstract]
- Rivera MN, Kim WJ, Wells J, et al.: An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315 (5812): 642-5, 2007.[PUBMED Abstract]
- Fukuzawa R, Anaka MR, Weeks RJ, et al.: Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene 28 (8): 1063-75, 2009.[PUBMED Abstract]
- Jenkins ZA, van Kogelenberg M, Morgan T, et al.: Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet 41 (1): 95-100, 2009.[PUBMED Abstract]
- Grohmann A, Tanneberger K, Alzner A, et al.: AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane. J Cell Sci 120 (Pt 21): 3738-47, 2007.[PUBMED Abstract]
- Satoh Y, Nakadate H, Nakagawachi T, et al.: Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer 95 (4): 541-7, 2006.[PUBMED Abstract]
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.[PUBMED Abstract]
- Lennerz JK, Timmerman RJ, Grange DK, et al.: Addition of H19 'loss of methylation testing' for Beckwith-Wiedemann syndrome (BWS) increases the diagnostic yield. J Mol Diagn 12 (5): 576-88, 2010.[PUBMED Abstract]
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.[PUBMED Abstract]
- Bliek J, Gicquel C, Maas S, et al.: Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J Pediatr 145 (6): 796-9, 2004.[PUBMED Abstract]
- Bjornsson HT, Brown LJ, Fallin MD, et al.: Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst 99 (16): 1270-3, 2007.[PUBMED Abstract]
- Fukuzawa R, Breslow NE, Morison IM, et al.: Epigenetic differences between Wilms' tumours in white and east-Asian children. Lancet 363 (9407): 446-51, 2004.[PUBMED Abstract]
- Gratias EJ, Dome JS, Jennings LJ, et al.: Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group. J Clin Oncol 34 (26): 3189-94, 2016.[PUBMED Abstract]
- Chagtai T, Zill C, Dainese L, et al.: Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol 34 (26): 3195-203, 2016.[PUBMED Abstract]
- Grundy PE, Breslow NE, Li S, et al.: Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 23 (29): 7312-21, 2005.[PUBMED Abstract]
- Messahel B, Williams R, Ridolfi A, et al.: Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study. Eur J Cancer 45 (5): 819-26, 2009.[PUBMED Abstract]
- Spreafico F, Gamba B, Mariani L, et al.: Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian Association of Pediatric Hematology and Oncology. J Urol 189 (1): 260-6, 2013.[PUBMED Abstract]
- Gratias EJ, Jennings LJ, Anderson JR, et al.: Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children's Oncology Group. Cancer 119 (21): 3887-94, 2013.[PUBMED Abstract]
- Hohenstein P, Pritchard-Jones K, Charlton J: The yin and yang of kidney development and Wilms' tumors. Genes Dev 29 (5): 467-82, 2015.[PUBMED Abstract]
- Foulkes WD, Priest JR, Duchaine TF: DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14 (10): 662-72, 2014.[PUBMED Abstract]
- Wu MK, Sabbaghian N, Xu B, et al.: Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230 (2): 154-64, 2013.[PUBMED Abstract]
- Palculict TB, Ruteshouser EC, Fan Y, et al.: Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet 53 (6): 385-8, 2016.[PUBMED Abstract]
- Chang HM, Triboulet R, Thornton JE, et al.: A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497 (7448): 244-8, 2013.[PUBMED Abstract]
- Alessandri JL, Cuillier F, Ramful D, et al.: Perlman syndrome: report, prenatal findings and review. Am J Med Genet A 146A (19): 2532-7, 2008.[PUBMED Abstract]
- Bardeesy N, Falkoff D, Petruzzi MJ, et al.: Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7 (1): 91-7, 1994.[PUBMED Abstract]
- el Bahtimi R, Hazen-Martin DJ, Re GG, et al.: Immunophenotype, mRNA expression, and gene structure of p53 in Wilms' tumors. Mod Pathol 9 (3): 238-44, 1996.[PUBMED Abstract]
- Wallkamm V, Dörlich R, Rahm K, et al.: Live imaging of Xwnt5A-ROR2 complexes. PLoS One 9 (10): e109428, 2014.[PUBMED Abstract]
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.[PUBMED Abstract]
- Williams RD, Al-Saadi R, Chagtai T, et al.: Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms' tumor. Clin Cancer Res 16 (7): 2036-45, 2010.[PUBMED Abstract]
- Muller E, Hudgins L: 9q22.3 Microdeletion. In: Pagon RA, Adam MP, Bird TD, et al., eds.: GeneReviews. Seattle, Wash: University of Washington, 1993-2018, pp. Available online. Last accessed June 08, 2020.[PUBMED Abstract]
- Garavelli L, Piemontese MR, Cavazza A, et al.: Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A 161A (11): 2894-901, 2013.[PUBMED Abstract]
- Cajaiba MM, Bale AE, Alvarez-Franco M, et al.: Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol 3 (10): 575-80, 2006.[PUBMED Abstract]
- Williams RD, Chagtai T, Alcaide-German M, et al.: Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 6 (9): 7232-43, 2015.[PUBMED Abstract]
- Fievet A, Belaud-Rotureau MA, Dugay F, et al.: Involvement of germline DDX1-MYCN duplication in inherited nephroblastoma. Eur J Med Genet 56 (12): 643-7, 2013.[PUBMED Abstract]
- Martins AG, Pinto AT, Domingues R, et al.: Identification of a novel CTR9 germline mutation in a family with Wilms tumor. Eur J Med Genet 61 (5): 294-299, 2018.[PUBMED Abstract]
- Charlton J, Irtan S, Bergeron C, et al.: Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med 19: e8, 2017.[PUBMED Abstract]
- Hu M, Fletcher J, McCahon E, et al.: Bilateral Wilms tumor and early presentation in pediatric patients is associated with the truncation of the Wilms tumor 1 protein. J Pediatr 163 (1): 224-9, 2013.[PUBMED Abstract]
- Murphy AJ, Davidoff AM: Bilateral Wilms Tumor: A Surgical Perspective. Children (Basel) 5 (10): , 2018.[PUBMED Abstract]
- Kalish JM, Doros L, Helman LJ, et al.: Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res 23 (13): e115-e122, 2017.[PUBMED Abstract]
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.[PUBMED Abstract]
- Mussa A, Duffy KA, Carli D, et al.: The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J Cancer Res Clin Oncol 145 (12): 3115-3123, 2019.[PUBMED Abstract]
- Hingorani M, Hanson I, van Heyningen V: Aniridia. Eur J Hum Genet 20 (10): 1011-7, 2012.[PUBMED Abstract]
- van Heyningen V, Hoovers JM, de Kraker J, et al.: Raised risk of Wilms tumour in patients with aniridia and submicroscopic WT1 deletion. J Med Genet 44 (12): 787-90, 2007.[PUBMED Abstract]
- Greene AK, Kieran M, Burrows PE, et al.: Wilms tumor screening is unnecessary in Klippel-Trenaunay syndrome. Pediatrics 113 (4): e326-9, 2004.[PUBMED Abstract]
- Schultz KAP, Rednam SP, Kamihara J, et al.: PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res 23 (12): e76-e82, 2017.[PUBMED Abstract]
- Schultz KAP, Williams GM, Kamihara J, et al.: DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res 24 (10): 2251-2261, 2018.[PUBMED Abstract]
- Mitchell SG, Pencheva B, Porter CC: Germline Genetics and Childhood Cancer: Emerging Cancer Predisposition Syndromes and Psychosocial Impacts. Curr Oncol Rep 21 (10): 85, 2019.[PUBMED Abstract]
- Green DM: Wilms' tumor. In: Greem DM: Diagnosis and Management of Malignant Solid Tumors in Infants and Children. Boston, Ma: Martinus Nijhoff Publishing, 1985, pp 129-86.[PUBMED Abstract]
- Servaes S, Khanna G, Naranjo A, et al.: Comparison of diagnostic performance of CT and MRI for abdominal staging of pediatric renal tumors: a report from the Children's Oncology Group. Pediatr Radiol 45 (2): 166-72, 2015.[PUBMED Abstract]
- Khanna G, Naranjo A, Hoffer F, et al.: Detection of preoperative wilms tumor rupture with CT: a report from the Children's Oncology Group. Radiology 266 (2): 610-7, 2013.[PUBMED Abstract]
- McDonald K, Duffy P, Chowdhury T, et al.: Added value of abdominal cross-sectional imaging (CT or MRI) in staging of Wilms' tumours. Clin Radiol 68 (1): 16-20, 2013.[PUBMED Abstract]
- Ritchey ML, Shamberger RC, Hamilton T, et al.: Fate of bilateral renal lesions missed on preoperative imaging: a report from the National Wilms Tumor Study Group. J Urol 174 (4 Pt 2): 1519-21; discussion 1521, 2005.[PUBMED Abstract]
- Khanna G, Rosen N, Anderson JR, et al.: Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: a report from the Children's Oncology Group. Pediatr Blood Cancer 58 (4): 551-5, 2012.[PUBMED Abstract]
- Begent J, Sebire NJ, Levitt G, et al.: Pilot study of F(18)-Fluorodeoxyglucose Positron Emission Tomography/computerised tomography in Wilms' tumour: correlation with conventional imaging, pathology and immunohistochemistry. Eur J Cancer 47 (3): 389-96, 2011.[PUBMED Abstract]
- Callaghan MU, Wong TE, Federici AB: Treatment of acquired von Willebrand syndrome in childhood. Blood 122 (12): 2019-22, 2013.[PUBMED Abstract]
- Shamberger RC, Guthrie KA, Ritchey ML, et al.: Surgery-related factors and local recurrence of Wilms tumor in National Wilms Tumor Study 4. Ann Surg 229 (2): 292-7, 1999.[PUBMED Abstract]
- Ehrlich P, Chi YY, Chintagumpala MM, et al.: Results of the First Prospective Multi-institutional Treatment Study in Children With Bilateral Wilms Tumor (AREN0534): A Report From the Children's Oncology Group. Ann Surg 266 (3): 470-478, 2017.[PUBMED Abstract]
- Hamilton TE, Green DM, Perlman EJ, et al.: Bilateral Wilms' tumor with anaplasia: lessons from the National Wilms' Tumor Study. J Pediatr Surg 41 (10): 1641-4, 2006.[PUBMED Abstract]
- Othersen HB, DeLorimer A, Hrabovsky E, et al.: Surgical evaluation of lymph node metastases in Wilms' tumor. J Pediatr Surg 25 (3): 330-1, 1990.[PUBMED Abstract]
- Green DM: Controversies in the management of Wilms tumour - immediate nephrectomy or delayed nephrectomy? Eur J Cancer 43 (17): 2453-6, 2007.[PUBMED Abstract]
- Shamberger RC, Ritchey ML, Haase GM, et al.: Intravascular extension of Wilms tumor. Ann Surg 234 (1): 116-21, 2001.[PUBMED Abstract]
