

ご利用について
医療専門家向けの本PDQがん情報要約では、小児星細胞腫の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児星細胞腫に関する一般情報
-
星細胞腫を含む原発性脳腫瘍は、小児の最も一般的な充実性腫瘍をともに構成する多様な疾病からなる1つのグループである。脳腫瘍はその組織像および分子的特徴によって分類されるが、腫瘍部位およびその拡がりの範囲も治療および予後に影響する重要な因子である。腫瘍の診断と分類には、免疫組織化学的分析、細胞遺伝学的ならびに分子遺伝学的所見、および細胞分裂能の測定が用いられる。
グリオーマは、脳および脊髄にみられるグリア前駆細胞から発生すると考えられている。グリオーマは、推定される臨床病理学的および組織学的サブタイプに従って命名される。星細胞腫は、小児のグリオーマで最も多く診断されるタイプである。
脳腫瘍の世界保健機関(WHO)分類に従うと、グリオーマはさらに低悪性度(悪性度IおよびII)または高悪性度(悪性度IIIおよびIV)の腫瘍に分類される。腫瘍が低悪性度の小児は予後が比較的良好であり、特に腫瘍が完全切除できる場合に予後が優れている。腫瘍が高悪性度の小児は一般的に予後が比較的不良であるが、これはサブタイプにいくぶん依存している。
PDQ小児脳腫瘍の治療要約は主に、神経系腫瘍に関するWHOの分類に従って構成されている。[ 1 ][ 2 ]神経系腫瘍の分類の詳しい説明と各種の脳腫瘍に対応する治療要約へのリンクについては、小児脳腫瘍および脊髄腫瘍の治療の概要に関するPDQ要約を参照のこと。
解剖学
小児星細胞腫は、中枢神経系(CNS)のいずれの部位にも発生する可能性がある(図を参照のこと)。腫瘍タイプごとのCNSの最好発部位については、表3を参照のこと。
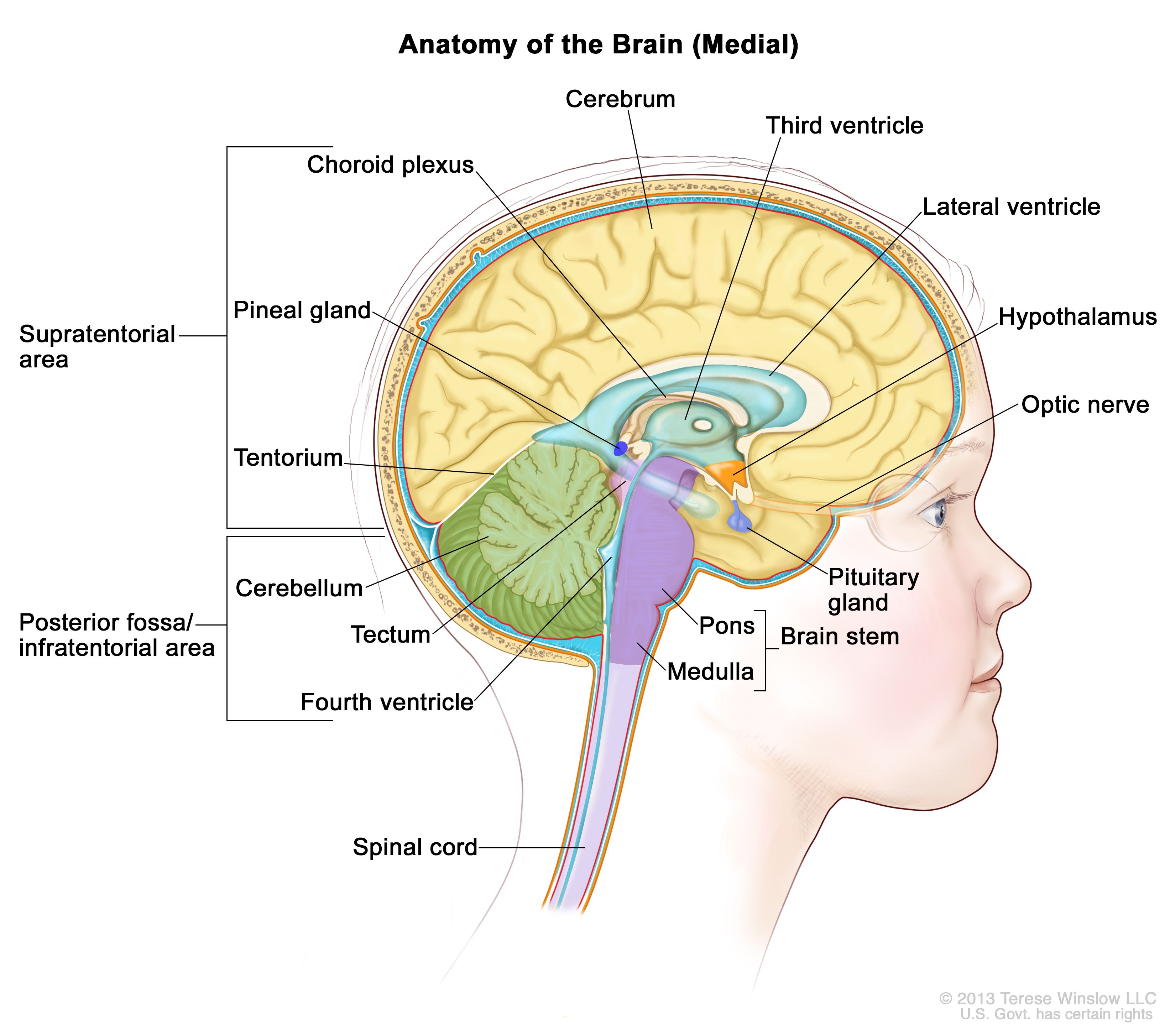
脳内部の解剖図で、大脳、小脳、脳幹、脊髄、視神経、視床下部、およびその他の脳組織を示す。 臨床的特徴
小児星細胞腫の主症状は、以下により異なる:
乳児および幼児では、視床下部に発生した低悪性度星細胞腫により間脳症候群を発症することがあり、やせほそっているが外見的には幸せそうにみえる小児で成長障害が認められる。こうした小児は、それ以外の神経学的所見に乏しいが、巨頭症、間欠的な嗜眠状態、および視覚障害を呈すことがある。[ 3 ]
診断的評価
星細胞腫の診断的評価には、脳または脊椎の磁気共鳴画像法(MRI)が含まれる。脳の原発腫瘍に対する脊髄MRIは通常、脳脊髄軸転移を除外するために初回評価としての脳MRIとの併用で施行される。
循環している腫瘍細胞がないか、脳脊髄液を検査する腰椎穿刺は、この疾患の小児においてはあまり実施されない。
小児星細胞腫および他の膠細胞由来の腫瘍の臨床病理学的分類
小児脳腫瘍の病理学的分類は、進展が続いている専門領域である。この領域において特別な専門知識を有する神経病理医による診断組織の検査が強く推奨される。
腫瘍タイプは、以下のように起源と推定されているグリア細胞を基にしている:
星細胞腫に対するWHOの組織学的悪性度
小児星細胞腫および他の膠細胞由来の腫瘍はWHOによるCNS腫瘍の組織型分類に従って、臨床病理学的および組織学的サブタイプに基づいて分類され、悪性度(悪性度I~IV)が決定される。[ 1 ]
WHOの組織学的悪性度では、低悪性度グリオーマまたは高悪性度グリオーマとして呼ばれることが多い(表1を参照のこと)。
表1.世界保健機関(WHO)の組織学的悪性度と中枢神経系腫瘍の分類との対応 WHOの組織学的悪性度 悪性度分類 I 低悪性度 II 低悪性度 III 高悪性度 IV 高悪性度 腫瘍の挙動は典型的に、一般的な生物学的変化により導かれるという証拠が発表され蓄積されているため、2016年のWHO基準では一部の腫瘍の診断で分子データが利用され始めた(表2を参照のこと)。このことは、CNSグリア系腫瘍では、びまん性グリオーマの分類における変化において最も明らかであり、びまん性グリオーマは病理組織学的類似性よりもむしろ遺伝的なドライバー変異に基づいて一緒に分類された。[ 2 ]次の2つのタイプのびまん性グリオーマはもはや別の疾患実体とは考えられていない:原線維性星細胞腫および原形質性星細胞腫。類上皮性膠芽腫はIDH野生型膠芽腫の1つのサブタイプとして分類された、新たに暫定的に含められた異型である。
表2.星細胞腫の2016年の世界保健機関(WHO)分類と組織学的悪性度a タイプ WHOの組織学的悪性度 a出典:Louis et al.[ 2 ] b2007年に、WHOは毛様細胞性星細胞腫の毛様類粘液性異型は、播種する可能性の高い侵攻性異型である可能性があると判断し、悪性度IIの腫瘍として再分類した。[ 1 ][ 2 ][ 4 ][ 5 ]2016年に、WHOはさらなる研究で毛様類粘液性異型の挙動が明らかにされるまでこれらの悪性度分類は行わないように提唱した。[ 1 ][ 2 ] びまん性星細胞腫: -びまん性星細胞腫、IDH変異型 II -退形成性星細胞腫、IDH変異型 III -膠芽腫、IDH野生型 IV -膠芽腫、IDH変異型 IV -びまん性正中グリオーマ、H3 K27M変異型 IV 他の星細胞腫: -毛様細胞性星細胞腫 I -毛様類粘液性星細胞腫 悪性度不明b -多形黄色星細胞腫 II -退形成性多形黄色星細胞腫 III -上衣下巨細胞星細胞腫 I 他のグリオーマ: -血管中心性膠腫 I -第三脳室の脈絡叢グリオーマ II —星芽腫 悪性度不明 CNSの発生部位
小児星細胞腫および他の膠細胞由来の腫瘍は、CNSのいずれの部位にも発生する可能性があるが、腫瘍タイプごとにCNS好発部位が異なる傾向がみられる(表3を参照のこと)。
表3.小児星細胞腫および他の膠細胞由来の腫瘍における中枢神経系(CNS)の好発部位 腫瘍のタイプ CNSの好発部位 毛様細胞性星細胞腫 視神経、視交叉/視床下部、視床および基底核、大脳半球、小脳、および脳幹;他に脊髄(まれ) 多形黄色星細胞腫 大脳の表在性部位(優先的に側頭葉) びまん性星細胞腫 大脳(前頭葉および側頭葉)、脳幹、脊髄、視神経、視交叉、視経路、視床下部および視床 退形成性星細胞腫、膠芽腫 大脳;ときに小脳、脳幹および脊髄 びまん性正中グリオーマ、H3 K27M変異型 橋、視床、脊髄、および他の正中線上の構造物 小脳:小脳に位置する星細胞腫の80%以上が低悪性度(毛様細胞性悪性度I)で、しばしば嚢胞性である;残りのほとんどがびまん性悪性度IIの星細胞腫である。小脳の悪性星細胞腫はまれである。[ 1 ][ 2 ]小脳または他の部位に発生する毛様細胞性星細胞腫についてイベントフリー生存を予測するために、特定の組織像(例えば、MIB-1標識率、退形成)の存在がレトロスペクティブに使用されている。[ 6 ][ 7 ][ 8 ]
脳幹:脳幹に発生する星細胞腫は、高悪性度の場合も低悪性度の場合もあり、いずれのタイプの頻度が高いかは、脳幹内の腫瘍部位に大きく依存している。[ 9 ][ 10 ]橋に浸潤していない腫瘍の大半は低悪性度グリオーマ(例、中脳視蓋グリオーマ)であり、一方でもっぱら橋に位置しているが外方増殖性要素のない腫瘍はほとんどがびまん性正中グリオーマ(例、H3 K27M変異型の遺伝子型を有するびまん性内在性橋グリオーマ)である。[ 9 ][ 10 ](詳しい情報については、小児脳幹グリオーマの治療に関するPDQ要約を参照のこと。)
大脳:高悪性度星細胞腫はしばしば局所浸潤性および広範性であり、大脳のテント上に発生する傾向がみられる。[ 11 ]くも膜下腔を経て拡がる可能性がある。中枢神経系以外への転移が報告されているが、多発性の局所再燃が発生するまではきわめてまれである。
大脳グリオーマ症はもはや異なる疾患実体とは考えられておらず、むしろいくつかのびまん性星細胞腫および、ときに乏突起膠細胞腫瘍でみられる増殖パターンの1つであると考えられる。その増殖パターンは大脳半球が広範囲に侵されることを包含しており、しばしば尾側に伸展し、脳幹、小脳、および/または脊髄に影響を及ぼす。[ 1 ]この増殖パターンが小脳に発生し、吻側に拡がることはまれである。[ 12 ]大脳グリオーマ症患者は当初は治療に反応を示す場合があるが、全般的に予後不良である。[ 13 ]
神経線維腫症1型(NF1)
NF1の小児は、視経路にWHO悪性度Iおよび悪性度IIの星細胞腫を発症する傾向が高い;すべてのNF1患者のうち、実に20%もの患者が視経路グリオーマを発症する。これらの患者では、小児が無症状であるか一見安定した神経障害および/または視覚障害を呈する場合のスクリーニングで腫瘍が発見される。
無症状の患者では病理学的確認が実施されないことが多い;生検が実施された際に、これらの腫瘍はびまん性の星細胞腫というよりはむしろ圧倒的に毛様細胞性(悪性度I)の星細胞腫であると判明している。[ 2 ][ 5 ][ 14 ]
一般に、サーベイランスの神経画像検査によって偶発的に発見される腫瘍には、治療を要しない。しばしば視力障害を引き起こす症状のある病変、またはX線撮影で進行が認められる病変は治療を要する。[ 15 ]
結節性硬化症
結節性硬化症の患者は低悪性度グリオーマ、特に上衣下巨細胞星細胞腫を発症する傾向が強い。TSC1またはTSC2のいずれかの遺伝子における変異が、哺乳類ラパマイシン標的蛋白(mTOR)経路に影響する経路を変化させ、増殖増加につながる。上衣下巨細胞星細胞腫はmTOR経路の阻害を介する標的アプローチに対する感受性が高い。[ 16 ]
ゲノム変化
低悪性度グリオーマの分子的特徴
毛様細胞性およびびまん性星細胞腫
BRAFおよびERK/MAPK経路の活性化に関与するゲノム変化は、低悪性度グリオーマの一種である毛様細胞性星細胞腫の散発例に非常に多くみられる。
BRAF-KIAA1549の変化
毛様細胞性星細胞腫におけるBRAFの活性化は、BRAF-KIAA1549の遺伝子融合によりBRAFの調節領域を欠く融合蛋白が産生されることで最も一般的に生じる。[ 17 ][ 18 ][ 19 ][ 20 ][ 21 ]この遺伝子融合は、テント下および正中線上の毛様細胞性星細胞腫のほとんどでみられるが、テント上(大脳半球)腫瘍で認められる頻度は低い。[ 17 ][ 18 ][ 22 ][ 23 ][ 24 ][ 25 ][ 26 ][ 27 ]
低悪性度のグリオーマを不完全切除した小児について著した1件の報告では、BRAF-KIAA1549融合の存在により、良好な臨床転帰(無増悪生存[PFS]および全生存[OS])が予測された。[ 26 ]しかしながら、CDKN2Aの欠失、7番染色体全体の増加、腫瘍の位置などの他の因子によって、転帰に対するBRAF変異の影響は変わる可能性がある。[ 28 ];[ 29 ][証拠レベル:3iiiDiii]BRAF-KIAA1549融合が認められる小児低悪性度グリオーマが高悪性度グリオーマに進行することはまれである。[ 30 ]
BRAF-KIAA1549融合によるBRAFの活性化は、他の小児低悪性度グリオーマ(例えば、毛様類粘液性星細胞腫)でも報告されている。[ 25 ][ 26 ]毛様細胞性星細胞腫では、ERK/MAPK経路を活性化する可能性のある他のゲノム変化(例、代替BRAF遺伝子融合、RAF1遺伝子再構成、RAS変異、およびBRAF V600Eの点変異)がまれに観察される。[ 18 ][ 20 ][ 21 ][ 31 ]
血管中心性膠腫
血管中心性膠腫は一般的に、痙攣発作を呈する大脳腫瘍として小児および若年成人に発症する。[ 2 ]
2016年の2件の報告で、血管中心性膠腫と診断されたほぼすべての症例にMYB遺伝子の変化が認められることが確認され、融合パートナーの検査が可能であった症例においてQKIが主要な融合パートナーであった。[ 41 ][ 42 ]血管中心性膠腫は最も一般的にはテント上に発生するが、MYB-QKI融合を伴う脳幹の血管中心性膠腫も報告されている。[ 43 ][ 44 ]
星芽腫
星芽腫は、組織学的にはGFAP陽性細胞で構成される膠細胞性新生物と定義され、しばしば硬化症を示す星状芽細胞性偽ロゼットを含む。星芽腫は主として、小児期から若年成人期に診断される。[ 2 ]
以下の研究で、星芽腫に関連するゲノム変化が記述されている:
これらの報告から、星芽腫の組織学的診断にはゲノム的に定義される不均一な疾患実体グループが含まれること示唆されている;MN1融合を伴う星芽腫は組織学的に診断された異なる症例の異なるサブセットである。[ 49 ]
結節性硬化症
結節性硬化症患児のほとんどでは、2つの結節性硬化症遺伝子(TSC1/ハマルチンまたはTSC2/ツベリン)のどちらかに生殖細胞変異が認められる。これらの変異は、いずれも哺乳類ラパマイシン標的蛋白(mTOR)複合体1の活性化を引き起こす。これらの小児では、上衣下巨細胞星細胞腫、皮質結節、および上衣下結節を発症するリスクがある。上衣下巨細胞星細胞腫はmTORの活性化により推進されるため、mTOR阻害剤はこうした腫瘍を有する小児において腫瘍退縮を誘発できる活性のある薬物である。[ 50 ]
高悪性度グリオーマの分子的特徴
小児の高悪性度グリオーマで、特に多形性膠芽腫は、成人に発生する腫瘍と生物学的に異なっている。[ 51 ][ 52 ][ 53 ][ 54 ]
DNAメチル化パターンを用いて同定されたサブグループ
小児の高悪性度グリオーマは、エピジェネティックパターン(DNAメチル化)に基づいて別個のサブグループに分類でき、これらのサブグループは腫瘍における独特な染色体コピー数増加/減少および遺伝子変異を示す。[ 55 ][ 56 ][ 57 ]小児の高悪性度グリオーマの中で特に独特なサブタイプは、ヒストン遺伝子の特定のアミノ酸に反復性の変異がみられるグリオーマであり、これらは合わせて、小児高悪性度グリオーマの約1/2を占める。[ 57 ]
DNAメチル化パターンに基づいて、以下の小児高悪性度グリオーマサブグループが同定され、これらは独特な分子的および臨床的特徴を示す:[ 57 ]
- ヒストンK27変異:H3.3(H3F3A)およびH3.1(HIST1H3BおよびまれにHIST1H3C)のK27での変異:ヒストンK27変異症例は、主に小児期中期(年齢中央値が約10歳)に現れ、ほぼ例外なく正中線構造(視床、脳幹、および脊髄)にみられるもので、きわめて不良な予後をもたらす。2016 WHO分類では、これらのがんを単一の疾患実体であるびまん性正中グリオーマ、H3 K27M変異型に分類しているが、後述のようにH3.3とH3.1変異を有する症例では臨床的および生物学的な相違がみられる。[ 2 ]これらの症例はK27Mの存在を同定するための免疫組織化学を用いて診断可能である。
- H3.3(H3F3A)のG34での変異:H3.3G34サブタイプは、少し年長の小児および若年成人(年齢中央値が14~18歳)にみられ、発生は大脳皮質に限局している。[ 55 ][ 56 ]H3.3G34症例は一般的にTP53およびATRXの変異を有し、全ゲノムにわたる広範な低メチル化を示す。H3F3A変異を有する患者は治療失敗のリスクが高い[ 60 ]が、予後はヒストン3.1または3.3 K27M変異を有する患者ほど不良ではない。[ 56 ]O6-メチルグアニン-DNA-メチルトランスフェラーゼ(MGMT)メチル化が約2/3の症例で観察され、IDH1変異サブタイプ(下記を参照のこと)を除いて、H3.3G34サブタイプは20%を超えるMGMTメチル化の割合を示す唯一の小児高悪性度グリオーマサブタイプである。[ 57 ]
- IDH1変異:IDH1変異症例は小児高悪性度グリオーマのわずかな割合(約5%)を占めており、腫瘍にIDH1変異が認められる小児高悪性度グリオーマ患者は、ほとんど例外なく大脳半球腫瘍を有する年齢の高い青年(小児集団における年齢中央値、16歳)である。[ 57 ]IDH1変異症例はしばしば、TP53変異、MGMTプロモーターメチル化、およびグリオーマ-CpG island methylator phenotype(G-CIMP)を示す。[ 55 ][ 56 ]IDH1変異を有する小児患者は、他の小児多形性膠芽腫患者よりも良好な予後を示す;IDH1変異を有する小児患者では、5年全生存率(OS)が60%を超えるが、野生型IDH1患者では5年OS率が20%未満である。[ 57 ]
- 多形黄色星細胞腫(PXA)-like:小児高悪性度グリオーマの約10%に、PXA-likeのDNAメチル化パターンがみられる。[ 56 ]PXA-like症例は一般的にBRAF V600E変異を有し、転帰が比較的良好である(約50%の5年生存率)。[ 57 ][ 60 ]
- 低悪性度グリオーマ-like:高悪性度グリオーマの組織学的外観を有する小児脳腫瘍の小規模なサブセットは、低悪性度グリオーマに似たDNAメチル化パターンを示す。[ 56 ][ 57 ]これらの症例は主に年齢の低い患者(年齢中央値は4歳)で観察される;多形性膠芽腫を診断された乳児16人中10人が低悪性度グリオーマ-like集団に含まれていた。[ 57 ]これらの患者の予後は、他の小児高悪性度グリオーマのサブタイプよりもはるかに良好である。[ 60 ]乳児における多形性膠芽腫の追加の考察については、下記を参照のこと。
他の変異
腫瘍にヒストン変異もIDH1変異も認められない小児多形性膠芽腫の高悪性度グリオーマ患者は、小児多形性膠芽腫症例の約40%を占める。[ 57 ][ 61 ]これは、他の小児高悪性度グリオーマサブタイプよりも遺伝子増幅率が高い不均一な集団である。最も一般的に増幅が認められる遺伝子は、PDGFRA、EGFR、CCND/CDK、およびMYC/MYCNである[ 55 ][ 56 ];この集団ではMGMTプロモーターのメチル化の割合は低い。[ 61 ]1件の報告でこの集団が3つのサブタイプに分けられた。高いMYCN増幅率を特徴とするサブタイプが最も不良な予後を示した一方、TERTプロモーター変異およびEGFR増幅を特徴とするサブタイプは最も良好な予後を示した。3つ目のグループはPDGFRA増幅により特徴付けられた。[ 61 ]
乳児におけるグリオーマ
多形性膠芽腫が診断された乳児および幼児は、より年齢の高い小児および成人の腫瘍と比較して腫瘍の分子的特徴が異なるようである。小児の多形性膠芽腫に対してDNAメチル化解析を実施したところ、腫瘍の分子的特徴が低悪性度グリオーマと一致した患者のグループ(多形性膠芽腫が組織学的に診断された小児患者の約7%を占めた)が確認された。この患者集団の年齢中央値は1歳で、乳児10人中8人が低悪性度グリオーマ-likeプロファイルを示した。[ 56 ]低悪性度グリオーマ-likeサブタイプは予後良好であった(3年OS率、約90%)。[ 56 ][ 57 ]BRAF V600E変異は、低悪性度グリオーマ-like腫瘍13例中4例および3歳以下の患者の腫瘍15例中3例で観察された。[ 56 ]
2つ目の報告では、生後36ヵ月未満の小児からの多形性膠芽腫について遺伝子コピー数の増加と減少、および選択された遺伝子の変異状態が調査された。[ 62 ]年齢の高い小児では測定可能な割合で観察された分子的変化(例、K27M、CDKN2A喪失、PDGFRA増幅、およびTERTプロモーター変異)は、これらの幼児の腫瘍ではまれであり、新たな異常(例、染色体14q32におけるSNORDの喪失)が一部の症例で観察された。
ゲノム特性化のために腫瘍組織が利用可能であった乳児118人を対象にした研究において、乳児(生後12ヵ月未満)に発生するグリオーマの特有な分子的特徴がさらに明らかにされた。[ 63 ]症例の約75%が低悪性度に分類されたが、低悪性度コホートに対するOS率が比較的低く(71%)、高悪性度コホートに対する生存が比較的良好である(55%)ため、この年齢集団における組織学的分類の有用性は低いことが示された。ゲノム特性化により、グリオーマの乳児集団は以下の3つのグループに分類された:
続発性高悪性度グリオーマ
小児続発性高悪性度グリオーマ(低悪性度グリオーマが先行する高悪性度グリオーマ)はまれである(886人を対象にした研究で2.9%)。BRAF-KIAA1549融合が認められる小児低悪性度グリオーマが高悪性度グリオーマに形質転換した例はないが、BRAF V600E突然が認められる低悪性度グリオーマは形質転換するリスクが高い。続発性高悪性度グリオーマ患者18人中の7人(約40%)にBRAF V600E突然が認められ、症例14人中8人(57%)にCDKN2Aの変化が認められた。[ 30 ]
神経線維腫症1型(NF1)
NF1の小児において高悪性度グリオーマが発生することがあるが、低悪性度グリオーマの方がはるかに一般的である。高悪性度腫瘍が発生する場合は、成人期に発生することが最も多い。NF1関連高悪性度グリオーマを有する患者23人(年齢中央値、38.8歳)のゲノム特性化によって、低悪性度グリオーマを有するNF1患者と比較して変異の割合が高かったことが示された(それぞれ、21.5の変異 vs 6の変異)。[ 64 ]大多数の患者が、ヘテロ接合性の消失またはNF1の2つ目のアレルにおける不活性化変異のいずれかを伴うNF1の生殖細胞変異を示した。NF1関連低悪性度グリオーマとは対照的に、高悪性度グリオーマに関連するゲノム変化が一般的であった(CDKN2A[58%]、ATRX[38%]、およびTP53[29%])。[ 64 ]
神経細胞腫瘍および混合神経細胞・膠細胞腫瘍の分子的特徴
神経細胞腫瘍および混合神経細胞・膠細胞腫瘍は、悪性度IIIの退形成性神経節膠腫を除いて、一般的に低悪性度腫瘍である。2016年のWHO分類で認識されている組織型には以下のものがある:[ 2 ]
胚芽異形成性神経上皮腫瘍(DNET)
DNETは小児および成人に発症し、診断時年齢中央値は青年期中期から後期である。病理組織学的には、柱状の乏突起膠腫様細胞および粘液中に遊離した皮質神経節細胞の存在を特徴とする。[ 1 ]側頭葉は最も一般的な部位であり、薬剤抵抗性てんかんに関連する。[ 65 ][ 66 ]
DNETの60~80%にFGFR1の変化が報告されており、FGFR1活性化点変異、キナーゼドメインの遺伝子内縦列重複、および活性化遺伝子融合が含まれる。[ 42 ][ 67 ][ 68 ]DNETでは、BRAF変異はまれである。
透明中隔のDNET
中隔DNETは一般に、閉塞性水頭症に関係した症状を呈する。[ 69 ][ 70 ]中隔DNETは緩徐な臨床像を有し、ほとんどの腫瘍が手術以外の治療を必要としない。他の文献で報告された症例を組み込んだ単一施設のシリーズにおいて、発症時年齢中央値は青年の年齢層であった。[ 71 ]
低悪性度グリオーマ(例、BRAF V600E)および皮質DNET(FGFR1変異)に一般的な変異は、中隔DNETではまれである。[ 70 ][ 71 ][ 72 ]その代わり、K385残基のPDGFRAにおける変異は、中隔DNETのほとんどの症例で典型的に認められる。
18例の中隔DNETの分子生物学的検討に関する報告から、14例がPDGFRA変異を有し、1例を除く全例がK385残基における変異であり[ 71 ]、PDGFの結合と同時に二量体形成および活性化に必要となる受容体-受容体相互作用を媒介するPDGFRAの細胞外領域にこの変異が認められることが示された。残りの4例のうち、3例は皮質DNETで観察されたものと一致するFGFR1変異を有した。2つ目の報告では、中隔DNETの4症例それぞれのK385においてPDGFRA変異が観察された。[ 72 ]総合すると、2件の報告から、中隔DNETは、ありふれた解剖学的部位および、ほとんどの症例においてPDGFRA変異を特徴とする異なる疾患実体であることが示されている。K385のPDGFRA変異を有する低悪性度グリア神経細胞性腫瘍もまた、脳梁および側脳室の脳室周囲白質における発生が確認されたことから、粘液型グリア神経細胞性腫瘍、PDGFRA p.K385変異型を中枢神経系(CNS)腫瘍の異なる疾患実体として考慮すべきであると提案されている。[ 73 ]
神経節膠腫
神経節膠腫は小児期から成人期に発症する。神経節膠腫は最も一般的には大脳皮質に発生し、痙攣発作を伴うが、脊髄など、他の部位にも発生する。[ 65 ][ 74 ]
神経節膠腫の分子的発生機序に対する統一的なテーマは、MAPK経路の活性化につながるゲノム変化である。[ 42 ][ 75 ]BRAFの変化は神経節膠腫症例の約50%で観察され、V600Eは飛び抜けて多くみられる変化となっている;しかしながら、他のBRAF変異および遺伝子融合も観察されている。神経節膠腫における他のあまり一般的ではない変異遺伝子には、KRAS、FGFR1/2、RAF1、NTRK2、およびNF1がある。[ 42 ][ 75 ]
線維形成性乳児星細胞腫(DIA)および線維形成性乳児神経節膠腫(DIG)
DIAおよびDIGは生後1年の間に最もしばしば発症し、コントラスト増強性の充実性結節が大きな嚢胞性成分に伴って認められる特徴的な画像所見を示す。[ 76 ][ 77 ]DIGはDIAよりも一般的であり[ 76 ]、メチル化アレイ解析では、両方が同時に診断される場合がある。[ 78 ]一般に外科的切除で良好な生存転帰が得られる。[ 76 ]
DIAおよびDIGにおいて最も一般的に観察されるゲノム変化はV600が関与するBRAF変異である;キナーゼ遺伝子が関与する遺伝子融合が観察される頻度はあまり高くない。
乳頭状グリア神経細胞性腫瘍
乳頭状グリア神経細胞性腫瘍は、主にテント上部に発生し、星細胞腫と神経細胞腫分化を示す低悪性度の二相性新生物である。[ 2 ]発症時年齢中央値は20代前半であるが、小児期から成人期に観察される。
乳頭状グリア神経細胞性腫瘍に関連する主要なゲノム変化はSLC44A1-PRKCAの遺伝子融合で、t(9:17)(q31;q24)転座に関連している。[ 81 ][ 82 ]メチル化アレイ解析を用いて乳頭状グリア神経細胞性腫瘍と組織学的に診断された28症例を対象にした1件の研究において、11症例が特有のメチル化クラスに集中した一方、残りの症例は他の腫瘍の疾患実体に典型的なメチル化プロファイルを示した。特有のメチル化クラスターにおける症例の分子解析では、NOTCH1-PRKCA遺伝子融合を有した単一症例を除く全例がSLC44A1-PRKCA遺伝子融合を有したことが示された。[ 83 ]このことから、PRKCA融合の存在を確認するための分子的検査法は、乳頭状グリア神経細胞性腫瘍の診断において、形態学に基づく検査法よりも誤分類の可能性が低いことが示唆されている。
ロゼット形成性グリア神経細胞性腫瘍(RGNT)
RGNTは青年および成人で発症し、腫瘍は一般的にテント下に位置するが、中脳または間脳領域に発生することもある。[ 84 ]典型的な組織学的外観は、ロゼットまたは血管周囲偽ロゼットで配列されたグリア細胞成分および神経細胞成分の両方を示す。[ 2 ]RGNT患者の転帰は一般的に良好で、WHO悪性度Iの指定と一致する。[ 84 ]
DNAメチル化プロファイル解析により、RGNTは他の低悪性度グリア細胞/グリア神経細胞性腫瘍の疾患実体とは区別される別個のエピジェネティックなプロファイルを有することが示されている。[ 84 ]RGNT症例30例の研究で、解析されたすべての腫瘍でFGFR1のホットスポット変異が観察された。[ 84 ]さらに、30例中19例(63%)ではPIK3CA活性化変異が同時に観察された。30例中10例(33%)ではNF1におけるミスセンス変異または機能障害性変異(damaging mutation)が同定され、7例の腫瘍にFGFR1、PIK3CA、およびNF1における変異が認められた。MAPK経路とPI3K経路の両方を活性化する変異の同時発生は、星細胞腫およびグリア神経細胞性腫瘍において特有なRGNTの変異プロファイルとなっている。
びまん性軟髄膜グリア神経細胞性腫瘍(DLGNT)
DLGNTは、放射線学的には磁気共鳴画像法(MRI)での軟髄膜増強(後頭蓋窩、脳幹領域、および脊髄に病変がみられうる)を特徴としているまれなCNS腫瘍である。[ 85 ]実質内病変は、認められる場合には一般的に脊髄にみられる[ 85 ];軟髄膜播種が認められず、組織形態学(histomorphologic)、免疫表現型、およびゲノムの特徴がDLGNTに類似した限局性の脊髄内グリア神経細胞性腫瘍が報告されている。[ 86 ]
DLGNTは、DNAメチル化アレイ解析で特有のエピジェネティックなプロファイルを示し、30例に適用されたアレイデータの教師なしクラスタリングにより、DLGNTの次の2つのサブクラスが明らかにされた:メチル化クラス(MC)-1(n = 17)およびMC-2(n = 13)。[ 85 ]注目すべきこととして、アレイ解析で明らかにされた症例の多くが当初は別の疾患実体(例、原始神経外胚葉性腫瘍、毛様細胞性星細胞腫、および退形成性星細胞腫)として診断されていた。DLGNT-MC-1を有する患者は、DLGNT-MC-2を有する患者よりも若年で診断された(それぞれ、5歳 vs 14歳)。5年全生存率は、DLGNT-MC-1を有する患者の方がDLGNT-MC-2を有する患者よりも高かった(それぞれ、100% vs 43%)。メチル化アレイ解析で明らかにされたDLGNT症例30例のゲノム所見を以下に示す:
脳室外神経細胞腫
脳室外神経細胞腫は組織学的に中心性神経細胞腫に類似しており、神経細胞分化を示す均一な小型細胞で構成されるが、脳室系に関連してというよりもむしろ脳実質に発生する。[ 2 ]脳室外神経細胞腫は小児期から成人期に発症する。