- Servaes SE, Hoffer FA, Smith EA, et al.: Imaging of Wilms tumor: an update. Pediatr Radiol 49 (11): 1441-1452, 2019.[PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Green DM, Breslow NE, Beckwith JB, et al.: Effect of duration of treatment on treatment outcome and cost of treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (12): 3744-51, 1998.[PUBMED Abstract]
- Kalapurakal JA, Dome JS, Perlman EJ, et al.: Management of Wilms' tumour: current practice and future goals. Lancet Oncol 5 (1): 37-46, 2004.[PUBMED Abstract]
- Ehrlich PF: Wilms tumor: progress and considerations for the surgeon. Surg Oncol 16 (3): 157-71, 2007.[PUBMED Abstract]
- Dome JS, Cotton CA, Perlman EJ, et al.: Treatment of anaplastic histology Wilms' tumor: results from the fifth National Wilms' Tumor Study. J Clin Oncol 24 (15): 2352-8, 2006.[PUBMED Abstract]
- Shamberger RC, Anderson JR, Breslow NE, et al.: Long-term outcomes for infants with very low risk Wilms tumor treated with surgery alone in National Wilms Tumor Study-5. Ann Surg 251 (3): 555-8, 2010.[PUBMED Abstract]
- Hol JA, Lopez-Yurda MI, Van Tinteren H, et al.: Prognostic significance of age in 5631 patients with Wilms tumour prospectively registered in International Society of Paediatric Oncology (SIOP) 93-01 and 2001. PLoS One 14 (8): e0221373, 2019.[PUBMED Abstract]
- Mitry E, Ciccolallo L, Coleman MP, et al.: Incidence of and survival from Wilms' tumour in adults in Europe: data from the EUROCARE study. Eur J Cancer 42 (14): 2363-8, 2006.[PUBMED Abstract]
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.[PUBMED Abstract]
- Kalapurakal JA, Nan B, Norkool P, et al.: Treatment outcomes in adults with favorable histologic type Wilms tumor-an update from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 60 (5): 1379-84, 2004.[PUBMED Abstract]
- Arrigo S, Beckwith JB, Sharples K, et al.: Better survival after combined modality care for adults with Wilms' tumor. A report from the National Wilms' Tumor Study. Cancer 66 (5): 827-30, 1990.[PUBMED Abstract]
- Byrd RL, Evans AE, D'Angio GJ: Adult Wilms tumor: effect of combined therapy on survival. J Urol 127 (4): 648-51, 1982.[PUBMED Abstract]
- de Vries-Brilland M, Sionneau B, Dutriaux C, et al.: Successful Treatment of Metastatic Adult Wilms Tumor With Anti-BRAF Treatment: A Case Report and a Brief Review of the Literature. Clin Genitourin Cancer 17 (4): e721-e723, 2019.[PUBMED Abstract]
- Segers H, van den Heuvel-Eibrink MM, Pritchard-Jones K, et al.: Management of adults with Wilms' tumor: recommendations based on international consensus. Expert Rev Anticancer Ther 11 (7): 1105-13, 2011.[PUBMED Abstract]
- Perlman EJ: Pediatric renal tumors: practical updates for the pathologist. Pediatr Dev Pathol 8 (3): 320-38, 2005 May-Jun.[PUBMED Abstract]
- Popov SD, Sebire NJ, Pritchard-Jones K, et al.: Renal tumors in children aged 10-16 Years: a report from the United Kingdom Children's Cancer and Leukaemia Group. Pediatr Dev Pathol 14 (3): 189-93, 2011 May-Jun.[PUBMED Abstract]
- Indolfi P, Jenkner A, Terenziani M, et al.: Synchronous bilateral Wilms tumor: a report from the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Cancer 119 (8): 1586-92, 2013.[PUBMED Abstract]
- Hamilton TE, Ritchey ML, Haase GM, et al.: The management of synchronous bilateral Wilms tumor: a report from the National Wilms Tumor Study Group. Ann Surg 253 (5): 1004-10, 2011.[PUBMED Abstract]
- Williams RD, Al-Saadi R, Natrajan R, et al.: Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer 50 (12): 982-95, 2011.[PUBMED Abstract]
- Vujanić GM, Harms D, Sandstedt B, et al.: New definitions of focal and diffuse anaplasia in Wilms tumor: the International Society of Paediatric Oncology (SIOP) experience. Med Pediatr Oncol 32 (5): 317-23, 1999.[PUBMED Abstract]
- Faria P, Beckwith JB, Mishra K, et al.: Focal versus diffuse anaplasia in Wilms tumor--new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. Am J Surg Pathol 20 (8): 909-20, 1996.[PUBMED Abstract]
- Beckwith JB: New developments in the pathology of Wilms tumor. Cancer Invest 15 (2): 153-62, 1997.[PUBMED Abstract]
- Beckwith JB: Precursor lesions of Wilms tumor: clinical and biological implications. Med Pediatr Oncol 21 (3): 158-68, 1993.[PUBMED Abstract]
- Perlman EJ, Faria P, Soares A, et al.: Hyperplastic perilobar nephroblastomatosis: long-term survival of 52 patients. Pediatr Blood Cancer 46 (2): 203-21, 2006.[PUBMED Abstract]
- Furtwängler R, Schmolze M, Gräber S, et al.: Pretreatment for bilateral nephroblastomatosis is an independent risk factor for progressive disease in patients with stage V nephroblastoma. Klin Padiatr 226 (3): 175-81, 2014.[PUBMED Abstract]
- Cooke A, Deshpande AV, La Hei ER, et al.: Ectopic nephrogenic rests in children: the clinicosurgical implications. J Pediatr Surg 44 (12): e13-6, 2009.[PUBMED Abstract]
- Wilms' tumor: status report, 1990. By the National Wilms' Tumor Study Committee. J Clin Oncol 9 (5): 877-87, 1991.[PUBMED Abstract]
- Green DM, Breslow NE, D'Angio GJ, et al.: Outcome of patients with Stage II/favorable histology Wilms tumor with and without local tumor spill: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 61 (1): 134-9, 2014.[PUBMED Abstract]
- Ehrlich PF, Anderson JR, Ritchey ML, et al.: Clinicopathologic findings predictive of relapse in children with stage III favorable-histology Wilms tumor. J Clin Oncol 31 (9): 1196-201, 2013.[PUBMED Abstract]
- D'Angio GJ, Breslow N, Beckwith JB, et al.: Treatment of Wilms' tumor. Results of the Third National Wilms' Tumor Study. Cancer 64 (2): 349-60, 1989.[PUBMED Abstract]
- Jereb B, Burgers JM, Tournade MF, et al.: Radiotherapy in the SIOP (International Society of Pediatric Oncology) nephroblastoma studies: a review. Med Pediatr Oncol 22 (4): 221-7, 1994.[PUBMED Abstract]
- Green DM: The treatment of stages I-IV favorable histology Wilms' tumor. J Clin Oncol 22 (8): 1366-72, 2004.[PUBMED Abstract]
- Graf N, Tournade MF, de Kraker J: The role of preoperative chemotherapy in the management of Wilms' tumor. The SIOP studies. International Society of Pediatric Oncology. Urol Clin North Am 27 (3): 443-54, 2000.[PUBMED Abstract]
- Vujanić GM, D'Hooghe E, Popov SD, et al.: The effect of preoperative chemotherapy on histological subtyping and staging of Wilms tumors: The United Kingdom Children's Cancer Study Group (UKCCSG) Wilms tumor trial 3 (UKW3) experience. Pediatr Blood Cancer 66 (3): e27549, 2019.[PUBMED Abstract]
- van den Heuvel-Eibrink MM, Hol JA, Pritchard-Jones K, et al.: Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat Rev Urol 14 (12): 743-752, 2017.[PUBMED Abstract]
- Green DM, Breslow NE, Beckwith JB, et al.: Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.[PUBMED Abstract]
- D'Angio GJ, Evans AE, Breslow N, et al.: The treatment of Wilms' tumor: Results of the national Wilms' tumor study. Cancer 38 (2): 633-46, 1976.[PUBMED Abstract]
- D'Angio GJ, Evans A, Breslow N, et al.: The treatment of Wilms' tumor: results of the Second National Wilms' Tumor Study. Cancer 47 (9): 2302-11, 1981.[PUBMED Abstract]
- Kieran K, Anderson JR, Dome JS, et al.: Lymph node involvement in Wilms tumor: results from National Wilms Tumor Studies 4 and 5. J Pediatr Surg 47 (4): 700-6, 2012.[PUBMED Abstract]
- Ritchey M, Daley S, Shamberger RC, et al.: Ureteral extension in Wilms' tumor: a report from the National Wilms' Tumor Study Group (NWTSG). J Pediatr Surg 43 (9): 1625-9, 2008.[PUBMED Abstract]
- Gow KW, Barnhart DC, Hamilton TE, et al.: Primary nephrectomy and intraoperative tumor spill: report from the Children's Oncology Group (COG) renal tumors committee. J Pediatr Surg 48 (1): 34-8, 2013.[PUBMED Abstract]
- McNeil DE, Langer JC, Choyke P, et al.: Feasibility of partial nephrectomy for Wilms' tumor in children with Beckwith-Wiedemann syndrome who have been screened with abdominal ultrasonography. J Pediatr Surg 37 (1): 57-60, 2002.[PUBMED Abstract]
- Scalabre A, Bergeron C, Brioude F, et al.: Is Nephron Sparing Surgery Justified in Wilms Tumor With Beckwith-Wiedemann Syndrome or Isolated Hemihypertrophy? Pediatr Blood Cancer 63 (9): 1571-7, 2016.[PUBMED Abstract]
- Auber F, Jeanpierre C, Denamur E, et al.: Management of Wilms tumors in Drash and Frasier syndromes. Pediatr Blood Cancer 52 (1): 55-9, 2009.[PUBMED Abstract]
- Neville H, Ritchey ML, Shamberger RC, et al.: The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg 37 (8): 1134-7, 2002.[PUBMED Abstract]
- Ferrer FA, Rosen N, Herbst K, et al.: Image based feasibility of renal sparing surgery for very low risk unilateral Wilms tumors: a report from the Children's Oncology Group. J Urol 190 (5): 1846-51, 2013.[PUBMED Abstract]
- Ritchey ML: Renal sparing surgery for Wilms tumor. J Urol 174 (4 Pt 1): 1172-3, 2005.[PUBMED Abstract]
- Cozzi DA, Zani A: Nephron-sparing surgery in children with primary renal tumor: indications and results. Semin Pediatr Surg 15 (1): 3-9, 2006.[PUBMED Abstract]
- Zhuge Y, Cheung MC, Yang R, et al.: Improved survival with lymph node sampling in Wilms tumor. J Surg Res 167 (2): e199-203, 2011.[PUBMED Abstract]
- Ritchey ML, Kelalis PP, Breslow N, et al.: Surgical complications after nephrectomy for Wilms' tumor. Surg Gynecol Obstet 175 (6): 507-14, 1992.[PUBMED Abstract]
- Ehrlich PF, Ferrer FA, Ritchey ML, et al.: Hepatic metastasis at diagnosis in patients with Wilms tumor is not an independent adverse prognostic factor for stage IV Wilms tumor: a report from the Children's Oncology Group/National Wilms Tumor Study Group. Ann Surg 250 (4): 642-8, 2009.[PUBMED Abstract]
- Ritchey ML: Primary nephrectomy for Wilms' tumor: approach of the National Wilms' Tumor Study Group. Urology 47 (6): 787-91, 1996.[PUBMED Abstract]
- Lall A, Pritchard-Jones K, Walker J, et al.: Wilms' tumor with intracaval thrombus in the UK Children's Cancer Study Group UKW3 trial. J Pediatr Surg 41 (2): 382-7, 2006.[PUBMED Abstract]
- Ritchey ML, Pringle KC, Breslow NE, et al.: Management and outcome of inoperable Wilms tumor. A report of National Wilms Tumor Study-3. Ann Surg 220 (5): 683-90, 1994.[PUBMED Abstract]
- Ritchey ML, Shamberger RC, Haase G, et al.: Surgical complications after primary nephrectomy for Wilms' tumor: report from the National Wilms' Tumor Study Group. J Am Coll Surg 192 (1): 63-8; quiz 146, 2001.[PUBMED Abstract]
- Tournade MF, Com-Nougué C, Voûte PA, et al.: Results of the Sixth International Society of Pediatric Oncology Wilms' Tumor Trial and Study: a risk-adapted therapeutic approach in Wilms' tumor. J Clin Oncol 11 (6): 1014-23, 1993.[PUBMED Abstract]
- Oberholzer HF, Falkson G, De Jager LC: Successful management of inferior vena cava and right atrial nephroblastoma tumor thrombus with preoperative chemotherapy. Med Pediatr Oncol 20 (1): 61-3, 1992.[PUBMED Abstract]
- Saarinen UM, Wikström S, Koskimies O, et al.: Percutaneous needle biopsy preceding preoperative chemotherapy in the management of massive renal tumors in children. J Clin Oncol 9 (3): 406-15, 1991.[PUBMED Abstract]
- Dykes EH, Marwaha RK, Dicks-Mireaux C, et al.: Risks and benefits of percutaneous biopsy and primary chemotherapy in advanced Wilms' tumour. J Pediatr Surg 26 (5): 610-2, 1991.[PUBMED Abstract]
- Thompson WR, Newman K, Seibel N, et al.: A strategy for resection of Wilms' tumor with vena cava or atrial extension. J Pediatr Surg 27 (7): 912-5, 1992.[PUBMED Abstract]
- Szavay P, Luithle T, Semler O, et al.: Surgery of cavoatrial tumor thrombus in nephroblastoma: a report of the SIOP/GPOH study. Pediatr Blood Cancer 43 (1): 40-5, 2004.[PUBMED Abstract]
- Powis M, Messahel B, Hobson R, et al.: Surgical complications after immediate nephrectomy versus preoperative chemotherapy in non-metastatic Wilms' tumour: findings from the 1991-2001 United Kingdom Children's Cancer Study Group UKW3 Trial. J Pediatr Surg 48 (11): 2181-6, 2013.[PUBMED Abstract]
- Rutigliano DN, Kayton ML, Steinherz P, et al.: The use of preoperative chemotherapy in Wilms' tumor with contained retroperitoneal rupture. J Pediatr Surg 42 (9): 1595-9, 2007.[PUBMED Abstract]
- Brisse HJ, Schleiermacher G, Sarnacki S, et al.: Preoperative Wilms tumor rupture: a retrospective study of 57 patients. Cancer 113 (1): 202-13, 2008.[PUBMED Abstract]
- Corn BW, Goldwein JW, Evans I, et al.: Outcomes in low-risk babies treated with half-dose chemotherapy according to the Third National Wilms' Tumor Study. J Clin Oncol 10 (8): 1305-9, 1992.[PUBMED Abstract]
- Morgan E, Baum E, Breslow N, et al.: Chemotherapy-related toxicity in infants treated according to the Second National Wilms' Tumor Study. J Clin Oncol 6 (1): 51-5, 1988.[PUBMED Abstract]
- Green DM, Norkool P, Breslow NE, et al.: Severe hepatic toxicity after treatment with vincristine and dactinomycin using single-dose or divided-dose schedules: a report from the National Wilms' Tumor Study. J Clin Oncol 8 (9): 1525-30, 1990.[PUBMED Abstract]
- Raine J, Bowman A, Wallendszus K, et al.: Hepatopathy-thrombocytopenia syndrome--a complication of dactinomycin therapy for Wilms' tumor: a report from the United Kingdom Childrens Cancer Study Group. J Clin Oncol 9 (2): 268-73, 1991.[PUBMED Abstract]
- Feusner JH, Ritchey ML, Norkool PA, et al.: Renal failure does not preclude cure in children receiving chemotherapy for Wilms tumor: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 50 (2): 242-5, 2008.[PUBMED Abstract]
- Veal GJ, English MW, Grundy RG, et al.: Pharmacokinetically guided dosing of carboplatin in paediatric cancer patients with bilateral nephrectomy. Cancer Chemother Pharmacol 54 (4): 295-300, 2004.[PUBMED Abstract]
- Dix DB, Fernandez CV, Chi YY, et al.: Augmentation of Therapy for Combined Loss of Heterozygosity 1p and 16q in Favorable Histology Wilms Tumor: A Children's Oncology Group AREN0532 and AREN0533 Study Report. J Clin Oncol 37 (30): 2769-2777, 2019.[PUBMED Abstract]
- Stokes CL, Stokes WA, Kalapurakal JA, et al.: Timing of Radiation Therapy in Pediatric Wilms Tumor: A Report From the National Cancer Database. Int J Radiat Oncol Biol Phys 101 (2): 453-461, 2018.[PUBMED Abstract]
- Dix DB, Seibel NL, Chi YY, et al.: Treatment of Stage IV Favorable Histology Wilms Tumor With Lung Metastases: A Report From the Children's Oncology Group AREN0533 Study. J Clin Oncol 36 (16): 1564-1570, 2018.[PUBMED Abstract]
- Daw NC, Chi YY, Kalapurakal JA, et al.: Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children's Oncology Group AREN0321 Study. J Clin Oncol 38 (14): 1558-1568, 2020.[PUBMED Abstract]
- Fernandez CV, Perlman EJ, Mullen EA, et al.: Clinical Outcome and Biological Predictors of Relapse After Nephrectomy Only for Very Low-risk Wilms Tumor: A Report From Children's Oncology Group AREN0532. Ann Surg 265 (4): 835-840, 2017.[PUBMED Abstract]
- Thomas PR, Tefft M, Compaan PJ, et al.: Results of two radiation therapy randomizations in the third National Wilms' Tumor Study. Cancer 68 (8): 1703-7, 1991.[PUBMED Abstract]
- Tefft M, D'Angio GJ, Beckwith B, et al.: Patterns of intra-abdominal relapse (IAR) in patients with Wilms' tumor who received radiation: analysis by histopathology. A report of National Wilms' Tumor Studies 1 and 2 (NWTS-1 & 2). Int J Radiat Oncol Biol Phys 6 (6): 663-7, 1980.[PUBMED Abstract]
- Thomas PR, Tefft M, Farewell VT, et al.: Abdominal relapses in irradiated second National Wilms' Tumor Study patients. J Clin Oncol 2 (10): 1098-101, 1984.[PUBMED Abstract]
- Meisel JA, Guthrie KA, Breslow NE, et al.: Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 44 (3): 579-85, 1999.[PUBMED Abstract]
- Daw NC, Chi YY, Kim Y, et al.: Treatment of stage I anaplastic Wilms' tumour: a report from the Children's Oncology Group AREN0321 study. Eur J Cancer 118: 58-66, 2019.[PUBMED Abstract]
- Green DM, Breslow NE, Beckwith JB, et al.: Treatment with nephrectomy only for small, stage I/favorable histology Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 19 (17): 3719-24, 2001.[PUBMED Abstract]
- Parsons LN, Mullen EA, Geller JI, et al.: Outcome analysis of stage I epithelial-predominant favorable-histology Wilms tumors: A report from Children's Oncology Group study AREN03B2. Cancer 126 (12): 2866-2871, 2020.[PUBMED Abstract]
- Kalapurakal JA, Li SM, Breslow NE, et al.: Intraoperative spillage of favorable histology wilms tumor cells: influence of irradiation and chemotherapy regimens on abdominal recurrence. A report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 76 (1): 201-6, 2010.[PUBMED Abstract]
- Fernandez CV, Mullen EA, Chi YY, et al.: Outcome and Prognostic Factors in Stage III Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group Study AREN0532. J Clin Oncol 36 (3): 254-261, 2018.[PUBMED Abstract]
- Grundy PE, Green DM, Dirks AC, et al.: Clinical significance of pulmonary nodules detected by CT and Not CXR in patients treated for favorable histology Wilms tumor on national Wilms tumor studies-4 and -5: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 631-5, 2012.[PUBMED Abstract]
- Verschuur A, Van Tinteren H, Graf N, et al.: Treatment of pulmonary metastases in children with stage IV nephroblastoma with risk-based use of pulmonary radiotherapy. J Clin Oncol 30 (28): 3533-9, 2012.[PUBMED Abstract]
- Breslow NE, Collins AJ, Ritchey ML, et al.: End stage renal disease in patients with Wilms tumor: results from the National Wilms Tumor Study Group and the United States Renal Data System. J Urol 174 (5): 1972-5, 2005.[PUBMED Abstract]
- Aronson DC, Slaar A, Heinen RC, et al.: Long-term outcome of bilateral Wilms tumors (BWT). Pediatr Blood Cancer 56 (7): 1110-3, 2011.[PUBMED Abstract]
- Zuppan CW, Beckwith JB, Weeks DA, et al.: The effect of preoperative therapy on the histologic features of Wilms' tumor. An analysis of cases from the Third National Wilms' Tumor Study. Cancer 68 (2): 385-94, 1991.[PUBMED Abstract]
- Ehrlich PF: Bilateral Wilms' tumor: the need to improve outcomes. Expert Rev Anticancer Ther 9 (7): 963-73, 2009.[PUBMED Abstract]
- Sudour H, Audry G, Schleimacher G, et al.: Bilateral Wilms tumors (WT) treated with the SIOP 93 protocol in France: epidemiological survey and patient outcome. Pediatr Blood Cancer 59 (1): 57-61, 2012.[PUBMED Abstract]
- Davidoff AM, Interiano RB, Wynn L, et al.: Overall Survival and Renal Function of Patients With Synchronous Bilateral Wilms Tumor Undergoing Surgery at a Single Institution. Ann Surg 262 (4): 570-6, 2015.[PUBMED Abstract]
- Kieran K, Williams MA, McGregor LM, et al.: Repeat nephron-sparing surgery for children with bilateral Wilms tumor. J Pediatr Surg 49 (1): 149-53, 2014.[PUBMED Abstract]
- Kist-van Holthe JE, Ho PL, Stablein D, et al.: Outcome of renal transplantation for Wilms' tumor and Denys-Drash syndrome: a report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Transplant 9 (3): 305-10, 2005.[PUBMED Abstract]
- Wong KF, Reulen RC, Winter DL, et al.: Risk of Adverse Health and Social Outcomes Up to 50 Years After Wilms Tumor: The British Childhood Cancer Survivor Study. J Clin Oncol 34 (15): 1772-9, 2016.[PUBMED Abstract]
- Breslow NE, Lange JM, Friedman DL, et al.: Secondary malignant neoplasms after Wilms tumor: an international collaborative study. Int J Cancer 127 (3): 657-66, 2010.[PUBMED Abstract]
- Termuhlen AM, Tersak JM, Liu Q, et al.: Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 57 (7): 1210-6, 2011.[PUBMED Abstract]
- Lange JM, Takashima JR, Peterson SM, et al.: Breast cancer in female survivors of Wilms tumor: a report from the national Wilms tumor late effects study. Cancer 120 (23): 3722-30, 2014.[PUBMED Abstract]