組織学的に脳室外神経細胞腫に分類され、メチル化アレイ解析に提出された40例の腫瘍に関する研究では、他の組織型の参照用腫瘍と異なる別個のクラスターを形成したのは26例のみであった。[ 88 ]メチル化アレイ解析で脳室外神経細胞腫と分類され、ゲノム特性化を実施できた症例について、15例中11例(73%)がFGFRファミリーメンバーに影響を及ぼす再構成を示し、FGFR1-TACC1が最も一般的な変化であった。[ 88 ]
予後
低悪性度星細胞腫
低悪性度星細胞腫(悪性度I[毛様細胞性]および悪性度II)は比較的に予後良好であり、全摘出を行える可能性がある境界明瞭な病変では特にその傾向が強い。[ 11 ][ 89 ][ 90 ][ 91 ][ 92 ]腫瘍の進展が生じた場合、通常は連続的な進展であり;他のCNS部位への播種はまれであるが、発生することはある。[ 93 ][ 94 ]転移はまれであるが、特にNF1に関連した腫瘍では、多巣性で発生する場合がある。
小児低悪性度星細胞腫に対する好ましくない予後的特徴には以下のものがある:[ 95 ][ 96 ][ 97 ][ 98 ]
毛様細胞性星細胞腫患者において、細胞増殖活性マーカーであるMIB-1標識指数の上昇はPFSの短縮に関連している。[ 8 ]毛様細胞性腫瘍に確認されたBRAF-KIAA1549融合は良好な臨床転帰をもたらす。[ 26 ]
視経路に腫瘍を有する小児の治療成績は、X線画像での疾患制御または生存だけでなく、視覚の転帰によっても評価される。孤立性の視神経腫瘍の患児では、病変が視交叉に及んでいる患児や視経路に沿って進展している患児よりも予後良好である。[ 100 ][ 101 ];[ 102 ][証拠レベル:3iiC]また、NF1小児の予後は、特にスクリーニング時に無症状の患者に腫瘍が発見された場合により良好である。[ 103 ]診断時の比較的良好な視力、診断時に比較的年齢が高いこと、およびNF1の存在が、良好な視覚の転帰に関連している。[ 104 ]
高悪性度星細胞腫
若年患者の高悪性度星細胞腫は、一般に不良な予後をもたらすが、肉眼的に全切除可能な退形成性星細胞腫の患者は予後がより良好な可能性があり[ 90 ][ 105 ][ 106 ]、H3 K27M変異のない腫瘍を有する患者も同様である。
小児多形性膠芽腫の分子的サブタイプは予後的意義を示す。[ 56 ]腫瘍にヒストンK27M変異が認められる患者は最も予後不良で、3年生存率は5%に満たない。視床における野生型高悪性度グリオーマは、H3.3変異を有するグリオーマよりいくぶん予後良好である。視床に浸潤する高悪性度グリオーマの場合、H3野生型腫瘍の患者の予後は、H3 K27M変異を有する患者(2年全生存率[OS]、13%)よりもやや良好である(2年OS、71%)。[ 107 ]腫瘍にIDH1変異が認められる患者は、小児多形性膠芽腫症例の中で最も予後良好なようであるが、ヒストンG34変異が認められる患者およびヒストンとIDH1変異の両方が認められない患者は中程度の予後を有する(3年OS率、約30%)。分子的因子と臨床因子の両方を含めた多変量解析では、遺伝子増幅およびK27M変異の存在はより不良な予後と関連する一方、IDH1変異の存在はより良好な予後と関連した。[ 56 ]
参考文献- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. Lyon, France: IARC Press, 2016.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Kilday JP, Bartels U, Huang A, et al.: Favorable survival and metabolic outcome for children with diencephalic syndrome using a radiation-sparing approach. J Neurooncol 116 (1): 195-204, 2014.[PUBMED Abstract]
- Louis DN, Ohgaki H, Wiestler OD, et al., eds.: WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon, France: IARC Press, 2007.[PUBMED Abstract]
- Komotar RJ, Burger PC, Carson BS, et al.: Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery 54 (1): 72-9; discussion 79-80, 2004.[PUBMED Abstract]
- Tibbetts KM, Emnett RJ, Gao F, et al.: Histopathologic predictors of pilocytic astrocytoma event-free survival. Acta Neuropathol 117 (6): 657-65, 2009.[PUBMED Abstract]
- Rodriguez FJ, Scheithauer BW, Burger PC, et al.: Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol 34 (2): 147-60, 2010.[PUBMED Abstract]
- Margraf LR, Gargan L, Butt Y, et al.: Proliferative and metabolic markers in incompletely excised pediatric pilocytic astrocytomas--an assessment of 3 new variables in predicting clinical outcome. Neuro Oncol 13 (7): 767-74, 2011.[PUBMED Abstract]
- Fried I, Hawkins C, Scheinemann K, et al.: Favorable outcome with conservative treatment for children with low grade brainstem tumors. Pediatr Blood Cancer 58 (4): 556-60, 2012.[PUBMED Abstract]
- Fisher PG, Breiter SN, Carson BS, et al.: A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocystic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer 89 (7): 1569-76, 2000.[PUBMED Abstract]
- Pfister S, Witt O: Pediatric gliomas. Recent Results Cancer Res 171: 67-81, 2009.[PUBMED Abstract]
- Rorke-Adams LB, Portnoy H: Long-term survival of an infant with gliomatosis cerebelli. J Neurosurg Pediatr 2 (5): 346-50, 2008.[PUBMED Abstract]
- Armstrong GT, Phillips PC, Rorke-Adams LB, et al.: Gliomatosis cerebri: 20 years of experience at the Children's Hospital of Philadelphia. Cancer 107 (7): 1597-606, 2006.[PUBMED Abstract]
- Allen JC: Initial management of children with hypothalamic and thalamic tumors and the modifying role of neurofibromatosis-1. Pediatr Neurosurg 32 (3): 154-62, 2000.[PUBMED Abstract]
- Molloy PT, Bilaniuk LT, Vaughan SN, et al.: Brainstem tumors in patients with neurofibromatosis type 1: a distinct clinical entity. Neurology 45 (10): 1897-902, 1995.[PUBMED Abstract]
- Franz DN, Weiss BD: Molecular therapies for tuberous sclerosis and neurofibromatosis. Curr Neurol Neurosci Rep 12 (3): 294-301, 2012.[PUBMED Abstract]
- Bar EE, Lin A, Tihan T, et al.: Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol 67 (9): 878-87, 2008.[PUBMED Abstract]
- Forshew T, Tatevossian RG, Lawson AR, et al.: Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218 (2): 172-81, 2009.[PUBMED Abstract]
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.[PUBMED Abstract]
- Jones DT, Kocialkowski S, Liu L, et al.: Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28 (20): 2119-23, 2009.[PUBMED Abstract]
- Pfister S, Janzarik WG, Remke M, et al.: BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118 (5): 1739-49, 2008.[PUBMED Abstract]
- Korshunov A, Meyer J, Capper D, et al.: Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 118 (3): 401-5, 2009.[PUBMED Abstract]
- Horbinski C, Hamilton RL, Nikiforov Y, et al.: Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol 119 (5): 641-9, 2010.[PUBMED Abstract]
- Yu J, Deshmukh H, Gutmann RJ, et al.: Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology 73 (19): 1526-31, 2009.[PUBMED Abstract]
- Lin A, Rodriguez FJ, Karajannis MA, et al.: BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol 71 (1): 66-72, 2012.[PUBMED Abstract]
- Hawkins C, Walker E, Mohamed N, et al.: BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17 (14): 4790-8, 2011.[PUBMED Abstract]
- Becker AP, Scapulatempo-Neto C, Carloni AC, et al.: KIAA1549: BRAF Gene Fusion and FGFR1 Hotspot Mutations Are Prognostic Factors in Pilocytic Astrocytomas. J Neuropathol Exp Neurol 74 (7): 743-54, 2015.[PUBMED Abstract]
- Horbinski C, Nikiforova MN, Hagenkord JM, et al.: Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol 14 (6): 777-89, 2012.[PUBMED Abstract]
- Roth JJ, Fierst TM, Waanders AJ, et al.: Whole Chromosome 7 Gain Predicts Higher Risk of Recurrence in Pediatric Pilocytic Astrocytomas Independently From KIAA1549-BRAF Fusion Status. J Neuropathol Exp Neurol 75 (4): 306-15, 2016.[PUBMED Abstract]
- Mistry M, Zhukova N, Merico D, et al.: BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33 (9): 1015-22, 2015.[PUBMED Abstract]
- Janzarik WG, Kratz CP, Loges NT, et al.: Further evidence for a somatic KRAS mutation in a pilocytic astrocytoma. Neuropediatrics 38 (2): 61-3, 2007.[PUBMED Abstract]
- López GY, Van Ziffle J, Onodera C, et al.: The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol 137 (1): 139-150, 2019.[PUBMED Abstract]
- Dougherty MJ, Santi M, Brose MS, et al.: Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol 12 (7): 621-30, 2010.[PUBMED Abstract]