- Green DM, Grigoriev YA, Nan B, et al.: Congestive heart failure after treatment for Wilms' tumor: a report from the National Wilms' Tumor Study group. J Clin Oncol 19 (7): 1926-34, 2001.[PUBMED Abstract]
- Green DM, Lange JM, Peabody EM, et al.: Pregnancy outcome after treatment for Wilms tumor: a report from the national Wilms tumor long-term follow-up study. J Clin Oncol 28 (17): 2824-30, 2010.[PUBMED Abstract]
- 腎細胞がん(RCC)
-
RCCの発生率
小児の腎臓に発生する悪性上皮性腫瘍は新たな小児腎腫瘍の5%を超える;したがって、これらの腫瘍は腎明細胞肉腫または腎ラブドイド腫瘍よりも一般的である。年間発生率は小児100万人当たり約4例であるのに対し、腎臓のウィルムス腫瘍の発生率はその29倍以上である。[ 1 ]
RCCは、成人で最も一般的な腎臓の原発性悪性腫瘍であるが、15歳未満の小児ではまれである。より年齢の高い青年(15~19歳)のグループでは、腎臓の悪性腫瘍の約3分の2がRCCである。[ 2 ]RCCの小児および青年(n = 515)では、21~30歳の患者よりも病勢が進行している。[ 1 ]
RCCに関連する病態
RCCに関連する病態には以下のものがある:
RCCの小児および青年に対する遺伝子検査
RCCの小児および青年を対象に、関連する症候群について調べる生殖細胞系遺伝子検査の適応を、表8に記載している。
表8.腎細胞がん(RCC)の小児および青年を対象とした生殖細胞系遺伝子検査(スクリーニング)の適応 a 検査の適応 腫瘍組織型 遺伝子検査 関連する症候群 VHL = フォン・ヒッペル-リンダウ病。 a 出典:Linehan et al.[ 26 ] 多巣性RCCまたはVHL病変 明細胞型 VHL遺伝子 フォン・ヒッペル-リンダウ症候群 明細胞型RCCの家族歴、またはVHL変異を認めない多巣性RCC 明細胞型 3番染色体の遺伝子転座 遺伝性非VHL明細胞型RCC症候群 多巣性乳頭状RCC、または乳頭状RCCの家族歴 乳頭状 MET遺伝子 遺伝性乳頭状RCC症候群 多巣性RCC、皮膚の線維毛包腫、肺の嚢胞、または自然気胸 嫌色素性細胞型、膨大細胞型、または明細胞型 生殖細胞系配列BHD遺伝子 バート・ホッグ・デュベ症候群 早発型子宮平滑筋腫の個人歴もしくは家族歴、または皮膚平滑筋腫 2型乳頭状がんまたは集合管がん FH遺伝子 遺伝性平滑筋腫/RCC症候群 多巣性RCC、早発性RCC、傍神経節腫/褐色細胞腫の存在、または傍神経節腫/褐色細胞腫の家族歴 明細胞型または嫌色素性細胞型 SDHB遺伝子、SDHC遺伝子、SDHD遺伝子 遺伝性傍神経節腫/褐色細胞腫症候群 RCCのゲノム情報
転座陽性腎がん(translocation-positive carcinomas of the kidney)は、腎細胞がん(RCC)の別の形態として認識されており、小児におけるRCCで最も一般的な形態と考えられ、小児RCCの40~50%を占めている。[ 27 ]小児および青年のRCC患者120人を対象にした小児腫瘍学グループ(COG)のプロスペクティブ臨床試験では、半数近い患者が転座陽性RCCを有した。[ 28 ][ 29 ]これらのがんの特徴は、Xp11.2に位置するTFE3遺伝子を巻き込んだ転座である。TFE3遺伝子は、以下のいずれかの遺伝子と組になる場合がある:
他のまれにみられる転座のサブタイプで、TFEB遺伝子融合を伴うt(6;11)(p21;q12)は、TFEBの過剰発現を引き起こす。TFE3およびTFEBを巻き込んだ転座は、これらの蛋白の過剰発現を引き起こすため、免疫組織化学検査で同定可能である。[ 30 ]
Xp11転座型RCCを発症する危険因子として、過去の化学療法への曝露のみが知られている。ある研究では、化学療法後から発症までの期間は、4~13年の範囲に及んでいた。報告されている患者では、すべてDNAトポイソメラーゼII阻害薬および/またはアルキル化剤が投与された。[ 31 ][ 32 ]
小児および若年成人における転座型RCCの生物学的挙動に関しては見解が分かれている。転座と関連するRCCよりも進行した病期(III期/IV期)で発見されたにもかかわらず、手術単独でRCCを治療した場合は予後良好であることを示唆しているシリーズが数件ある一方で、このような患者では転帰が劣っていることを報告しているメタアナリシスも1件ある。[ 33 ][ 34 ][ 35 ]これらの患者の転帰については、現在実施中のCOG AREN03B2(NCT00898365) Biology and Classification Studyで検討されている。Xp11転座を有する転移性RCCでは、血管内皮増殖因子受容体標的療法および哺乳類ラパマイシン標的蛋白(mTOR)阻害薬に活性があるとみられている。[ 36 ]転座を伴ったRCCの初回切除から20~30年後でも、再発が報告されている。[ 22 ]
Xp11転座を伴うRCCの診断では、TFE3免疫組織化学単独で報告されている症例で転座が認められていないことから、これを用いるよりも分子遺伝学アプローチによる確認が必要である。TFE3が陽性で、TFE3転座を認めず、代わりにALK転座を示すまれなRCC症例のサブセットがある。この症例のサブセットは、RCC内で新たに認識されたサブグループで、未分類の小児RCCの15~20%が含まれると推定されている。6~16歳の小児で報告されている8症例で、以下が観察された:[ 37 ][ 38 ][ 39 ][ 40 ]
RCCの組織型
小児のRCCは成人のRCCと組織学的に異なる。主に乳頭状および明細胞型の2つの形態学的サブグループが同定されているが、RCCの約25%はこれらのカテゴリーのどちらにも適合しない異質な特徴を示す。[ 3 ]小児RCCは乳頭状型のサブタイプがより高頻度にみられ(小児RCCの20~50%)、ときにウィルムス腫瘍、後腎腺腫、および後腎腺線維腫(metanephric adenofibroma)の状況で発生することがある。[ 41 ]
小児および若年患者におけるRCCでは、より高齢の成人とは遺伝的および形態学的範囲が異なる。[ 3 ][ 32 ][ 41 ][ 42 ]
RCCの予後および予後因子
RCCの予後因子には以下のものがある:
RCCの主要な予後因子は病期である。National Cancer Data Baseで確認されたRCCの小児および青年304人の年齢中央値は13歳であった;患者の39%が限局病期I期疾患、16%がII期疾患、33%がIII期疾患、および12%がIV期疾患を呈した。5年全生存(OS)率はI期およびII期疾患の患者で100%、III期疾患の患者で71%、およびIV期疾患の患者で8%であった。[ 43 ]年齢および性別は生存に有意な影響を及ぼさなかった。生存は、腫瘍の大きさの増加(P < 0.001)、リンパ節転移陽性状態(P = 0.001)、およびより高い病理学的病期(P < 0.001)によるネガティブな影響を受けた。[ 43 ]一部の患者の詳細は入手できず、追跡が不完全であるため、National Cancer Data Baseからのこの論説で得られたデータには限界がある。4cm以下の腫瘍の大きさは生存に影響する場合も影響しない場合もあり、小児における局所リンパ節転移は重要ではない可能性がある。
RCCの小児と成人における転帰の重要な差は、局所リンパ節転移の予後的意義である。RCCでリンパ節転移を認める成人の5年OS率は約20%であるが、文献ではRCCで診断時に局所リンパ節転移を認める(遠隔転移は認めない)小児の72%がこの疾患を生き抜くことが示唆されている。[ 27 ]集団ベースのがん登録からの患者49人を対象とした別のシリーズにおいても、同等の知見が確認された。このシリーズでは、患者の33%が乳頭状型、22%が転座型、6%が明細胞サブタイプ、16%が未分類であった。5年生存率は限局性疾患の患者で96%、所属リンパ節転移陽性の患者で75%、遠隔転移を来したRCCの患者で33%であった。[ 44 ]
RCCの臨床的特徴および診断的評価
RCCは、以下の症状を呈することがある:
(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)原発腫瘍が小さく(7cm)、リンパ節が十分に採取された40人の患者を対象にしたCOGのプロスペクティブ臨床試験では、19人がリンパ節転移陽性であった。[ 28 ]この試験の転帰の結果が待たれる。(病期評価に関する詳しい情報については、成人の腎細胞がんの治療の要約に関するPDQ要約の腎細胞がんの病期情報のセクションを参照のこと。)
RCCの治療
RCC患者の生存は、診察時の疾患の病期と根治的腎摘出術時の切除の完全性による影響を受ける。すべてのRCC患者のOS率は64~87%である。小児RCCの5年生存率は、I期で90%以上、II期で80%以上、III期で70%であるが、IV期では15%未満である。[ 27 ]レトロスペクティブ解析であることおよび罹患患者が少数であることから、この治療分野における証拠レベルには限界がある。
RCCの標準治療法には、以下の選択肢がある:
リンパ節郭清を伴う根治的腎摘出術
RCCに対する一次治療には、腎完全切除および関連するリンパ節郭清がある。[ 27 ]
リンパ節郭清を伴う腎温存手術。
腎温存手術は、容積が小さい限局性の病変で、慎重に選択された患者に検討される場合がある。2件の小規模シリーズにおいて、腎部分切除術を受けた患者では、根治的腎摘出術を受けた患者と転帰が同程度と考えられた。[ 32 ][ 45 ]
他のアプローチ
成人のRCCと同様に、小児でも切除不能な転移性病変に対する標準治療は存在しない。放射線に対する反応は乏しく、化学療法も有効ではない。インターフェロンαおよびインターロイキン2などの薬剤による免疫療法は、がんの制御にある程度の効果がある。[ 46 ][ 47 ]原発腫瘍の切除に伴い、まれに肺転移巣が自然退縮することがある。
いくつかの標的療法薬(例えば、ソラフェニブ、スニチニブ、ベバシズマブ、テムシロリムス、パゾパニブ、アキシチニブ、およびエベロリムス)がRCCの成人向けの用途で承認されている;ただし、これらの薬物はRCCの患児では検証されていない。TFE3転座陽性RCCの小児および青年患者に関する症例報告から、複数のチロシンキナーゼ阻害薬への反応性が示唆されている。[ 29 ][ 48 ][ 49 ]METを発現している転座陽性RCCの小児患者にカボザンチニブを使用して、病変の縮小および症状の改善が報告されている。[ 50 ]TFE3が陽性で、転座を認めないあらゆるRCCは、ALK発現および転座について検査すべきである。このサブタイプの認識によりALK阻害薬療法の考慮に至ることがある。[ 37 ]
(標的療法の実施に関する詳しい情報については、成人の腎細胞がんの治療に関するPDQ要約を参照のこと。)
RCCに対して臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
現在実施されている国および/または施設の臨床試験の例を次に掲げる:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Akhavan A, Richards M, Shnorhavorian M, et al.: Renal cell carcinoma in children, adolescents and young adults: a National Cancer Database study. J Urol 193 (4): 1336-41, 2015.[PUBMED Abstract]
- Bernstein L, Linet M, Smith MA, et al.: Renal tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 79-90. Also available online. Last accessed June 08, 2020.[PUBMED Abstract]
- Bruder E, Passera O, Harms D, et al.: Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol 28 (9): 1117-32, 2004.[PUBMED Abstract]
- Schimke RN, Collins DL, Stolle CA: Von Hippel-Lindau syndrome. In: Pagon RA, Adam MP, Bird TD, et al., eds.: GeneReviews. Seattle, Wash: University of Washington, 1993-2018, pp. Available online. Last accessed June 08, 2020.[PUBMED Abstract]
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.[PUBMED Abstract]
- Park HK, Zhang S, Wong MK, et al.: Clinical presentation of epithelioid angiomyolipoma. Int J Urol 14 (1): 21-5, 2007.[PUBMED Abstract]
- Pea M, Bonetti F, Martignoni G, et al.: Apparent renal cell carcinomas in tuberous sclerosis are heterogeneous: the identification of malignant epithelioid angiomyolipoma. Am J Surg Pathol 22 (2): 180-7, 1998.[PUBMED Abstract]
- Wang N, Perkins KL: Involvement of band 3p14 in t(3;8) hereditary renal carcinoma. Cancer Genet Cytogenet 11 (4): 479-81, 1984.[PUBMED Abstract]
- Ricketts C, Woodward ER, Killick P, et al.: Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 100 (17): 1260-2, 2008.[PUBMED Abstract]
- Linehan WM, Bratslavsky G, Pinto PA, et al.: Molecular diagnosis and therapy of kidney cancer. Annu Rev Med 61: 329-43, 2010.[PUBMED Abstract]
- Swartz MA, Karth J, Schneider DT, et al.: Renal medullary carcinoma: clinical, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology 60 (6): 1083-9, 2002.[PUBMED Abstract]
- Sandberg JK, Mullen EA, Cajaiba MM, et al.: Imaging of renal medullary carcinoma in children and young adults: a report from the Children's Oncology Group. Pediatr Radiol 47 (12): 1615-1621, 2017.[PUBMED Abstract]
- Hakimi AA, Koi PT, Milhoua PM, et al.: Renal medullary carcinoma: the Bronx experience. Urology 70 (5): 878-82, 2007.[PUBMED Abstract]
- Strouse JJ, Spevak M, Mack AK, et al.: Significant responses to platinum-based chemotherapy in renal medullary carcinoma. Pediatr Blood Cancer 44 (4): 407-11, 2005.[PUBMED Abstract]
- Rathmell WK, Monk JP: High-dose-intensity MVAC for Advanced Renal Medullary Carcinoma: Report of Three Cases and Literature Review. Urology 72 (3): 659-63, 2008.[PUBMED Abstract]
- Ezekian B, Englum B, Gilmore BF, et al.: Renal medullary carcinoma: A national analysis of 159 patients. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- Alrashdi I, Levine S, Paterson J, et al.: Hereditary leiomyomatosis and renal cell carcinoma: very early diagnosis of renal cancer in a paediatric patient. Fam Cancer 9 (2): 239-43, 2010.[PUBMED Abstract]
- Bayley JP, Launonen V, Tomlinson IP: The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 9: 20, 2008.[PUBMED Abstract]
- Wilson CL, Ness KK, Neglia JP, et al.: Renal carcinoma after childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst 105 (7): 504-8, 2013.[PUBMED Abstract]
- Dhall D, Al-Ahmadie HA, Dhall G, et al.: Pediatric renal cell carcinoma with oncocytoid features occurring in a child after chemotherapy for cardiac leiomyosarcoma. Urology 70 (1): 178.e13-5, 2007.[PUBMED Abstract]
- Schafernak KT, Yang XJ, Hsueh W, et al.: Pediatric renal cell carcinoma as second malignancy: reports of two cases and a review of the literature. Can J Urol 14 (6): 3739-44, 2007.[PUBMED Abstract]
- Rais-Bahrami S, Drabick JJ, De Marzo AM, et al.: Xp11 translocation renal cell carcinoma: delayed but massive and lethal metastases of a chemotherapy-associated secondary malignancy. Urology 70 (1): 178.e3-6, 2007.[PUBMED Abstract]
- Brassesco MS, Valera ET, Bonilha TA, et al.: Secondary PSF/TFE3-associated renal cell carcinoma in a child treated for genitourinary rhabdomyosarcoma. Cancer Genet 204 (2): 108-10, 2011.[PUBMED Abstract]
- Breslow NE, Lange JM, Friedman DL, et al.: Secondary malignant neoplasms after Wilms tumor: an international collaborative study. Int J Cancer 127 (3): 657-66, 2010.[PUBMED Abstract]
- Falzarano SM, McKenney JK, Montironi R, et al.: Renal Cell Carcinoma Occurring in Patients With Prior Neuroblastoma: A Heterogenous Group of Neoplasms. Am J Surg Pathol 40 (7): 989-97, 2016.[PUBMED Abstract]
- Linehan WM, Pinto PA, Bratslavsky G, et al.: Hereditary kidney cancer: unique opportunity for disease-based therapy. Cancer 115 (10 Suppl): 2252-61, 2009.[PUBMED Abstract]
- Geller JI, Dome JS: Local lymph node involvement does not predict poor outcome in pediatric renal cell carcinoma. Cancer 101 (7): 1575-83, 2004.[PUBMED Abstract]
- Geller JI, Ehrlich PF, Cost NG, et al.: Characterization of adolescent and pediatric renal cell carcinoma: A report from the Children's Oncology Group study AREN03B2. Cancer 121 (14): 2457-64, 2015.[PUBMED Abstract]
- Ambalavanan M, Geller JI: Treatment of advanced pediatric renal cell carcinoma. Pediatr Blood Cancer 66 (8): e27766, 2019.[PUBMED Abstract]
- Argani P, Hicks J, De Marzo AM, et al.: Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol 34 (9): 1295-303, 2010.[PUBMED Abstract]
- Argani P, Laé M, Ballard ET, et al.: Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol 24 (10): 1529-34, 2006.[PUBMED Abstract]
- Ramphal R, Pappo A, Zielenska M, et al.: Pediatric renal cell carcinoma: clinical, pathologic, and molecular abnormalities associated with the members of the mit transcription factor family. Am J Clin Pathol 126 (3): 349-64, 2006.[PUBMED Abstract]
- Geller JI, Argani P, Adeniran A, et al.: Translocation renal cell carcinoma: lack of negative impact due to lymph node spread. Cancer 112 (7): 1607-16, 2008.[PUBMED Abstract]
- Camparo P, Vasiliu V, Molinie V, et al.: Renal translocation carcinomas: clinicopathologic, immunohistochemical, and gene expression profiling analysis of 31 cases with a review of the literature. Am J Surg Pathol 32 (5): 656-70, 2008.[PUBMED Abstract]
- Qiu Rao, Bing Guan, Zhou XJ: Xp11.2 Translocation renal cell carcinomas have a poorer prognosis than non-Xp11.2 translocation carcinomas in children and young adults: a meta-analysis. Int J Surg Pathol 18 (6): 458-64, 2010.[PUBMED Abstract]
- Malouf GG, Camparo P, Oudard S, et al.: Targeted agents in metastatic Xp11 translocation/TFE3 gene fusion renal cell carcinoma (RCC): a report from the Juvenile RCC Network. Ann Oncol 21 (9): 1834-8, 2010.[PUBMED Abstract]
- Thorner PS, Shago M, Marrano P, et al.: TFE3-positive renal cell carcinomas are not always Xp11 translocation carcinomas: Report of a case with a TPM3-ALK translocation. Pathol Res Pract 212 (10): 937-942, 2016.[PUBMED Abstract]
- Cajaiba MM, Jennings LJ, Rohan SM, et al.: ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer 55 (5): 442-51, 2016.[PUBMED Abstract]
- Smith NE, Deyrup AT, Mariño-Enriquez A, et al.: VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol 38 (6): 858-63, 2014.[PUBMED Abstract]
- Cajaiba MM, Jennings LJ, George D, et al.: Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer 55 (10): 814-7, 2016.[PUBMED Abstract]
- Estrada CR, Suthar AM, Eaton SH, et al.: Renal cell carcinoma: Children's Hospital Boston experience. Urology 66 (6): 1296-300, 2005.[PUBMED Abstract]
- Carcao MD, Taylor GP, Greenberg ML, et al.: Renal-cell carcinoma in children: a different disorder from its adult counterpart? Med Pediatr Oncol 31 (3): 153-8, 1998.[PUBMED Abstract]
- Rialon KL, Gulack BC, Englum BR, et al.: Factors impacting survival in children with renal cell carcinoma. J Pediatr Surg 50 (6): 1014-8, 2015.[PUBMED Abstract]
- Selle B, Furtwängler R, Graf N, et al.: Population-based study of renal cell carcinoma in children in Germany, 1980-2005: more frequently localized tumors and underlying disorders compared with adult counterparts. Cancer 107 (12): 2906-14, 2006.[PUBMED Abstract]
- Cook A, Lorenzo AJ, Salle JL, et al.: Pediatric renal cell carcinoma: single institution 25-year case series and initial experience with partial nephrectomy. J Urol 175 (4): 1456-60; discussion 1460, 2006.[PUBMED Abstract]
- Fyfe G, Fisher RI, Rosenberg SA, et al.: Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol 13 (3): 688-96, 1995.[PUBMED Abstract]
- Coppin C, Porzsolt F, Awa A, et al.: Immunotherapy for advanced renal cell cancer. Cochrane Database Syst Rev (1): CD001425, 2005.[PUBMED Abstract]
- De Pasquale MD, Pessolano R, Boldrini R, et al.: Continuing response to subsequent treatment lines with tyrosine kinase inhibitors in an adolescent with metastatic renal cell carcinoma. J Pediatr Hematol Oncol 33 (5): e176-9, 2011.[PUBMED Abstract]
- Chowdhury T, Prichard-Jones K, Sebire NJ, et al.: Persistent complete response after single-agent sunitinib treatment in a case of TFE translocation positive relapsed metastatic pediatric renal cell carcinoma. J Pediatr Hematol Oncol 35 (1): e1-3, 2013.[PUBMED Abstract]
- Wedekind MF, Ranalli M, Shah N: Clinical efficacy of cabozantinib in two pediatric patients with recurrent renal cell carcinoma. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- 腎ラブドイド腫瘍
-
腎ラブドイド腫瘍に関する一般情報
ラブドイド腫瘍は、一般的に乳児および幼児に起こるきわめて侵攻性の悪性腫瘍である。最も一般的な発生部位は、腎臓(悪性ラブドイド腫瘍と呼ばれる)および中枢神経系(CNS)(非定型奇形腫様/ラブドイド腫瘍)であるが、ラブドイド腫瘍はまた軟部組織のほとんどの部位に発生しうる。(中枢神経系疾患の治療に関する情報については、小児中枢神経系非定型奇形腫様/ラブドイド腫瘍の治療に関するPDQ要約を参照のこと。)再燃は早期に現れる(診断からの期間中央値、8ヵ月)。[ 1 ][ 2 ]
腎ラブドイド腫瘍の診断を示唆する特徴的な臨床所見には、次のものがある:[ 3 ]
(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
約3分の2の患者が進行期で発見される。両側性の症例が報告されている。[ 1 ]腎ラブドイド腫瘍は肺と脳に転移する傾向がある。腎ラブドイド腫瘍患者の実に10~15%は、CNS病変を併発している。[ 4 ]腎ラブドイド腫瘍に用いられる病期分類システムは、ウィルムス腫瘍用のシステムと同じである。(詳しい情報については、本要約のウィルムス腫瘍の病期情報のセクションを参照のこと。)
組織学的に、腎ラブドイド腫瘍の最もはっきりした特徴は、大きな胞状核、明瞭な単一の核小体をもち、いくつかの細胞では球状の好酸性細胞質封入体が存在する大型の細胞型である。
腎ラブドイド腫瘍のゲノム情報
どの解剖学的な部位に存在するラブドイド腫瘍にも共通して、染色体22q11.2に位置するSMARCB1(INI1/SNF5/BAF47)遺伝子の機能喪失という遺伝子異常が認められる。以下の本文では原発部位に関係なく、ラブドイド腫瘍について述べる。SMARCB1は、遺伝子の転写制御に重要な役割を果たすSWItch/Sucrose NonFermentable(SWI/SNF)クロマチンリモデリング複合体を構成する蛋白をコードする。[ 5 ][ 6 ]機能喪失は、SMARCB1遺伝子の一部またはすべての喪失につながる欠失、および一般に、SMARCB1蛋白のpremature truncationにつながるフレームシフトまたはナンセンス変異である変異により生じる。[ 6 ][ 7 ]ごく一部のラブドイド腫瘍は、SWI/SNF複合体の一次ATPアーゼであるSMARCA4の変異により引き起こされる。[ 8 ][ 9 ]ラブドイド腫瘍35例のエクソーム配列解析では変異率がごく低いことが明らかになっており、SMARCB1以外に反復変異を生じている遺伝子はなく、これが腫瘍形成に寄与しているとみられる。[ 10 ]
脳および/または腎臓に原発腫瘍が1つまたは複数認められる患者においてSMARCB1の生殖細胞変異が確認されており、ラブドイド腫瘍発症の遺伝的素因と一致している。[ 11 ][ 12 ]ラブドイド腫瘍患者の約3分の1に、SMARCB1の生殖細胞変異が認められる。[ 6 ][ 13 ]ほとんどの症例において変異はde novoであり、遺伝性ではない。ラブドイド腫瘍と生殖細胞の変異または欠失が認められる患児の診断時の年齢中央値は6ヵ月で、明らかな散発性病変の患児の中央値(18ヵ月)よりも若い。[ 14 ]生殖細胞系モザイク現象(germline mosaicism)は、複数の罹患した同胞を有するいくつかの家族に対して示唆されている。生殖細胞変異がある患者は、予後が最も悪い可能性があるとみられている。[ 15 ][ 16 ]ラブドイド腫瘍患者ではSMARCA4の生殖細胞変異も報告されている。[ 8 ][ 17 ]
ラブドイド腫瘍素因症候群
早期発症、多巣性病変、および家族性症例により、ラブドイド腫瘍素因症候群の可能性が強く裏付けられる。このことは、まれな家族性症例、および散発性ラブドイド腫瘍とみられる患者サブセットにおけるSMARCB1の生殖細胞変異の存在によって確認されている。これらの症例は、ラブドイド腫瘍素因症候群1型と呼ばれている。脳、腎臓、軟部組織にラブドイド腫瘍を有する患者35人(N = 100)に、SMARCB1の生殖細胞異常が認められた。これらの異常には、点変異およびフレームシフト変異、遺伝子内欠失および重複のほか、より大きな欠失などがあった。9例で、変異を含むSMARCB1のコピーが親から子に伝播していることが示された。この9例のうち8例では、1人以上の家族もラブドイド腫瘍またはシュワン腫と診断されていた;8家系のうち2家系には、複数の患児が存在し、性腺モザイク現象との一致がみられた。[ 6 ]
SMARCA4遺伝子における不活性型生殖細胞変異の2例が、関連のない2家系の小児3人に認められ、ラブドイド腫瘍素因症候群2型と呼ばれる類似した表現型の症候群を呈した。[ 8 ][ 9 ]これらの症例でSMARCA4は古典的な腫瘍抑制遺伝子のように振る舞い、一方の症例では生殖細胞系の有害な変異が遺伝し、他方では腫瘍において獲得された。他の報告には、エクソーム配列解析プロジェクトで発見された常染色体優性遺伝パターンについての記述がある。[ 18 ]
腎ラブドイド腫瘍の遺伝子検査とサーベイランス
全年齢のラブドイド腫瘍患者に対して生殖細胞系検査が推奨される。また、少ないが現実に存在する家族性再発のリスクを考慮して、遺伝カウンセリングも治療計画に含められる。変異が認められた場合は親のスクリーニングを検討すべきであるが、ただ、そのようなスクリーニングで陽性となる可能性は低い。家系内に特定のSMARCB1変異または欠失が確認されている場合は、出生前診断を実施することができる。[ 6 ]
現在のところ、SMARCB1の機能喪失性生殖細胞変異を原因とするラブドイド腫瘍素因症候群1型を有する患者にサーベイランスを実施する有効性に関する証拠はほとんど得られていない。しかしながら、切断型変異を有するSMARCB1キャリアではかなりの致死性および若年での発症を伴う腫瘍の侵攻性の性質のために、コンセンサスに基づく推奨が策定されている。これらの推奨は小児がん遺伝子の専門家集団(腫瘍医、放射線科医、遺伝医を含む)によって開発された。これらの推奨は、SMARCB1の生殖細胞変異を有する患者にモニタリングを実施する有益性を確認するために、公式には研究されていない。外科的に切除可能な疾患の潜在的な生存利益を考慮すると、早期発見により全生存(OS)が改善しうると仮定されている。[ 19 ][ 20 ][ 21 ]
SMARCB1生殖細胞変異を認める患者のサーベイランスには以下が含まれる:
腎ラブドイド腫瘍の予後および予後因子
腎ラブドイド腫瘍患者の予後は、依然として不良である。National Wilms Tumor Study (NWTS)(NWTS-1、NWTS-2、NWTS-3、NWTS-4、およびNWTS-5 [COG-Q9401/NCT00002611])に参加した患者142人のレビューによると、以下のように年齢および病期が重要な予後因子であることが確認された:[ 4 ]
腎ラブドイド腫瘍の治療
比較的まれな腫瘍であるため、腎ラブドイド腫瘍の患者はすべて登録して臨床試験に組み入れることを考えるべきである。至適治療法を決定し実施するには、腎腫瘍の治療に経験のあるがん専門医(小児外科医または小児泌尿器科専門医、小児放射線腫瘍医、および小児腫瘍医)からなる集学的チームによる治療計画の策定が必要である。
腎ラブドイド腫瘍に対する標準治療法は存在しない。[ 22 ]
腎ラブドイド腫瘍の研究では、以下の結果が観察されている:
- ビンクリスチン/ダクチノマイシン vs ビンクリスチン/ダクチノマイシン/ドキソルビシンによる術前治療に対する腫瘍反応のレトロスペクティブ比較に基づき、ドキソルビシンは腎臓の悪性ラブドイド腫瘍に対して活性がある薬剤とみなされている。[ 23 ][証拠レベル:3iiiDiv]
- NWTS-5試験では、転帰不良のため、シクロホスファミド、エトポシド、およびカルボプラチンを併用するラブドイド腫瘍の治療群を閉鎖した。エトポシドとシスプラチンとの併用;エトポシドとイホスファミドとの併用;およびイホスファミド、カルボプラチンおよびエトポシドの併用(ICE化学療法)が用いられている。[ 24 ][ 25 ]
- 大量アルキル化剤療法とその後の大量化学療法による地固め療法に加え、場合によってX線像での寛解達成後に自家幹細胞移植を施行する治療は、多少の長期生存をもたらした(患者13人中5人)。この小規模シリーズ(N = 21)において、切除不能な原発腫瘍を患い、生存した患者はいなかった。[ 26 ]
- International Society of Pediatric Oncology(SIOP)、Gesellschaft für Pädiatrische Onkologie und Hämatologie(GPOH)、およびEuropean Rhabdoid Tumor Registryからの腎臓の悪性ラブドイド腫瘍患者58人を対象にレトロスペクティブ解析が実施された。[ 27 ]
腎ラブドイド腫瘍に対して臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- van den Heuvel-Eibrink MM, van Tinteren H, Rehorst H, et al.: Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr Blood Cancer 56 (5): 733-7, 2011.[PUBMED Abstract]
- Reinhard H, Reinert J, Beier R, et al.: Rhabdoid tumors in children: prognostic factors in 70 patients diagnosed in Germany. Oncol Rep 19 (3): 819-23, 2008.[PUBMED Abstract]
- Amar AM, Tomlinson G, Green DM, et al.: Clinical presentation of rhabdoid tumors of the kidney. J Pediatr Hematol Oncol 23 (2): 105-8, 2001.[PUBMED Abstract]
- Tomlinson GE, Breslow NE, Dome J, et al.: Rhabdoid tumor of the kidney in the National Wilms' Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol 23 (30): 7641-5, 2005.[PUBMED Abstract]
- Imbalzano AN, Jones SN: Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell 7 (4): 294-5, 2005.[PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.[PUBMED Abstract]
- Versteege I, Sévenet N, Lange J, et al.: Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394 (6689): 203-6, 1998.[PUBMED Abstract]
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.[PUBMED Abstract]
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.[PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.[PUBMED Abstract]