- Dias-Santagata D, Lam Q, Vernovsky K, et al.: BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One 6 (3): e17948, 2011.[PUBMED Abstract]
- Schindler G, Capper D, Meyer J, et al.: Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 121 (3): 397-405, 2011.[PUBMED Abstract]
- Lassaletta A, Zapotocky M, Mistry M, et al.: Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J Clin Oncol 35 (25): 2934-2941, 2017.[PUBMED Abstract]
- Ho CY, Mobley BC, Gordish-Dressman H, et al.: A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol 130 (4): 575-85, 2015.[PUBMED Abstract]
- Jones DT, Hutter B, Jäger N, et al.: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45 (8): 927-32, 2013.[PUBMED Abstract]
- Zhang J, Wu G, Miller CP, et al.: Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45 (6): 602-12, 2013.[PUBMED Abstract]
- Ramkissoon LA, Horowitz PM, Craig JM, et al.: Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 110 (20): 8188-93, 2013.[PUBMED Abstract]
- Bandopadhayay P, Ramkissoon LA, Jain P, et al.: MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48 (3): 273-82, 2016.[PUBMED Abstract]
- Qaddoumi I, Orisme W, Wen J, et al.: Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131 (6): 833-45, 2016.[PUBMED Abstract]
- D'Aronco L, Rouleau C, Gayden T, et al.: Brainstem angiocentric gliomas with MYB-QKI rearrangements. Acta Neuropathol 134 (4): 667-669, 2017.[PUBMED Abstract]
- Chan E, Bollen AW, Sirohi D, et al.: Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 134 (4): 671-673, 2017.[PUBMED Abstract]
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.[PUBMED Abstract]
- Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.[PUBMED Abstract]
- Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.[PUBMED Abstract]
- Hirose T, Nobusawa S, Sugiyama K, et al.: Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 28 (5): 684-694, 2018.[PUBMED Abstract]
- Lucas CG, Solomon DA, Perry A: A review of recently described genetic alterations in central nervous system tumors. Hum Pathol 96: 56-66, 2020.[PUBMED Abstract]
- Franz DN, Belousova E, Sparagana S, et al.: Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 381 (9861): 125-32, 2013.[PUBMED Abstract]
- Paugh BS, Qu C, Jones C, et al.: Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28 (18): 3061-8, 2010.[PUBMED Abstract]
- Bax DA, Mackay A, Little SE, et al.: A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res 16 (13): 3368-77, 2010.[PUBMED Abstract]
- Ward SJ, Karakoula K, Phipps KP, et al.: Cytogenetic analysis of paediatric astrocytoma using comparative genomic hybridisation and fluorescence in-situ hybridisation. J Neurooncol 98 (3): 305-18, 2010.[PUBMED Abstract]
- Pollack IF, Hamilton RL, Sobol RW, et al.: IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27 (1): 87-94, 2011.[PUBMED Abstract]
- Sturm D, Witt H, Hovestadt V, et al.: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 (4): 425-37, 2012.[PUBMED Abstract]
- Korshunov A, Ryzhova M, Hovestadt V, et al.: Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129 (5): 669-78, 2015.[PUBMED Abstract]
- Mackay A, Burford A, Carvalho D, et al.: Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4): 520-537.e5, 2017.[PUBMED Abstract]
- Buczkowicz P, Hoeman C, Rakopoulos P, et al.: Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46 (5): 451-6, 2014.[PUBMED Abstract]
- Taylor KR, Mackay A, Truffaux N, et al.: Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46 (5): 457-61, 2014.[PUBMED Abstract]
- Mackay A, Burford A, Molinari V, et al.: Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 33 (5): 829-842.e5, 2018.[PUBMED Abstract]
- Korshunov A, Schrimpf D, Ryzhova M, et al.: H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134 (3): 507-516, 2017.[PUBMED Abstract]
- Gielen GH, Gessi M, Buttarelli FR, et al.: Genetic Analysis of Diffuse High-Grade Astrocytomas in Infancy Defines a Novel Molecular Entity. Brain Pathol 25 (4): 409-17, 2015.[PUBMED Abstract]
- Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al.: Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10 (1): 4343, 2019.[PUBMED Abstract]
- D'Angelo F, Ceccarelli M, Tala, et al.: The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med 25 (1): 176-187, 2019.[PUBMED Abstract]
- Blumcke I, Spreafico R, Haaker G, et al.: Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med 377 (17): 1648-1656, 2017.[PUBMED Abstract]
- Stone TJ, Keeley A, Virasami A, et al.: Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol 135 (1): 115-129, 2018.[PUBMED Abstract]
- Rivera B, Gayden T, Carrot-Zhang J, et al.: Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol 131 (6): 847-63, 2016.[PUBMED Abstract]
- Matsumura N, Nobusawa S, Ito J, et al.: Multiplex ligation-dependent probe amplification analysis is useful for detecting a copy number gain of the FGFR1 tyrosine kinase domain in dysembryoplastic neuroepithelial tumors. J Neurooncol 143 (1): 27-33, 2019.[PUBMED Abstract]
- Baisden BL, Brat DJ, Melhem ER, et al.: Dysembryoplastic neuroepithelial tumor-like neoplasm of the septum pellucidum: a lesion often misdiagnosed as glioma: report of 10 cases. Am J Surg Pathol 25 (4): 494-9, 2001.[PUBMED Abstract]
- Gessi M, Hattingen E, Dörner E, et al.: Dysembryoplastic Neuroepithelial Tumor of the Septum Pellucidum and the Supratentorial Midline: Histopathologic, Neuroradiologic, and Molecular Features of 7 Cases. Am J Surg Pathol 40 (6): 806-11, 2016.[PUBMED Abstract]
- Chiang JCH, Harreld JH, Tanaka R, et al.: Septal dysembryoplastic neuroepithelial tumor: a comprehensive clinical, imaging, histopathologic, and molecular analysis. Neuro Oncol 21 (6): 800-808, 2019.[PUBMED Abstract]
- Solomon DA, Korshunov A, Sill M, et al.: Myxoid glioneuronal tumor of the septum pellucidum and lateral ventricle is defined by a recurrent PDGFRA p.K385 mutation and DNT-like methylation profile. Acta Neuropathol 136 (2): 339-343, 2018.[PUBMED Abstract]
- Lucas CG, Villanueva-Meyer JE, Whipple N, et al.: Myxoid glioneuronal tumor, PDGFRA p.K385-mutant: clinical, radiologic, and histopathologic features. Brain Pathol 30 (3): 479-494, 2020.[PUBMED Abstract]
- Becker AJ: Ganglioglioma. In: Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. Lyon, France: IARC Press, 2016, pp 138-41.[PUBMED Abstract]
- Pekmezci M, Villanueva-Meyer JE, Goode B, et al.: The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6 (1): 47, 2018.[PUBMED Abstract]