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.[PUBMED Abstract]
- Biegel JA: Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 20 (1): E11, 2006.[PUBMED Abstract]
- Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.[PUBMED Abstract]
- Geller JI, Roth JJ, Biegel JA: Biology and Treatment of Rhabdoid Tumor. Crit Rev Oncog 20 (3-4): 199-216, 2015.[PUBMED Abstract]
- Janson K, Nedzi LA, David O, et al.: Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47 (3): 279-84, 2006.[PUBMED Abstract]
- Sévenet N, Sheridan E, Amram D, et al.: Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65 (5): 1342-8, 1999.[PUBMED Abstract]
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.[PUBMED Abstract]
- Witkowski L, Lalonde E, Zhang J, et al.: Familial rhabdoid tumour 'avant la lettre'--from pathology review to exome sequencing and back again. J Pathol 231 (1): 35-43, 2013.[PUBMED Abstract]
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.[PUBMED Abstract]
- Mitchell SG, Pencheva B, Porter CC: Germline Genetics and Childhood Cancer: Emerging Cancer Predisposition Syndromes and Psychosocial Impacts. Curr Oncol Rep 21 (10): 85, 2019.[PUBMED Abstract]
- Foulkes WD, Kamihara J, Evans DGR, et al.: Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res 23 (12): e62-e67, 2017.[PUBMED Abstract]
- Ahmed HU, Arya M, Levitt G, et al.: Part II: Treatment of primary malignant non-Wilms' renal tumours in children. Lancet Oncol 8 (9): 842-8, 2007.[PUBMED Abstract]
- Furtwängler R, Nourkami-Tutdibi N, Leuschner I, et al.: Malignant rhabdoid tumor of the kidney: significantly improved response to pre-operative treatment intensified with doxorubicin. Cancer Genet 207 (9): 434-6, 2014.[PUBMED Abstract]
- Waldron PE, Rodgers BM, Kelly MD, et al.: Successful treatment of a patient with stage IV rhabdoid tumor of the kidney: case report and review. J Pediatr Hematol Oncol 21 (1): 53-7, 1999 Jan-Feb.[PUBMED Abstract]
- Wagner L, Hill DA, Fuller C, et al.: Treatment of metastatic rhabdoid tumor of the kidney. J Pediatr Hematol Oncol 24 (5): 385-8, 2002 Jun-Jul.[PUBMED Abstract]
- Venkatramani R, Shoureshi P, Malvar J, et al.: High dose alkylator therapy for extracranial malignant rhabdoid tumors in children. Pediatr Blood Cancer 61 (8): 1357-61, 2014.[PUBMED Abstract]
- Furtwängler R, Kager L, Melchior P, et al.: High-dose treatment for malignant rhabdoid tumor of the kidney: No evidence for improved survival-The Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) experience. Pediatr Blood Cancer 65 (1): , 2018.[PUBMED Abstract]
- 腎明細胞肉腫
-
腎明細胞肉腫に関する一般情報
腎明細胞肉腫はウィルムス腫瘍の異型ではないが、予後良好な組織型(FH)のウィルムス腫瘍よりも再燃率および死亡率が高い重要な原発腎腫瘍である。[ 1 ]古典的パターンの腎明細胞肉腫は規則的に拡がる線維血管壁で区切られる細胞巣または細胞索によって定義されている。明細胞肉腫は肺転移のほか、骨、脳、および軟部組織にも拡がる。[ 1 ](小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
年齢が低いことおよび病期がIV期であることが、イベントフリー生存(EFS)に対する予後不良因子として特定された。[ 2 ]
以前から、再燃は化学療法完了後、長期間の間隔(最大14年)を置いて起こっていたが、現在の治療法を用いると、3年後の再燃はまれである。[ 3 ]脳は再発が頻繁に起こる部位であり、患者が受けている強化化学療法から細胞が守られる聖域になっていることが示唆される。[ 2 ][ 3 ][ 4 ][ 5 ]脳で起こる再発の臨床徴候を認識することは、定期的なフォローアップにおいて重要である。フォローアップ中に実施する脳画像検査の頻度については、標準の推奨事項が存在しない。
腎明細胞肉腫のゲノム情報
腎明細胞肉腫はまれな腎腫瘍で、小児におけるすべての原発性腎悪性腫瘍の約5%を占め、米国では毎年約20例の新規症例が発生し、3歳前に最も多くみられる。[ 1 ]腎明細胞肉腫はまれであり、実験モデルがないため、その分子的背景はほとんど解明されていない。
腎明細胞肉腫には、次のようないくつかの生物学的特徴が報告されている:
腎明細胞肉腫の治療
比較的まれな腫瘍であるので、腎明細胞肉腫の患者はすべて登録して臨床試験に組み入れることを考えるべきである。至適治療法を決定し実施するには、腎腫瘍の治療に経験のあるがん専門医(小児外科医または小児泌尿器科専門医、小児放射線腫瘍医、および小児腫瘍医)からなる集学的チームによる治療計画の策定が必要である。
腎明細胞肉腫の患児の全生存(OS)率は予後良好な組織型のウィルムス腫瘍の患児より低いため、治療のアプローチがウィルムス腫瘍の場合とは異なる。すべての患者に腫瘍床への術後放射線照射が行われ、化学療法レジメンの一部としてドキソルビシンが投与される。
腎明細胞肉腫の標準治療法には、以下の選択肢がある:
手術、化学療法、および放射線療法。
証拠(手術、化学療法、および放射線療法):
- National Wilms Tumor Study-3(NWTS-3)試験では、ビンクリスチン、ダクチノマイシン、放射線療法の併用にドキソルビシンを追加することにより、腎明細胞肉腫患者の無病生存率の改善が得られた。[ 1 ]
- NWTS-4試験では、ビンクリスチン、ダクチノマイシン、およびドキソルビシンの15ヵ月間の投与で構成されるレジメンDD-4A、および放射線療法を使用した。[ 14 ]
- NWTS-5(COG-Q9401/NCT00002611)試験では、I~IV期の腎明細胞肉腫の患児が、こうした高リスク群のさらなる生存率改善を目的として、ビンクリスチン、ドキソルビシン、シクロホスファミド、およびエトポシドを併用した新しい化学療法レジメンを用いて治療された。すべての小児が腫瘍床に放射線療法を受けた。[ 3 ]
- NWTS-1、NWTS-2、NWTS-3、NWTS-4、およびNWTS-5試験で治療が行われたI期腎明細胞肉腫患者のレビューでは、化学療法および放射線療法の多様なレジメンにより、優れた100%のOS率が示された。[ 15 ]
(再発疾患に関する情報については、本要約の再発腎明細胞肉腫の治療のセクションを参照のこと。)
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Argani P, Perlman EJ, Breslow NE, et al.: Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 24 (1): 4-18, 2000.[PUBMED Abstract]
- Furtwängler R, Gooskens SL, van Tinteren H, et al.: Clear cell sarcomas of the kidney registered on International Society of Pediatric Oncology (SIOP) 93-01 and SIOP 2001 protocols: a report of the SIOP Renal Tumour Study Group. Eur J Cancer 49 (16): 3497-506, 2013.[PUBMED Abstract]
- Seibel NL, Chi YY, Perlman EJ, et al.: Impact of cyclophosphamide and etoposide on outcome of clear cell sarcoma of the kidney treated on the National Wilms Tumor Study-5 (NWTS-5). Pediatr Blood Cancer 66 (1): e27450, 2019.[PUBMED Abstract]
- Radulescu VC, Gerrard M, Moertel C, et al.: Treatment of recurrent clear cell sarcoma of the kidney with brain metastasis. Pediatr Blood Cancer 50 (2): 246-9, 2008.[PUBMED Abstract]
- Gooskens SL, Furtwängler R, Spreafico F, et al.: Treatment and outcome of patients with relapsed clear cell sarcoma of the kidney: a combined SIOP and AIEOP study. Br J Cancer 111 (2): 227-33, 2014.[PUBMED Abstract]
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.[PUBMED Abstract]
- Argani P, Kao YC, Zhang L, et al.: Primary Renal Sarcomas With BCOR-CCNB3 Gene Fusion: A Report of 2 Cases Showing Histologic Overlap With Clear Cell Sarcoma of Kidney, Suggesting Further Link Between BCOR-related Sarcomas of the Kidney and Soft Tissues. Am J Surg Pathol 41 (12): 1702-1712, 2017.[PUBMED Abstract]
- Karlsson J, Valind A, Gisselsson D: BCOR internal tandem duplication and YWHAE-NUTM2B/E fusion are mutually exclusive events in clear cell sarcoma of the kidney. Genes Chromosomes Cancer 55 (2): 120-3, 2016.[PUBMED Abstract]
- Astolfi A, Melchionda F, Perotti D, et al.: Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 6 (38): 40934-9, 2015.[PUBMED Abstract]
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.[PUBMED Abstract]
- Wong MK, Ng CCY, Kuick CH, et al.: Clear cell sarcomas of the kidney are characterised by BCOR gene abnormalities, including exon 15 internal tandem duplications and BCOR-CCNB3 gene fusion. Histopathology 72 (2): 320-329, 2018.[PUBMED Abstract]
- Kao YC, Sung YS, Zhang L, et al.: Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney. Am J Surg Pathol 40 (8): 1009-20, 2016.[PUBMED Abstract]
- Argani P, Pawel B, Szabo S, et al.: Diffuse Strong BCOR Immunoreactivity Is a Sensitive and Specific Marker for Clear Cell Sarcoma of the Kidney (CCSK) in Pediatric Renal Neoplasia. Am J Surg Pathol 42 (8): 1128-1131, 2018.[PUBMED Abstract]
- Seibel NL, Li S, Breslow NE, et al.: Effect of duration of treatment on treatment outcome for patients with clear-cell sarcoma of the kidney: a report from the National Wilms' Tumor Study Group. J Clin Oncol 22 (3): 468-73, 2004.[PUBMED Abstract]
- Kalapurakal JA, Perlman EJ, Seibel NL, et al.: Outcomes of patients with revised stage I clear cell sarcoma of kidney treated in National Wilms Tumor Studies 1-5. Int J Radiat Oncol Biol Phys 85 (2): 428-31, 2013.[PUBMED Abstract]
- 先天性間葉芽腎腫
-
先天性間葉芽腎腫に関する一般情報
間葉芽腎腫は小児腎腫瘍の約5%を占め、90%を超える症例が生後1年以内に発症する。症例の15%以上が出生前に発見される。[ 1 ]間葉芽腎腫は生後6ヵ月に満たない乳児では最もよくみられる腎腫瘍である。[ 2 ]診断時年齢中央値は生後1~2ヵ月である。男児の方が女児よりも2倍多く診断される。2歳を過ぎた小児が診断された場合は、その診断を疑問視すべきである。[ 1 ]
生後7ヵ月で診断された患者では、5年イベントフリー生存率が94%で、5年全生存(OS)率が96%である。[ 3 ]英国の臨床試験で研究された間葉芽腎腫の小児50人および同時期に国内登録から得られた80例を対象にした報告では、死亡例はみられなかった。[ 1 ]しかしながら、文献の包括的レビューで12例の死亡が報告された;これら12例の死亡のうち、7例は乳児における手術合併症によるものであった。[ 4 ][証拠レベル:3iiiDii]
肉眼的に間葉芽腎腫は、腎芽腫と鑑別することができない孤立性の片側性腫瘤として現れる。顕微鏡的に、これらは紡錘型間葉細胞で構成される。間葉芽腎腫は以下の3つの組織学的サブタイプに分けることができる:
頻度の高い遺伝子変化はt(12;15)(q13;q25)転座で、15p15上のETV6およびNTRK3遺伝子の融合をもたらし、ほとんど細胞性間葉芽腎腫のみに発生する。転座について分析された間葉芽腎腫79例のコホートでは、すべての古典的(n = 38)および混合型(n = 12)間葉芽腎腫が転座陰性であった。[ 8 ]この同じ転座は乳児性線維肉腫で最初に報告されたが、形態学的外観が類似していることに加えて、細胞性間葉芽腎腫および乳児性線維肉腫の症例は11番染色体の増加など、他の遺伝的変化も共有している。[ 9 ]
間葉芽腎腫患者の再発リスクは、細胞性サブタイプの存在およびIII期疾患と密接に関連している。[ 5 ]先天性間葉芽腎腫患者79人を対象にしたInternational Society of Pediatric Oncology(SIOP)のシリーズでは、細胞性サブグループで転座陽性腫瘍を有した患者ではこの遺伝子融合を有さなかった患者と比較して、無再燃生存(RFS)率が有意に優れていた(それぞれ100% vs 73%)。[ 8 ]
(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
先天性間葉芽腎腫の治療
先天性間葉芽腎腫患者のOSはきわめて優れている;しかしながら、症例の約半数で報告されている死因が治療関連であり、これらの患者のほとんどが非常に若年であった(年齢中央値、1歳未満)。[ 4 ]このことは、治療の時期と種類に関して、および献身的で専門的な小児腫瘍学が重要な状況で腎腫瘍の乳児には特別な配慮が必要であるということを強調している。
I期とII期(患者の80%)およびIII期(古典的および混合型サブタイプ)先天性間葉芽腎腫に対する標準治療法の選択肢には以下のものがある:
III期(細胞性サブタイプ)先天性間葉芽腎腫に対する治療法の選択肢には以下のものがある:
- 腎摘出術。
- 化学療法。
腎摘出術
証拠(腎摘出術):
- SIOP/Gesellschaft für Pädiatrische Hämatologie und Onkologie(GPOH)の腎芽細胞腫に関する研究では、先天性間葉芽腎腫を有する患者111人が93.2%の5年RFS率および96.8%の5年OS率を示した。[ 8 ]
補助化学療法
補助化学療法は、診断時に生後3ヵ月以上で細胞性サブタイプのIII期間葉芽腎腫を有する患者に推奨されている。[ 5 ]III期の細胞性サブタイプの先天性間葉芽腎腫に関する研究において、手術単独で治療された12人の患者のうち7人が再燃した一方、補助化学療法(主としてダクチノマイシン/ビンクリスチンおよびときにドキソルビシン)で治療された14人の患者では4人が再燃を来した。[ 1 ][ 5 ][ 10 ]シクロホスファミドおよびイホスファミドがこれらの薬物と併用され、活性を示している。[ 11 ]
不完全切除を受けた生後2ヵ月未満のIII期の乳児には、化学療法が不要な場合がある。[ 1 ]
(再発疾患に関する情報については、本要約の再発先天性間葉芽腎腫の治療のセクションを参照のこと。)
先天性間葉芽腎腫に対して臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
現在実施されている国および/または施設の臨床試験の例を次に掲げる:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- England RJ, Haider N, Vujanic GM, et al.: Mesoblastic nephroma: a report of the United Kingdom Children's Cancer and Leukaemia Group (CCLG). Pediatr Blood Cancer 56 (5): 744-8, 2011.[PUBMED Abstract]
- Jehangir S, Kurian JJ, Selvarajah D, et al.: Recurrent and metastatic congenital mesoblastic nephroma: where does the evidence stand? Pediatr Surg Int 33 (11): 1183-1188, 2017.[PUBMED Abstract]