- Bianchi F, Tamburrini G, Massimi L, et al.: Supratentorial tumors typical of the infantile age: desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA). A review. Childs Nerv Syst 32 (10): 1833-8, 2016.[PUBMED Abstract]
- Trehan G, Bruge H, Vinchon M, et al.: MR imaging in the diagnosis of desmoplastic infantile tumor: retrospective study of six cases. AJNR Am J Neuroradiol 25 (6): 1028-33, 2004 Jun-Jul.[PUBMED Abstract]
- Wang AC, Jones DTW, Abecassis IJ, et al.: Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities with Frequent BRAFV600 Mutations. Mol Cancer Res 16 (10): 1491-1498, 2018.[PUBMED Abstract]
- Blessing MM, Blackburn PR, Krishnan C, et al.: Desmoplastic Infantile Ganglioglioma: A MAPK Pathway-Driven and Microglia/Macrophage-Rich Neuroepithelial Tumor. J Neuropathol Exp Neurol 78 (11): 1011-1021, 2019.[PUBMED Abstract]
- Greer A, Foreman NK, Donson A, et al.: Desmoplastic infantile astrocytoma/ganglioglioma with rare BRAF V600D mutation. Pediatr Blood Cancer 64 (6): , 2017.[PUBMED Abstract]
- Pages M, Lacroix L, Tauziede-Espariat A, et al.: Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3: 85, 2015.[PUBMED Abstract]
- Bridge JA, Liu XQ, Sumegi J, et al.: Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23 (2): 121-8, 2013.[PUBMED Abstract]
- Hou Y, Pinheiro J, Sahm F, et al.: Papillary glioneuronal tumor (PGNT) exhibits a characteristic methylation profile and fusions involving PRKCA. Acta Neuropathol 137 (5): 837-846, 2019.[PUBMED Abstract]
- Sievers P, Appay R, Schrimpf D, et al.: Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol 138 (3): 497-504, 2019.[PUBMED Abstract]
- Deng MY, Sill M, Chiang J, et al.: Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136 (2): 239-253, 2018.[PUBMED Abstract]
- Chiang JCH, Harreld JH, Orr BA, et al.: Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol 134 (1): 159-162, 2017.[PUBMED Abstract]
- Chiang J, Dalton J, Upadhyaya SA, et al.: Chromosome arm 1q gain is an adverse prognostic factor in localized and diffuse leptomeningeal glioneuronal tumors with BRAF gene fusion and 1p deletion. Acta Neuropathol 137 (1): 179-181, 2019.[PUBMED Abstract]
- Sievers P, Stichel D, Schrimpf D, et al.: FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol 136 (2): 293-302, 2018.[PUBMED Abstract]
- Fisher PG, Tihan T, Goldthwaite PT, et al.: Outcome analysis of childhood low-grade astrocytomas. Pediatr Blood Cancer 51 (2): 245-50, 2008.[PUBMED Abstract]
- Qaddoumi I, Sultan I, Gajjar A: Outcome and prognostic features in pediatric gliomas: a review of 6212 cases from the Surveillance, Epidemiology, and End Results database. Cancer 115 (24): 5761-70, 2009.[PUBMED Abstract]
- Wisoff JH, Sanford RA, Heier LA, et al.: Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the Children's Oncology Group. Neurosurgery 68 (6): 1548-54; discussion 1554-5, 2011.[PUBMED Abstract]
- Bandopadhayay P, Bergthold G, London WB, et al.: Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr Blood Cancer 61 (7): 1173-9, 2014.[PUBMED Abstract]
- von Hornstein S, Kortmann RD, Pietsch T, et al.: Impact of chemotherapy on disseminated low-grade glioma in children and adolescents: report from the HIT-LGG 1996 trial. Pediatr Blood Cancer 56 (7): 1046-54, 2011.[PUBMED Abstract]
- Mazloom A, Hodges JC, Teh BS, et al.: Outcome of patients with pilocytic astrocytoma and leptomeningeal dissemination. Int J Radiat Oncol Biol Phys 84 (2): 350-4, 2012.[PUBMED Abstract]
- Stokland T, Liu JF, Ironside JW, et al.: A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol 12 (12): 1257-68, 2010.[PUBMED Abstract]
- Mirow C, Pietsch T, Berkefeld S, et al.: Children <1 year show an inferior outcome when treated according to the traditional LGG treatment strategy: a report from the German multicenter trial HIT-LGG 1996 for children with low grade glioma (LGG). Pediatr Blood Cancer 61 (3): 457-63, 2014.[PUBMED Abstract]
- Rakotonjanahary J, De Carli E, Delion M, et al.: Mortality in Children with Optic Pathway Glioma Treated with Up-Front BB-SFOP Chemotherapy. PLoS One 10 (6): e0127676, 2015.[PUBMED Abstract]
- Gnekow AK, Walker DA, Kandels D, et al.: A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer 81: 206-225, 2017.[PUBMED Abstract]
- Chamdine O, Broniscer A, Wu S, et al.: Metastatic Low-Grade Gliomas in Children: 20 Years' Experience at St. Jude Children's Research Hospital. Pediatr Blood Cancer 63 (1): 62-70, 2016.[PUBMED Abstract]
- Due-Tønnessen BJ, Helseth E, Scheie D, et al.: Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg 37 (2): 71-80, 2002.[PUBMED Abstract]
- Massimi L, Tufo T, Di Rocco C: Management of optic-hypothalamic gliomas in children: still a challenging problem. Expert Rev Anticancer Ther 7 (11): 1591-610, 2007.[PUBMED Abstract]
- Campagna M, Opocher E, Viscardi E, et al.: Optic pathway glioma: long-term visual outcome in children without neurofibromatosis type-1. Pediatr Blood Cancer 55 (6): 1083-8, 2010.[PUBMED Abstract]
- Hernáiz Driever P, von Hornstein S, Pietsch T, et al.: Natural history and management of low-grade glioma in NF-1 children. J Neurooncol 100 (2): 199-207, 2010.[PUBMED Abstract]
- Falzon K, Drimtzias E, Picton S, et al.: Visual outcomes after chemotherapy for optic pathway glioma in children with and without neurofibromatosis type 1: results of the International Society of Paediatric Oncology (SIOP) Low-Grade Glioma 2004 trial UK cohort. Br J Ophthalmol 102 (10): 1367-1371, 2018.[PUBMED Abstract]
- Finlay JL, Boyett JM, Yates AJ, et al.: Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol 13 (1): 112-23, 1995.[PUBMED Abstract]
- Villano JL, Seery TE, Bressler LR: Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother Pharmacol 64 (4): 647-55, 2009.[PUBMED Abstract]
- Karremann M, Gielen GH, Hoffmann M, et al.: Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro Oncol 20 (1): 123-131, 2018.[PUBMED Abstract]
- 小児星細胞腫の病期情報
-
小児星細胞腫に対して認められている病期分類システムはない。本要約において、小児星細胞腫の治療は以下の分類を用いて記述される:
- 小児星細胞腫に対する治療法選択肢の概要
-
小児および青年のがん患者の生存において、劇的な改善が達成されている。1975年から2010年の間に、小児がんの死亡率は50%を超える低下を示した。[ 1 ]最も利用可能で受け入れられている治療法を向上させるために種々の臨床試験が実施されており、その結果として、小児がんにおける生存率が幾度も改善されてきた。小児科での臨床試験は、新たな治療法と現在標準とされている治療法とを比較するようデザインされる。こうした比較は、2つの治療群のランダム化研究で実施されるが、それ以外にも1つの新たな治療法を評価し、その結果を従来の治療法を評価して既に得られている結果と比較することによって行われる。小児のがんは比較的まれであるため、小児脳腫瘍の患児はすべて臨床試験への登録を検討すべきである。現在実施中の米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトから入手することができる。