- van den Heuvel-Eibrink MM, Grundy P, Graf N, et al.: Characteristics and survival of 750 children diagnosed with a renal tumor in the first seven months of life: A collaborative study by the SIOP/GPOH/SFOP, NWTSG, and UKCCSG Wilms tumor study groups. Pediatr Blood Cancer 50 (6): 1130-4, 2008.[PUBMED Abstract]
- Gooskens SL, Houwing ME, Vujanic GM, et al.: Congenital mesoblastic nephroma 50 years after its recognition: A narrative review. Pediatr Blood Cancer 64 (7): , 2017.[PUBMED Abstract]
- Furtwaengler R, Reinhard H, Leuschner I, et al.: Mesoblastic nephroma--a report from the Gesellschaft fur Pädiatrische Onkologie und Hämatologie (GPOH). Cancer 106 (10): 2275-83, 2006.[PUBMED Abstract]
- El Demellawy D, Cundiff CA, Nasr A, et al.: Congenital mesoblastic nephroma: a study of 19 cases using immunohistochemistry and ETV6-NTRK3 fusion gene rearrangement. Pathology 48 (1): 47-50, 2016.[PUBMED Abstract]
- Argani P, Ladanyi M: Recent advances in pediatric renal neoplasia. Adv Anat Pathol 10 (5): 243-60, 2003.[PUBMED Abstract]
- Vokuhl C, Nourkami-Tutdibi N, Furtwängler R, et al.: ETV6-NTRK3 in congenital mesoblastic nephroma: A report of the SIOP/GPOH nephroblastoma study. Pediatr Blood Cancer 65 (4): , 2018.[PUBMED Abstract]
- Knezevich SR, Garnett MJ, Pysher TJ, et al.: ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res 58 (22): 5046-8, 1998.[PUBMED Abstract]
- Bayindir P, Guillerman RP, Hicks MJ, et al.: Cellular mesoblastic nephroma (infantile renal fibrosarcoma): institutional review of the clinical, diagnostic imaging, and pathologic features of a distinctive neoplasm of infancy. Pediatr Radiol 39 (10): 1066-74, 2009.[PUBMED Abstract]
- McCahon E, Sorensen PH, Davis JH, et al.: Non-resectable congenital tumors with the ETV6-NTRK3 gene fusion are highly responsive to chemotherapy. Med Pediatr Oncol 40 (5): 288-92, 2003.[PUBMED Abstract]
- 腎臓のユーイング肉腫
-
腎臓のユーイング肉腫に関する一般情報
腎臓のユーイング肉腫(以前は神経上皮腫瘍として知られていた)は、きわめてまれで、若年成人にみられるという特有な傾向を示す。非常に侵攻性の強い腫瘍であり、大きな腫瘍を形成し、腎被膜を越えて腎静脈内に進展することが多く、症例の40%で転移所見が認められる。[ 1 ][ 2 ][ 3 ]
腎臓のユーイング肉腫は、CD99(MIC-2)陽性およびEWS/FLI-1融合転写産物の検出を特徴とする。腎臓のユーイング肉腫内には、明細胞肉腫、ラブドイド腫瘍、悪性末梢神経鞘由来腫瘍、および副神経節細胞腫にもみられる巣状性異型性組織学的特徴が認められる。[ 1 ][ 4 ](詳しい情報については、ユーイング肉腫の治療に関するPDQ要約を参照のこと。)
腎臓のユーイング肉腫の治療
腎臓のユーイング肉腫に対する標準治療法は存在しない。しかし、化学療法および放射線療法による治療に加え、積極的な手術アプローチによって、過去の報告よりも転帰良好であると考えられる。[ 2 ]腎摘出術を受けた後の患者には、イホスファミドの代わりにシクロホスファミドを投与することも検討するべきである。[ 2 ][ 3 ]
ユーイング肉腫プロトコルに基づく治療を検討すべきである。[ 1 ]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Parham DM, Roloson GJ, Feely M, et al.: Primary malignant neuroepithelial tumors of the kidney: a clinicopathologic analysis of 146 adult and pediatric cases from the National Wilms' Tumor Study Group Pathology Center. Am J Surg Pathol 25 (2): 133-46, 2001.[PUBMED Abstract]
- Tagarelli A, Spreafico F, Ferrari A, et al.: Primary renal soft tissue sarcoma in children. Urology 80 (3): 698-702, 2012.[PUBMED Abstract]
- Rowe RG, Thomas DG, Schuetze SM, et al.: Ewing sarcoma of the kidney: case series and literature review of an often overlooked entity in the diagnosis of primary renal tumors. Urology 81 (2): 347-53, 2013.[PUBMED Abstract]
- Ellison DA, Parham DM, Bridge J, et al.: Immunohistochemistry of primary malignant neuroepithelial tumors of the kidney: a potential source of confusion? A study of 30 cases from the National Wilms Tumor Study Pathology Center. Hum Pathol 38 (2): 205-11, 2007.[PUBMED Abstract]
- 原発性腎筋上皮がん
-
原発性腎筋上皮がんに関する一般情報
筋上皮がんは、主に軟部組織に現れる侵攻性の悪性腫瘍で、ときに内臓器官にみられる。報告症例全体の約20%が小児であり、特に予後不良な転帰、高頻度の転移発生、および短い全生存期間と関係している。[ 1 ]
原発性腎筋上皮がんの2症例が小児にみられ、いずれの症例にもEWSR1と腎臓内で特有な機能を果たす転写因子である新規の融合パートナーKLF15との転座が認められた。診断確定に有用な特徴としては、サイトケラチン、S-100、および平滑筋マーカーの共発現、EWSR1再構成の報告などがある。[ 2 ]
原発性腎筋上皮がんの治療
標準療法は確立されていないが、化学療法および放射線療法に加えて、腫瘍原発巣および肺結節(存在する場合)の外科的切除が使用されている。[ 2 ]
参考文献- Gleason BC, Fletcher CD: Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol 31 (12): 1813-24, 2007.[PUBMED Abstract]
- Cajaiba MM, Jennings LJ, Rohan SM, et al.: Expanding the Spectrum of Renal Tumors in Children: Primary Renal Myoepithelial Carcinomas With a Novel EWSR1-KLF15 Fusion. Am J Surg Pathol 40 (3): 386-94, 2016.[PUBMED Abstract]
- 嚢胞性部分的分化型腎芽細胞腫
-
嚢胞性部分的分化型腎芽細胞腫に関する一般情報
嚢胞性部分的分化型腎芽細胞腫は、ウィルムス腫瘍のまれな嚢胞性の変異型であり(1%)、独特の病理学的および臨床的特徴を伴う。この腫瘍は全体的に嚢胞で構成されており、その薄い中隔は腫瘍の唯一の固形部分である。中隔は、胚性の間質細胞型または上皮細胞型を認めるまたは認めないあらゆる量の芽体細胞(blastemal cells)を含む。いくつかの病理学的特徴により、この腫瘍は標準的なウィルムス腫瘍と区別される。嚢胞性部分的分化型腎芽腫でDICER1変異が報告されておらず、このことは嚢胞性部分的分化型腎芽細胞腫と多房性嚢胞性腎腫を区別する考えを支持するものである。[ 1 ]
手術時の腫瘍漏出(tumor spillage)後の再発が報告されている。[ 2 ][証拠レベル:3iiiA]
(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
嚢胞性部分的分化型腎芽細胞腫の治療
嚢胞性部分的分化型腎芽細胞腫の標準治療法には、以下の選択肢がある:
参考文献- Cajaiba MM, Khanna G, Smith EA, et al.: Pediatric cystic nephromas: distinctive features and frequent DICER1 mutations. Hum Pathol 48: 81-7, 2016.[PUBMED Abstract]
- Baker JM, Viero S, Kim PC, et al.: Stage III cystic partially differentiated nephroblastoma recurring after nephrectomy and chemotherapy. Pediatr Blood Cancer 50 (1): 129-31, 2008.[PUBMED Abstract]
- Blakely ML, Shamberger RC, Norkool P, et al.: Outcome of children with cystic partially differentiated nephroblastoma treated with or without chemotherapy. J Pediatr Surg 38 (6): 897-900, 2003.[PUBMED Abstract]
- 多房性嚢胞性腎腫
-
多房性嚢胞性腎腫に関する一般情報
多房性嚢胞性腎腫は、腎上皮により裏打ちされた嚢胞からなるまれな良性病変である。2峰性の発症年齢分布を特徴とし、乳児/若年小児または成人のいずれかの女性に多くみられる。これらの病変は両側性に生じることがあり、家族性パターンが報告されている。
多房嚢腫性腎腫瘍は、胸膜肺芽腫およびDICER1変異に関連している。腎未熟型肉腫もまた、DICER1変異に関連している。[ 1 ]これは、DICER1変異を認めない成人の嚢胞性腎腫と対照的であることから、成人と小児症例の違いが裏付けられる。遺伝カウンセリング、DICER1変異検査、固形または嚢胞性の肺病変に対するスクリーニングを検討すべきである。[ 2 ][ 3 ][ 4 ][ 5 ]
(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
多房嚢腫性腎腫瘍の治療
多房性嚢胞性腎腫に対する標準治療法の選択肢は手術である。
参考文献- Wu MK, Goudie C, Druker H, et al.: Evolution of Renal Cysts to Anaplastic Sarcoma of Kidney in a Child With DICER1 Syndrome. Pediatr Blood Cancer 63 (7): 1272-5, 2016.[PUBMED Abstract]
- Dehner LP, Messinger YH, Schultz KA, et al.: Pleuropulmonary Blastoma: Evolution of an Entity as an Entry into a Familial Tumor Predisposition Syndrome. Pediatr Dev Pathol 18 (6): 504-11, 2015 Nov-Dec.[PUBMED Abstract]
- Doros LA, Rossi CT, Yang J, et al.: DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 27 (9): 1267-80, 2014.[PUBMED Abstract]
- Cajaiba MM, Khanna G, Smith EA, et al.: Pediatric cystic nephromas: distinctive features and frequent DICER1 mutations. Hum Pathol 48: 81-7, 2016.[PUBMED Abstract]
- Li Y, Pawel BR, Hill DA, et al.: Pediatric Cystic Nephroma Is Morphologically, Immunohistochemically, and Genetically Distinct From Adult Cystic Nephroma. Am J Surg Pathol 41 (4): 472-481, 2017.[PUBMED Abstract]
- 原発性腎滑膜肉腫
-
原発性腎滑膜肉腫に関する一般情報
原発性腎滑膜肉腫は腎胎児性肉腫のサブセットであり、特徴としてt(x;18)(p11;q11)のSS18-SSX転座がみられる。単相性紡錘細胞滑膜肉腫(monophasic spindle cell synovial sarcoma)と組織学が類似しており、侵攻性の腫瘍と考えられ、症例の50%以上で患者の転帰は不良である(n = 16)。[ 1 ]原発性腎滑膜肉腫において、2つ目の新たな遺伝子融合多様体、SS18-NEDD4が同定されている。[ 2 ]原発性腎滑膜肉腫は閉じ込められた拡張型腎尿細管に由来する嚢腫様の構造を含む。原発性腎滑膜肉腫は若年成人でより高頻度に起こる。
(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
原発性腎滑膜肉腫の治療
原発性腎滑膜肉腫に対する標準治療法の選択肢は化学療法である。原発性腎滑膜肉腫に用いられる化学療法レジメンは、ウィルムス腫瘍に対して使用されてきた化学療法レジメンとは異なる。[ 3 ]
参考文献- Schoolmeester JK, Cheville JC, Folpe AL: Synovial sarcoma of the kidney: a clinicopathologic, immunohistochemical, and molecular genetic study of 16 cases. Am J Surg Pathol 38 (1): 60-5, 2014.[PUBMED Abstract]
- Argani P, Zhang L, Sung YS, et al.: Novel SS18-NEDD4 gene fusion in a primary renal synovial sarcoma. Genes Chromosomes Cancer 59 (3): 203-208, 2020.[PUBMED Abstract]
- Argani P, Faria PA, Epstein JI, et al.: Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol 24 (8): 1087-96, 2000.[PUBMED Abstract]
- 腎未熟型肉腫
-
腎未熟型肉腫に関する一般情報
腎未熟型肉腫(anaplastic sarcoma of the kidney)は、主に15歳未満の患者に同定されているまれな腎腫瘍である。
患者は腎腫瘤を呈し、最も一般的な転移部位は、肺、肝、および骨である。(小児腎腫瘍の臨床的特徴および診断的評価に関する詳しい情報については、本要約のウィルムス腫瘍の臨床的特徴およびウィルムス腫瘍の診断的評価および病期評価のセクションを参照のこと。)
10q21と18p11.2の間の再構成などの細胞遺伝学異常が報告されている。[ 1 ]これらの腫瘍は小児胸膜肺芽腫(詳しい情報については小児胸膜肺芽腫の治療に関するPDQ要約を参照のこと)および肝未分化胎児性肉腫(詳しい情報については小児肝がんの治療に関するPDQ要約の肝未分化胎児性肉腫に対する治療法の選択肢のセクションを参照のこと)に類似した病理学的特徴を示す。胸膜肺芽腫と腎肉腫の間には関係があるため、遺伝カウンセリングとDICER1の生殖細胞変異に対する検査を検討すべきである。年齢とDICER1変異検査に基づいて、固形または嚢胞性の肺病変に対するスクリーニングも検討すべきである。[ 2 ]
腎未熟型肉腫の治療
腎未熟型肉腫に対する標準治療法の選択肢は存在しない。従来、これらの腫瘍は退形成型ウィルムス腫瘍として同定され、それに応じて治療されてきた。[ 3 ]
参考文献- Gomi K, Hamanoue S, Tanaka M, et al.: Anaplastic sarcoma of the kidney with chromosomal abnormality: first report on cytogenetic findings. Hum Pathol 41 (10): 1495-9, 2010.[PUBMED Abstract]
- Doros LA, Rossi CT, Yang J, et al.: DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 27 (9): 1267-80, 2014.[PUBMED Abstract]
- Vujanić GM, Kelsey A, Perlman EJ, et al.: Anaplastic sarcoma of the kidney: a clinicopathologic study of 20 cases of a new entity with polyphenotypic features. Am J Surg Pathol 31 (10): 1459-68, 2007.[PUBMED Abstract]
- 腎芽腫症
-
腎芽腫症(びまん性過形成腎葉周囲腎芽腫症)に関する一般情報
多巣性のnephrogenic restの中には過形成となるものがあり、芽体細胞(blastemal cell)または尿細管細胞の皮質を厚くして腎臓を拡張させることがある。びまん性過形成腎葉周囲腎芽腫症とウィルムス腫瘍の困難な鑑別を下すのに、放射線学的検査が有用な場合がある。磁気共鳴画像法でnephrogenic restは造影剤を用いて均質かつ低強度の像が認められるのに対し、ウィルムス腫瘍ではエコー輝度の混合した不均質な像が得られる。切開生検は解釈が困難で、生検には病変部と周囲の腎実質との接合部を含めることが不可欠である。[ 1 ]化学療法の実施後に分化することがある。
腎芽腫症(びまん性過形成腎葉周囲腎芽腫症)の治療
びまん性過形成腎葉周囲腎芽腫症に対する治療には、以下の選択肢がある:
- 術前化学療法。
- 腎温存手術。両側例の頻度の高さとその後のウィルムス腫瘍の可能性から、腎温存手術が適応となりうる。[ 1 ]
証拠(術前化学療法および手術):
参考文献- Perlman EJ, Faria P, Soares A, et al.: Hyperplastic perilobar nephroblastomatosis: long-term survival of 52 patients. Pediatr Blood Cancer 46 (2): 203-21, 2006.[PUBMED Abstract]
- 再発小児腎腫瘍の治療
-
再発した腎ラブドイド腫瘍、腎明細胞肉腫、腎臓の神経上皮腫瘍、および腎細胞がんの患者では、第I相および第II相臨床試験が利用できる場合、これに基づく治療を考慮すべきである。
進行時に疾患に向けた治療を行う決定がなされたかどうかに関係なく、緩和ケアは依然として管理の中心となっている。これにより、QOLを最大化しながら、末期疾患に関連する症状とストレスを緩和する試みが確保される。
表9に、新たに診断された再発小児腎腫瘍に対する治療法の選択肢を記述する。
表9.再発小児腎腫瘍に対する治療法の選択肢 腫瘍型 治療法の選択肢 通常リスクの再燃ウィルムス腫瘍 手術、放射線療法、および化学療法 高リスクおよび超高リスクの再燃ウィルムス腫瘍 化学療法、手術、および/または放射線療法 造血幹細胞移植 再発腎明細胞肉腫 化学療法、手術、および/または放射線療法 再発先天性間葉芽腎腫 手術、化学療法、および放射線療法。 再発ウィルムス腫瘍の予後、予後因子、およびリスクカテゴリー