至適治療法を決定し実施するには、小児脳腫瘍の治療経験を有するがん専門医からなる集学的チームによる計画が必要である。小児脳腫瘍に対する放射線照射はきわめて高度な技術が必要とされ、この専門領域での経験を有し、最適な結果が確実に得られる施設で実施すべきである。
脳腫瘍患者の長期にわたる管理は複雑であり、集学的アプローチが必要となる。(小児および青年がん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)のPDQ要約を参照のこと。)
表4には、低悪性度および高悪性度小児星細胞腫に対する標準治療法の選択肢が記述されている。
表4.小児星細胞腫に対する標準治療法の選択肢 治療群 標準治療法の選択肢 小児低悪性度星細胞腫: 新規診断小児低悪性度星細胞腫 介入なしの経過観察 手術 補助療法(切除不十分な腫瘍を対象): —手術後の経過観察 —化学療法 —放射線療法 —標的療法 進行性/再発小児低悪性度星細胞腫 再度の手術 放射線療法 化学療法 化学療法を併用するまたは併用しない標的療法 小児高悪性度星細胞腫: 新規診断小児高悪性度星細胞腫 手術 補助療法: —放射線療法 —化学療法 再発小児高悪性度星細胞腫 手術(標準治療とみなされない) 幹細胞移植(SCT)を伴う大量化学療法(標準治療とみなされない) 放射線療法(標準治療とみなされない) BRAF V600E変異を認める患者に対してBRAF阻害薬を用いた標的療法(標準治療とみなされない) 参考文献- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- 小児低悪性度星細胞腫の治療
-
至適管理を決定しこれを実施するために、治療はしばしば、小児脳腫瘍の治療経験があるがん専門医の集学的チームによって導かれる。
低悪性度視経路星細胞腫の小児に対する治療法の選択肢は、生存率を改善するだけでなく視機能を安定させるために検討すべきである。[ 1 ][ 2 ]
新規診断小児低悪性度星細胞腫に対する標準治療法の選択肢には以下のものがある:
介入なしの経過観察
神経線維腫症1型(NF1)または偶然発見された症状を伴わない腫瘤を有する患者に対して何らかの介入を行わない場合には、経過観察が選択肢の1つである。[ 3 ]視経路グリオーマの自然退縮は、NF1を伴う小児と伴わない小児で報告されている。[ 4 ][ 5 ]
手術
小児低悪性度星細胞腫の一次治療は外科的切除であり[ 6 ][ 7 ]、手術の実施可能性は腫瘍の部位により決定される。乳児および幼児では、視交叉/視床下部に発生した低悪性度星細胞腫の手術は困難となる;そのため、生検が常に実施されるわけではない。これは、特にNF1の患者に当てはまる。[ 6 ]NF1と関連している場合、腫瘍は多巣性の場合がある。びまん性星細胞腫(世界保健機関[WHO]分類の悪性度II)は、全摘が困難な場合があるため、これらの患者ではこのことがより不良な転帰の一因となることがある。
切除後は、直ちに(小児腫瘍学グループ[COG]の基準では切除後48時間以内に)術後MRIを施行する。完全切除例にはその後も定期的にサーベイランス検査を施行するが、術後3~6ヵ月間における価値は明らかではない。[ 13 ];[ 14 ][証拠レベル:3iiiDiii]
低悪性度グリオーマで、手術による治療後に経過観察を行った小児の転帰に関連する因子が、評価可能患者518人を含むCOGの研究で特定された。[ 7 ]全群における全体的な転帰は、8年無増悪生存(PFS)率が78%、8年全生存(OS)率が96%であった。以下の因子が予後に関係していた:[ 7 ]
小脳毛様細胞性星細胞腫に対する長期の機能的な結果は比較的良好である。手術単独で治療された低悪性度グリオーマ患者におけるフルスケール知能指数(IQ)の平均値は標準的集団に近い。しかしながら、これらの患者には長期にわたって医学的、心理学的、および教育的障害がみられることがある。[ 16 ];[ 17 ][ 18 ][証拠レベル:3iiiC]
補助療法
低悪性度グリオーマの完全切除後の補助療法は、その後に再発がみられない限り、一般に必要ない。亜全摘患者には治療法の選択肢は個々の患者で判断する必要があり、以下の治療の1つ以上が行われる:
シャントや他の脳脊髄液の分流手技が必要になることもある。
手術後の経過観察
腫瘍の一部が切除されている患者で、特に腫瘍の再増殖速度が非常に遅いと予想される場合は、病変に向けた治療をさらに実施することなく、患者を観察してもよい。肉眼的全切除に至らない患者でも、約50%は5~8年まで無増悪を持続することがあるため、選択された患者では経過観察の戦略が支持される。[ 7 ]
化学療法
放射線療法に伴う長期的副作用を考慮すると、術後化学療法が最初に推奨される場合がある。
化学療法により客観的な腫瘍退縮が得られる可能性があるため、ほとんどの患者で放射線療法が必要になる時期が遅くなる。[ 19 ][ 20 ][ 21 ][ 22 ]化学療法は、視経路グリオーマの青年において放射線療法を遅らせたり、避けたりできる可能性のある選択肢でもある。[ 23 ][証拠レベル:3iiDii]化学療法は視床下部グリオーマおよび間脳症候群を有する小児における腫瘍を収縮させ、治療に反応する小児において体重増加が得られることが示されている。[ 24 ]
腫瘍進行または症状を有する切除不能な低悪性度グリオーマを治療するために最も広く使用されているレジメンは、以下の通りである:
COGは、10歳未満のNF1を有さない低悪性度視交叉/視床下部グリオーマの小児を、カルボプラチンおよびビンクリスチンの併用(CV)またはTPCVのいずれかのレジメンで治療した第III相ランダム化試験(COG-A9952)の結果を報告した。5年イベントフリー生存(EFS)率は、CVレジメンでは39%(±4%)、TPCVレジメンでは52%(±5%)であった。2つのレジメン間で毒性作用の割合は比較的同等であった。[ 27 ]同じ研究において、NF1の小児がCVによる治療を受ける群に非ランダムに割り付けられた。NF1の小児に対する5年EFS率は69%(±4%)で、CVを受けたNF1ではない小児よりも顕著に優れていた。多変量解析において、NF1は優れたEFS(ただし、OSではない)の独立した予測因子であった。[ 28 ]
低悪性度グリオーマの小児においてビンクリスチン/カルボプラチンによる治療とビンクリスチン/カルボプラチン + エトポシドによる治療を比較した1件の多施設プロスペクティブ・ランダム化試験では、2つのレジメン間でPFSおよびOSにおける差を実証できなかった。[ 29 ][証拠レベル:1iiD]
進行性または症候性の切除不可能な低悪性度星細胞腫の患児に対する化学療法アプローチでは、これら以外のレジメンも用いられており、以下が挙げられる:
視経路グリオーマに対して化学療法を受けた小児において、NF1を有さない小児はNF1を有する小児よりも疾患進行の割合が高く、乳児は1歳を超える小児よりも疾患進行の割合が高い。[ 20 ][ 21 ][ 30 ][ 35 ]視機能の状態(視力および視野を含む)は治療後の転帰および改善の重要な判断基準であり、X線画像で反応が得られている患者においてでさえ多様である。散発性の視経路グリオーマの小児は、NF1の小児よりも視覚の転帰が不良である。[ 35 ];[ 38 ][ 39 ][証拠レベル:3iiiC]初期の比較的良好な視力、比較的年齢が高いこと、および視交叉部後部に病変が認められないことが、化学療法後の視力の改善または安定に関連していた。[ 40 ]
放射線療法
病勢の進行が明らかになるまで、通常は放射線療法を保留し[ 41 ][ 42 ]、化学療法を用いることでさらに遅らせることも可能である。[ 19 ][ 20 ]
放射線療法が適応される低悪性度グリオーマの小児では、放射線分布を腫瘍の輪郭に合わせて照射し、正常脳組織を避けるアプローチ(3次元原体照射療法、強度変調放射線療法、定位放射線療法、および陽子線治療[荷電粒子線療法])がいずれも有効と考えられ、このような放射線療法に伴う急性および長期の毒性を潜在的に減らすことができる。[ 43 ][ 44 ];[ 45 ][証拠レベル:3iDiii]一般的に1.8Gy分割で54Gyの放射線量が用いられる。[ 46 ][ 47 ]
放射線療法実施後は、放射線により生じる画像上の変化と疾患増悪(これは通常、放射線療法後最初の1年間に生じるが、それ以降も生じる可能性がある)を、特に毛様細胞性星細胞腫の患者では、注意深く区別しなければならない。[ 48 ][ 49 ][ 50 ][ 51 ];[ 52 ][証拠レベル:2A];[ 53 ][証拠レベル:2C];[ 54 ][証拠レベル:3iiiDi];[ 55 ][証拠レベル:3iiiDii];[ 12 ][ 56 ][証拠レベル:3iiiDiii]
視交叉部のグリオーマおよび後部視経路視交叉部のグリオーマを有する患児の大半は放射線療法により長期の疾患制御が得られるが、重大な知的および内分泌系障害および脳血管障害および晩期死亡を来すことがあり、二次腫瘍のリスクも増大する可能性がある。[ 57 ][ 58 ][ 59 ];[ 53 ][証拠レベル:2C]1件の集団ベース研究により、放射線療法は晩期死亡に関連する最も重要な危険因子であることが確認されたが、放射線療法が必要な患者はリスクが比較的高い集団を反映している可能性がある。[ 59 ]
NF1の小児は、放射線関連二次腫瘍および血管の変化に起因する罹病に対するリスクがより高い。これらの患者では、放射線療法およびアルキル化剤が理論的に神経毒性作用および二次悪性腫瘍を誘発するリスクを高めることを考慮して、最終手段として用いられる。[ 60 ]
標的療法
症候性の上衣下巨細胞星細胞腫(SEGA)の小児に対しては、哺乳類ラパマイシン標的蛋白(mTOR)を阻害する薬物(例、エベロリムスおよびシロリムス)が研究されている。
証拠(mTOR阻害剤を用いたSEGAの治療):
臨床評価段階にある治療法の選択肢
特定の患者には、初期段階の臨床試験が利用できる場合がある。これらの試験は、COG、Pediatric Brain Tumor Consortium、または他の団体を通して利用できる場合がある。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Nicolin G, Parkin P, Mabbott D, et al.: Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer 53 (7): 1231-7, 2009.[PUBMED Abstract]
- Kramm CM, Butenhoff S, Rausche U, et al.: Thalamic high-grade gliomas in children: a distinct clinical subset? Neuro Oncol 13 (6): 680-9, 2011.[PUBMED Abstract]
- Listernick R, Ferner RE, Liu GT, et al.: Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol 61 (3): 189-98, 2007.[PUBMED Abstract]
- Albright AL: Feasibility and advisability of resections of thalamic tumors in pediatric patients. J Neurosurg 100 (5 Suppl Pediatrics): 468-72, 2004.[PUBMED Abstract]
- Piccirilli M, Lenzi J, Delfinis C, et al.: Spontaneous regression of optic pathways gliomas in three patients with neurofibromatosis type I and critical review of the literature. Childs Nerv Syst 22 (10): 1332-7, 2006.[PUBMED Abstract]
- Due-Tønnessen BJ, Helseth E, Scheie D, et al.: Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg 37 (2): 71-80, 2002.[PUBMED Abstract]
- Wisoff JH, Sanford RA, Heier LA, et al.: Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the Children's Oncology Group. Neurosurgery 68 (6): 1548-54; discussion 1554-5, 2011.[PUBMED Abstract]
- Tseng JH, Tseng MY: Survival analysis of 81 children with primary spinal gliomas: a population-based study. Pediatr Neurosurg 42 (6): 347-53, 2006.[PUBMED Abstract]
- Ahmed R, Menezes AH, Torner JC: Role of resection and adjuvant therapy in long-term disease outcomes for low-grade pediatric intramedullary spinal cord tumors. J Neurosurg Pediatr 18 (5): 594-601, 2016.[PUBMED Abstract]
- Milano MT, Johnson MD, Sul J, et al.: Primary spinal cord glioma: a Surveillance, Epidemiology, and End Results database study. J Neurooncol 98 (1): 83-92, 2010.[PUBMED Abstract]
- Scheinemann K, Bartels U, Huang A, et al.: Survival and functional outcome of childhood spinal cord low-grade gliomas. Clinical article. J Neurosurg Pediatr 4 (3): 254-61, 2009.[PUBMED Abstract]