予後良好な組織型(FH)のウィルムス腫瘍患者の約15%および退形成組織型ウィルムス腫瘍患者の50%が再発を経験する。[ 1 ]最も一般的な再燃部位は肺で、腹部/側腹部および肝がこれに続く。ウィルムス腫瘍の小児における再発は、脳(0.5%)または骨でまれである。[ 2 ][ 3 ]以前は、予後良好な組織型のウィルムス腫瘍が再発した患者の救助率は25~40%であった。現代の併用療法の結果として、再発後の転帰は60%まで改善した。[ 4 ][ 5 ]
再発後の転帰に影響する可能性がある多くの予後的特徴が解析されているが、これらの特徴が互いに独立しているかどうかを明らかにするのは困難である。また、以下の予後因子は、原発および再発ウィルムス腫瘍に対する治療が進化するにつれて変化していくと考えられる:
National Wilms Tumor Study-5(NWTS-5)試験(NWTS-5 [COG-Q9401/NCT00002611])では、無再発期間および再発部位はもはや予後に関して有意ではないことが示された。[ 4 ][ 7 ]しかし、International Society of Pediatric Oncology(SIOP)の研究では、診断から12ヵ月以内に肺の再燃が認められた患者の予後(5年全生存[OS]率が47%)が、診断後12ヵ月以上経過してから肺の再燃が認められた患者の予後(5年OS率が75%)より悪かった。[ 8 ]
上記の結果を基に、以下の3つのリスクカテゴリーが識別された:
通常リスクの再燃ウィルムス腫瘍の治療
ウィルムス腫瘍が小さくI期で、手術のみの治療を受けた患児におけるEFS率は84%であった。1例以外すべての再燃した小児が、再発箇所に応じた治療により救助された。[ 7 ][ 10 ]
初期治療が即時腎摘出術に続いてビンクリスチンおよびダクチノマイシンによる化学療法で構成され、その後に再燃するウィルムス腫瘍患者は再治療が成功する可能性がある。
再燃した標準リスクのウィルムス腫瘍に対する治療法には、以下の選択肢がある:
手術、放射線療法、および化学療法
証拠(手術、放射線療法、および化学療法):
- 外科的切除(可能な場合)、放射線療法、ならびにビンクリスチン、ドキソルビシン、およびシクロホスファミドとエトポシドおよびシクロホスファミドとの交互のコースを用いるNWTS-5再燃プロトコルで58人の患者が治療された。[ 7 ]
高リスクおよび超高リスクの再燃ウィルムス腫瘍の治療
再燃した高リスクおよび超高リスクのウィルムス腫瘍に対する治療法には、以下の選択肢がある:
化学療法、手術、および/または放射線療法
証拠(化学療法、手術、および/または放射線療法):
- ビンクリスチン、ダクチノマイシン、およびドキソルビシンと放射線療法による初回治療後に再燃または進行した片側性ウィルムス腫瘍患者の約50%は、再治療が有効な可能性がある。片側性ウィルムス腫瘍患者60人が、シクロホスファミド/エトポシドとカルボプラチン/エトポシドとの交互のコース、手術、および放射線療法によるNWTS-5の再燃プロトコルによる治療を受けた。[ 4 ][証拠レベル:2A]
診断時にII期、III期、およびIV期の退形成型腫瘍を有する患者は、再発すると予後がきわめて不良である。[ 9 ]この患者集団では、イホスファミド、エトポシド、およびカルボプラチンによる併用療法の有効性が認められているが、重大な血液学的毒性作用が観察されている。[ 11 ]
HSCT
大量化学療法とその後の自家HSCTは、再発した高リスク患者に対して用いられている。[ 12 ][ 13 ];[ 14 ][証拠レベル:3ii]
証拠(HSCT):
- 1990年から2013年の間に大量化学療法とその後の自家HSCTを受けた再燃ウィルムス腫瘍患者253人の転帰がCenter for International Blood and Marrow Transplantation Researchに報告され、レビューが行われた。[ 15 ]
- 再燃したおよび難治性ウィルムス腫瘍で、大量化学療法とその後の自家幹細胞救助(HD-ASCR)で治療された患者24人を対象にした単一施設のシリーズにおいて、以下の結果が報告された:[ 14 ][証拠レベル:3ii]
化学療法と移植を比較したランダム化試験は報告されておらず、ケースシリーズには選択バイアスがかかっている。
このような救済療法の試みが失敗した場合にはできる限り第I相または第II相試験に基づく治療を提供すべきである。
再発ウィルムス腫瘍に対して臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
現在実施されている国および/または施設の臨床試験の例を次に掲げる:
再発腎明細胞肉腫の治療
腎明細胞肉腫は晩期再燃を特徴とする。しかし1992年以後の試験では、3年以内の再燃が最も多く、再発の好発部位は脳と肺であった。[ 16 ][ 17 ]再燃した腎明細胞肉腫患者37人のシリーズでは、再燃後の5年EFS率が18%、再燃後のOS率が26%であった。[ 17 ]
再燃した腎明細胞肉腫の至適治療法は確立されていない。再発腎明細胞肉腫患者の治療は、初期治療および再発部位により異なる。
再発腎明細胞肉腫に対する標準治療法には、以下の選択肢がある:
- 化学療法、完全外科切除(可能な場合)、および/または放射線療法。
シクロホスファミドおよびカルボプラチンを、初期に使用していない場合は、検討すべきである。脳に発生した症例も含む再発腎明細胞肉腫の患者では、イホスファミド、カルボプラチン、およびエトポシド(ICE)と外科的切除、放射線療法、またはその両方からなる局所制御との併用療法で効果が認められている。[ 17 ];[ 18 ][証拠レベル:2A]
再発腎明細胞肉腫の患者に大量化学療法とその後のHSCTを実施するかどうかは確定していない。再燃した腎明細胞肉腫の患者、計24人が大量化学療法とその後の自家HSCTを受けた。これらの患者のうち、12人(50%)が中央値52ヵ月後に無病生存していた。既に第二完全寛解に達していた患者は大量化学療法を受ける傾向が強かったことに注意すべきである。[ 13 ][ 17 ][ 18 ]
再発腎明細胞肉腫に対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
現在実施されている国および/または施設の臨床試験の例を次に掲げる:
再発先天性間葉芽腎腫の治療
再燃は先天性間葉芽腎腫患者の4%で報告され、いずれの再燃も診断後12ヵ月以内に起こった。ほとんどの再燃が局所で発生するが、転移性の再燃も報告されている。[ 19 ]再燃した患者の約70%が、手術、化学療法、および放射線療法の併用で構成された個別化治療で生存した。[ 19 ]
ETV6-NTRK3融合を含む再発または難治性疾患の患者に対しては、標的療法を考慮すべきである。ラロトレクチニブおよびエヌトレクチニブは、既知の後天性の抵抗性変異が認められず、NTRK遺伝子融合が認められる固形腫瘍を有し、転移性であるかまたは外科的切除により重度の罹病を来す可能性が高く、満足できる代替治療選択肢がないか、がんが治療後に進行した成人および小児患者に対して承認されているNTRK阻害薬である。[ 20 ][ 21 ]
再発先天性間葉芽腎腫に対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Green DM, Breslow NE, Beckwith JB, et al.: Effect of duration of treatment on treatment outcome and cost of treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (12): 3744-51, 1998.[PUBMED Abstract]
- Venkatramani R, Chi YY, Coppes MJ, et al.: Outcome of patients with intracranial relapse enrolled on national Wilms Tumor Study Group clinical trials. Pediatr Blood Cancer 64 (7): , 2017.[PUBMED Abstract]
- Iaboni DSM, Chi YY, Kim Y, et al.: Outcome of Wilms tumor patients with bone metastasis enrolled on National Wilms Tumor Studies 1-5: A report from the Children's Oncology Group. Pediatr Blood Cancer 66 (1): e27430, 2019.[PUBMED Abstract]
- Malogolowkin M, Cotton CA, Green DM, et al.: Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D, and doxorubicin. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 50 (2): 236-41, 2008.[PUBMED Abstract]
- Reinhard H, Schmidt A, Furtwängler R, et al.: Outcome of relapses of nephroblastoma in patients registered in the SIOP/GPOH trials and studies. Oncol Rep 20 (2): 463-7, 2008.[PUBMED Abstract]
- Grundy P, Breslow N, Green DM, et al.: Prognostic factors for children with recurrent Wilms' tumor: results from the Second and Third National Wilms' Tumor Study. J Clin Oncol 7 (5): 638-47, 1989.[PUBMED Abstract]
- Green DM, Cotton CA, Malogolowkin M, et al.: Treatment of Wilms tumor relapsing after initial treatment with vincristine and actinomycin D: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 48 (5): 493-9, 2007.[PUBMED Abstract]
- Warmann SW, Furtwängler R, Blumenstock G, et al.: Tumor biology influences the prognosis of nephroblastoma patients with primary pulmonary metastases: results from SIOP 93-01/GPOH and SIOP 2001/GPOH. Ann Surg 254 (1): 155-62, 2011.[PUBMED Abstract]
- Dome JS, Cotton CA, Perlman EJ, et al.: Treatment of anaplastic histology Wilms' tumor: results from the fifth National Wilms' Tumor Study. J Clin Oncol 24 (15): 2352-8, 2006.[PUBMED Abstract]
- Shamberger RC, Anderson JR, Breslow NE, et al.: Long-term outcomes for infants with very low risk Wilms tumor treated with surgery alone in National Wilms Tumor Study-5. Ann Surg 251 (3): 555-8, 2010.[PUBMED Abstract]
- Abu-Ghosh AM, Krailo MD, Goldman SC, et al.: Ifosfamide, carboplatin and etoposide in children with poor-risk relapsed Wilms' tumor: a Children's Cancer Group report. Ann Oncol 13 (3): 460-9, 2002.[PUBMED Abstract]
- Garaventa A, Hartmann O, Bernard JL, et al.: Autologous bone marrow transplantation for pediatric Wilms' tumor: the experience of the European Bone Marrow Transplantation Solid Tumor Registry. Med Pediatr Oncol 22 (1): 11-4, 1994.[PUBMED Abstract]
- Pein F, Michon J, Valteau-Couanet D, et al.: High-dose melphalan, etoposide, and carboplatin followed by autologous stem-cell rescue in pediatric high-risk recurrent Wilms' tumor: a French Society of Pediatric Oncology study. J Clin Oncol 16 (10): 3295-301, 1998.[PUBMED Abstract]
- Rossoff J, Tse WT, Duerst RE, et al.: High-dose chemotherapy and autologous hematopoietic stem-cell rescue for treatment of relapsed and refractory Wilms tumor: Re-evaluating outcomes. Pediatr Hematol Oncol 35 (5-6): 316-321, 2018 Aug - Sep.[PUBMED Abstract]
- Malogolowkin MH, Hemmer MT, Le-Rademacher J, et al.: Outcomes following autologous hematopoietic stem cell transplant for patients with relapsed Wilms' tumor: a CIBMTR retrospective analysis. Bone Marrow Transplant 52 (11): 1549-1555, 2017.[PUBMED Abstract]
- Seibel NL, Sun J, Anderson JR, et al.: Outcome of clear cell sarcoma of the kidney (CCSK) treated on the National Wilms Tumor Study-5 (NWTS). [Abstract] J Clin Oncol 24 (Suppl 18): A-9000, 502s, 2006.[PUBMED Abstract]
- Gooskens SL, Furtwängler R, Spreafico F, et al.: Treatment and outcome of patients with relapsed clear cell sarcoma of the kidney: a combined SIOP and AIEOP study. Br J Cancer 111 (2): 227-33, 2014.[PUBMED Abstract]
- Radulescu VC, Gerrard M, Moertel C, et al.: Treatment of recurrent clear cell sarcoma of the kidney with brain metastasis. Pediatr Blood Cancer 50 (2): 246-9, 2008.[PUBMED Abstract]
- Gooskens SL, Houwing ME, Vujanic GM, et al.: Congenital mesoblastic nephroma 50 years after its recognition: A narrative review. Pediatr Blood Cancer 64 (7): , 2017.[PUBMED Abstract]
- Drilon A, Laetsch TW, Kummar S, et al.: Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 378 (8): 731-739, 2018.[PUBMED Abstract]
- Entrectinib Shows Pediatric Potential. Cancer Discov 9 (7): OF4, 2019.[PUBMED Abstract]
- 小児がん治療に関する特別な考慮事項
-
小児および青年のがんはまれであるが、1975年以降、小児がん全体の発生率は徐々に増加している。[ 1 ]小児および青年のがん患者は、小児期および青年期に発生するがんの治療経験を有する専門医で構成される集学的チームを擁する医療機関に紹介すべきである。この集学的チームのアプローチとは、至適な生存期間およびQOLを得られるような治療、支持療法、リハビリテーションを小児が必ず受けられるようにするため、以下の医療専門家およびその他の専門家の技能を集結したものである:
小児および青年のがんの支持療法に関する具体的な情報については、支持療法と緩和ケアに関するPDQ要約を参照のこと。
米国小児科学会は、小児がん施設とそれらが小児がん患者の治療において担う役割に関するガイドラインを概説している。[ 2 ]このような小児がん施設では、小児および青年に発症するほとんどの種類のがんに関する臨床試験が行われており、大半の患者およびその家族に参加する機会が与えられている。小児および青年のがんに関する臨床試験は一般に、現在標準とされている治療法と、それより効果的であると思われる治療法とを比較するようデザインされる。小児がんの治癒的療法の進歩のほとんどは、小児腫瘍学グループ(COG)、International Society of Pediatric Oncology(SIOP)などの共同グループの後援を通じて、そうした臨床試験によって達成されたものである。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
参考文献- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.[PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.[PUBMED Abstract]
- 本要約の変更点(06/08/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
ウィルムス腫瘍
表2が改訂され、レジメンUH1およびレジメンUH2の記述が追加された(引用、参考文献220としてDaw et al.)。
本文で、小児腫瘍学グループ(COG)により用いられた放射線量に関する記述が改訂された。また本文に以下の記述が追加された;COG-AREN0321試験で用いられた放射線療法レジメンは現在標準の治療となっている。
本文に、International Society of Pediatric Oncology(SIOP)により用いられた放射線療法アプローチに関する記述が追加された。
本文に、すべての年齢の予後良好である組織型(上皮優勢型を示す)のI期ウィルムス腫瘍患者の転帰を報告したCOG研究の結果に関する記述が追加された(引用、参考文献228としてParsons et al.および証拠レベル:3iiiA)。
表5が改訂され、COG AREN0321研究においてびまん性退形成型のII期ウィルムス腫瘍を有する患者に対する転帰の結果が追加された。
表6が改訂され、COG AREN0321研究においてびまん性退形成型のIII期ウィルムス腫瘍を有する患者に対する転帰の結果が追加された。
表7が改訂され、COG AREN0321研究においてびまん性退形成型のIV期ウィルムス腫瘍を有する患者に対する転帰の結果が追加された。
本文に、びまん性退形成型ウィルムス腫瘍で測定可能な病変を有する患者に対してビンクリスチンおよびイリノテカンの併用を初期治療段階で検証したAREN0321研究の結果に関する記述が追加された(証拠レベル:3iiiDiiが追加された)。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、ウィルムス腫瘍とその他の小児腎腫瘍の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Wilms Tumor and Other Childhood Kidney Tumors Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/kidney/hp/wilms-treatment-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389282]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。