- Sawamura Y, Kamada K, Kamoshima Y, et al.: Role of surgery for optic pathway/hypothalamic astrocytomas in children. Neuro Oncol 10 (5): 725-33, 2008.[PUBMED Abstract]
- Sutton LN, Cnaan A, Klatt L, et al.: Postoperative surveillance imaging in children with cerebellar astrocytomas. J Neurosurg 84 (5): 721-5, 1996.[PUBMED Abstract]
- Dorward IG, Luo J, Perry A, et al.: Postoperative imaging surveillance in pediatric pilocytic astrocytomas. J Neurosurg Pediatr 6 (4): 346-52, 2010.[PUBMED Abstract]
- Mirow C, Pietsch T, Berkefeld S, et al.: Children <1 year show an inferior outcome when treated according to the traditional LGG treatment strategy: a report from the German multicenter trial HIT-LGG 1996 for children with low grade glioma (LGG). Pediatr Blood Cancer 61 (3): 457-63, 2014.[PUBMED Abstract]
- Beebe DW, Ris MD, Armstrong FD, et al.: Cognitive and adaptive outcome in low-grade pediatric cerebellar astrocytomas: evidence of diminished cognitive and adaptive functioning in National Collaborative Research Studies (CCG 9891/POG 9130). J Clin Oncol 23 (22): 5198-204, 2005.[PUBMED Abstract]
- Turner CD, Chordas CA, Liptak CC, et al.: Medical, psychological, cognitive and educational late-effects in pediatric low-grade glioma survivors treated with surgery only. Pediatr Blood Cancer 53 (3): 417-23, 2009.[PUBMED Abstract]
- Daszkiewicz P, Maryniak A, Roszkowski M, et al.: Long-term functional outcome of surgical treatment of juvenile pilocytic astrocytoma of the cerebellum in children. Childs Nerv Syst 25 (7): 855-60, 2009.[PUBMED Abstract]
- Packer RJ, Ater J, Allen J, et al.: Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg 86 (5): 747-54, 1997.[PUBMED Abstract]
- Gnekow AK, Falkenstein F, von Hornstein S, et al.: Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol 14 (10): 1265-84, 2012.[PUBMED Abstract]
- Laithier V, Grill J, Le Deley MC, et al.: Progression-free survival in children with optic pathway tumors: dependence on age and the quality of the response to chemotherapy--results of the first French prospective study for the French Society of Pediatric Oncology. J Clin Oncol 21 (24): 4572-8, 2003.[PUBMED Abstract]
- Prados MD, Edwards MS, Rabbitt J, et al.: Treatment of pediatric low-grade gliomas with a nitrosourea-based multiagent chemotherapy regimen. J Neurooncol 32 (3): 235-41, 1997.[PUBMED Abstract]
- Chong AL, Pole JD, Scheinemann K, et al.: Optic pathway gliomas in adolescence--time to challenge treatment choices? Neuro Oncol 15 (3): 391-400, 2013.[PUBMED Abstract]
- Gropman AL, Packer RJ, Nicholson HS, et al.: Treatment of diencephalic syndrome with chemotherapy: growth, tumor response, and long term control. Cancer 83 (1): 166-72, 1998.[PUBMED Abstract]
- Gururangan S, Cavazos CM, Ashley D, et al.: Phase II study of carboplatin in children with progressive low-grade gliomas. J Clin Oncol 20 (13): 2951-8, 2002.[PUBMED Abstract]
- Dodgshun AJ, Maixner WJ, Heath JA, et al.: Single agent carboplatin for pediatric low-grade glioma: A retrospective analysis shows equivalent efficacy to multiagent chemotherapy. Int J Cancer 138 (2): 481-8, 2016.[PUBMED Abstract]
- Ater JL, Zhou T, Holmes E, et al.: Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children's Oncology Group. J Clin Oncol 30 (21): 2641-7, 2012.[PUBMED Abstract]
- Ater JL, Xia C, Mazewski CM, et al.: Nonrandomized comparison of neurofibromatosis type 1 and non-neurofibromatosis type 1 children who received carboplatin and vincristine for progressive low-grade glioma: A report from the Children's Oncology Group. Cancer 122 (12): 1928-36, 2016.[PUBMED Abstract]
- Gnekow AK, Walker DA, Kandels D, et al.: A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer 81: 206-225, 2017.[PUBMED Abstract]
- Massimino M, Spreafico F, Cefalo G, et al.: High response rate to cisplatin/etoposide regimen in childhood low-grade glioma. J Clin Oncol 20 (20): 4209-16, 2002.[PUBMED Abstract]
- Massimino M, Spreafico F, Riva D, et al.: A lower-dose, lower-toxicity cisplatin-etoposide regimen for childhood progressive low-grade glioma. J Neurooncol 100 (1): 65-71, 2010.[PUBMED Abstract]
- Mora J, Perez-Jaume S, Cruz O: Treatment of childhood astrocytomas with irinotecan and cisplatin. Clin Transl Oncol 20 (4): 500-507, 2018.[PUBMED Abstract]
- von Hornstein S, Kortmann RD, Pietsch T, et al.: Impact of chemotherapy on disseminated low-grade glioma in children and adolescents: report from the HIT-LGG 1996 trial. Pediatr Blood Cancer 56 (7): 1046-54, 2011.[PUBMED Abstract]
- Bouffet E, Jakacki R, Goldman S, et al.: Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol 30 (12): 1358-63, 2012.[PUBMED Abstract]
- Lassaletta A, Scheinemann K, Zelcer SM, et al.: Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. J Clin Oncol 34 (29): 3537-3543, 2016.[PUBMED Abstract]
- Gururangan S, Fisher MJ, Allen JC, et al.: Temozolomide in children with progressive low-grade glioma. Neuro Oncol 9 (2): 161-8, 2007.[PUBMED Abstract]
- Khaw SL, Coleman LT, Downie PA, et al.: Temozolomide in pediatric low-grade glioma. Pediatr Blood Cancer 49 (6): 808-11, 2007.[PUBMED Abstract]
- Moreno L, Bautista F, Ashley S, et al.: Does chemotherapy affect the visual outcome in children with optic pathway glioma? A systematic review of the evidence. Eur J Cancer 46 (12): 2253-9, 2010.[PUBMED Abstract]
- Shofty B, Ben-Sira L, Freedman S, et al.: Visual outcome following chemotherapy for progressive optic pathway gliomas. Pediatr Blood Cancer 57 (3): 481-5, 2011.[PUBMED Abstract]
- Falzon K, Drimtzias E, Picton S, et al.: Visual outcomes after chemotherapy for optic pathway glioma in children with and without neurofibromatosis type 1: results of the International Society of Paediatric Oncology (SIOP) Low-Grade Glioma 2004 trial UK cohort. Br J Ophthalmol 102 (10): 1367-1371, 2018.[PUBMED Abstract]
- Fisher BJ, Leighton CC, Vujovic O, et al.: Results of a policy of surveillance alone after surgical management of pediatric low grade gliomas. Int J Radiat Oncol Biol Phys 51 (3): 704-10, 2001.[PUBMED Abstract]
- Tsang DS, Murphy ES, Merchant TE: Radiation Therapy for Optic Pathway and Hypothalamic Low-Grade Gliomas in Children. Int J Radiat Oncol Biol Phys 99 (3): 642-651, 2017.[PUBMED Abstract]
- Greenberger BA, Pulsifer MB, Ebb DH, et al.: Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys 89 (5): 1060-8, 2014.[PUBMED Abstract]
- Paulino AC, Mazloom A, Terashima K, et al.: Intensity-modulated radiotherapy (IMRT) in pediatric low-grade glioma. Cancer 119 (14): 2654-9, 2013.[PUBMED Abstract]
- Müller K, Gnekow A, Falkenstein F, et al.: Radiotherapy in pediatric pilocytic astrocytomas. A subgroup analysis within the prospective multicenter study HIT-LGG 1996 by the German Society of Pediatric Oncology and Hematology (GPOH). Strahlenther Onkol 189 (8): 647-55, 2013.[PUBMED Abstract]
- Bitterman DS, MacDonald SM, Yock TI, et al.: Revisiting the Role of Radiation Therapy for Pediatric Low-Grade Glioma. J Clin Oncol 37 (35): 3335-3339, 2019.[PUBMED Abstract]
- Cherlow JM, Shaw DWW, Margraf LR, et al.: Conformal Radiation Therapy for Pediatric Patients with Low-Grade Glioma: Results from the Children's Oncology Group Phase 2 Study ACNS0221. Int J Radiat Oncol Biol Phys 103 (4): 861-868, 2019.[PUBMED Abstract]
- Chawla S, Korones DN, Milano MT, et al.: Spurious progression in pediatric brain tumors. J Neurooncol 107 (3): 651-7, 2012.[PUBMED Abstract]
- Marcus KJ, Goumnerova L, Billett AL, et al.: Stereotactic radiotherapy for localized low-grade gliomas in children: final results of a prospective trial. Int J Radiat Oncol Biol Phys 61 (2): 374-9, 2005.[PUBMED Abstract]
- Combs SE, Schulz-Ertner D, Moschos D, et al.: Fractionated stereotactic radiotherapy of optic pathway gliomas: tolerance and long-term outcome. Int J Radiat Oncol Biol Phys 62 (3): 814-9, 2005.[PUBMED Abstract]
- Naftel RP, Pollack IF, Zuccoli G, et al.: Pseudoprogression of low-grade gliomas after radiotherapy. Pediatr Blood Cancer 62 (1): 35-9, 2015.[PUBMED Abstract]
- Merchant TE, Kun LE, Wu S, et al.: Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol 27 (22): 3598-604, 2009.[PUBMED Abstract]
- Merchant TE, Conklin HM, Wu S, et al.: Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol 27 (22): 3691-7, 2009.[PUBMED Abstract]
- Kano H, Niranjan A, Kondziolka D, et al.: Stereotactic radiosurgery for pilocytic astrocytomas part 2: outcomes in pediatric patients. J Neurooncol 95 (2): 219-29, 2009.[PUBMED Abstract]
- Hallemeier CL, Pollock BE, Schomberg PJ, et al.: Stereotactic radiosurgery for recurrent or unresectable pilocytic astrocytoma. Int J Radiat Oncol Biol Phys 83 (1): 107-12, 2012.[PUBMED Abstract]
- Mansur DB, Rubin JB, Kidd EA, et al.: Radiation therapy for pilocytic astrocytomas of childhood. Int J Radiat Oncol Biol Phys 79 (3): 829-34, 2011.[PUBMED Abstract]
- Jenkin D, Angyalfi S, Becker L, et al.: Optic glioma in children: surveillance, resection, or irradiation? Int J Radiat Oncol Biol Phys 25 (2): 215-25, 1993.[PUBMED Abstract]
- Khafaga Y, Hassounah M, Kandil A, et al.: Optic gliomas: a retrospective analysis of 50 cases. Int J Radiat Oncol Biol Phys 56 (3): 807-12, 2003.[PUBMED Abstract]
- Krishnatry R, Zhukova N, Guerreiro Stucklin AS, et al.: Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: A population-based study. Cancer 122 (8): 1261-9, 2016.[PUBMED Abstract]
- Grill J, Couanet D, Cappelli C, et al.: Radiation-induced cerebral vasculopathy in children with neurofibromatosis and optic pathway glioma. Ann Neurol 45 (3): 393-6, 1999.[PUBMED Abstract]
- Franz DN, Agricola KD, Tudor CA, et al.: Everolimus for tumor recurrence after surgical resection for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. J Child Neurol 28 (5): 602-7, 2013.[PUBMED Abstract]
- Krueger DA, Care MM, Holland K, et al.: Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363 (19): 1801-11, 2010.[PUBMED Abstract]
- Weidman DR, Pole JD, Bouffet E, et al.: Dose-level response rates of mTor inhibition in tuberous sclerosis complex (TSC) related subependymal giant cell astrocytoma (SEGA). Pediatr Blood Cancer 62 (10): 1754-60, 2015.[PUBMED Abstract]
- Franz DN, Leonard J, Tudor C, et al.: Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 59 (3): 490-8, 2006.[PUBMED Abstract]
- Franz DN, Belousova E, Sparagana S, et al.: Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 381 (9861): 125-32, 2013.[PUBMED Abstract]
- Franz DN, Agricola K, Mays M, et al.: Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol 78 (6): 929-38, 2015.[PUBMED Abstract]
- 進行性/再発小児低悪性度星細胞腫の治療
-
至適管理を決定しこれを実施するために、治療はしばしば、小児脳腫瘍の治療経験があるがん専門医の集学的チームによって導かれる。
低悪性度視経路星細胞腫の小児に対する治療法の選択肢は、生存率を改善するだけでなく視機能を安定させるために検討すべきである。[ 1 ][ 2 ]
小児低悪性度星細胞腫は、最初の発症および初期治療後何年も経過してから進行または再発することがある。
個別の計画は、以下に基づいて調整する必要がある:
通常、再発病変は原発腫瘍部位に認められるが、多巣性疾患または頭蓋内の他の部位および脊髄軟髄膜に広く播種した疾患が報告されている。[ 3 ][ 4 ]低悪性度びまん性原線維性星細胞腫が再発したほとんどの小児において病変は低悪性度である;しかしながら、より高い悪性度の腫瘍に転換する可能性もある。サーベイランスの画像検査では、しばしば星細胞腫の再発が特定される。[ 5 ]再発時には、再燃腫瘍の範囲を確認するための完全な評価が適応とされる。
進行性/再発小児低悪性度星細胞腫に対する標準治療法の選択肢には以下のものがある:
再度の手術
低悪性度星細胞腫患者が、手術単独での治療後に再燃した場合は、再度外科的切除の候補となりうる。[ 6 ]外科的介入の必要性は、以下の点を基に個別に決定しなければならない:
再度の手術の有用性は、再発部位および重大な神経学的損傷を伴わずにほぼ完全な切除/肉眼的全切除が行えるかどうかによる影響を受ける[ 7 ]
放射線療法
放射線療法を使用する理論的根拠は、第一選択治療として使用した場合も再発の時点で使用した場合も基本的に同じである(本要約の小児低悪性度星細胞腫の治療のセクションの放射線療法のサブセクションを参照のこと)。放射線療法を受けたことのない患者であれば、局所放射線療法が治療選択肢となる場合があるが、患児の年齢ならびに腫瘍の範囲および位置によっては、放射線の代わりに化学療法を検討してもよい。[ 8 ][証拠レベル:3iA];[ 9 ][証拠レベル:3iiiDi]
放射線療法が適応となる低悪性度グリオーマの患児では、原体照射療法のアプローチが有効と考えられ、この放射線療法に伴う急性および長期の毒性が抑えられる可能性がある。[ 10 ][ 11 ][ 12 ][ 13 ]
化学療法
切除不能部位に再発が認められる場合は、化学療法を検討すべきである。
化学療法によって、比較的長期の疾患制御が得られる場合がある。[ 14 ][ 15 ]レジメンの選択は、以前に化学療法が利用されているかどうか次第である。カルボプラチンおよびビンクリスチン(CV);thioguanine、プロカルバジン、ロムスチン、およびビンクリスチンの併用(TPCV);ビンブラスチン単独;テモゾロミド単独;またはテモゾロミドとカルボプラチンおよびビンクリスチンとの併用など、数多くの選択肢を検討できる。[ 14 ][ 15 ][ 16 ][ 17 ]
化学療法を併用するまたは併用しない標的療法
ベバシズマブでは、イリノテカンと併用することで抗腫瘍活性も確認されており、一部の症例では臨床的改善または視力改善につながっている。[ 18 ]
証拠(標的療法[ベバシズマブ]):
- 再発低悪性度グリオーマの小児に対するベバシズマブ + イリノテカンの第II相研究で、以下の結果が観察された:[ 19 ]
- 再発低悪性度グリオーマの患者14人を対象としたパイロット研究でもベバシズマブをベースにした治療法が評価され、以下が観察された:[ 20 ][証拠レベル:3iiDi];[ 21 ][証拠レベル:3iiiDiv]
- ベバシズマブはまた、低悪性度グリオーマの小児で放射線により生じる腫瘍増大による症状がみられる場合にも用いられている。[ 22 ]
BRAF変異がかなりの割合の低悪性度グリオーマをもたらすことが同定されたことにより、複数の進行中の臨床試験でこの分子経路のさまざまな要素(例、MEKおよびBRAF)の阻害が活発に検証されており、初期の報告ではかなりの活性が示唆されている。ベムラフェニブおよびダブラフェニブといった第一世代のBRAF阻害薬はBRAF V600E変異腫瘍に対して活性がある一方で、BRAF遺伝子融合が認められる腫瘍にはMAPK経路の矛盾した活性化の可能性があるためこれらの薬物は禁忌である。[ 23 ][ 24 ]
BRAFおよびMEK阻害薬の研究には以下のものがある:
- 腫瘍にBRAF V600E変異がみられる患者に対する臨床研究の取り組みの焦点は、MEK阻害薬と併用するBRAF阻害薬の評価に当てられている。こうした併用は、BRAF V600E変異がみられる成人のがんの治療に承認されており、BRAF阻害薬またはMEK阻害薬のいずれかを単一薬として用いるよりも有効である。[ 25 ]
- MEK阻害薬のセルメチニブが、低悪性度グリオーマを有する小児に対して第I/II相臨床試験で研究されている(PBTC-029[NCT01089101])。
- PBTC-029試験の第I相の構成要素から、以下の結果が示された:[ 32 ]
- 本試験の第II相構成要素の1層はBRAFゲノム変化を有する患者であった。[ 33 ]
- この試験の第II相構成要素の3層はNF1関連低悪性度グリオーマを有する患者であった。[ 33 ]
すべての層における最も一般的な毒性作用は、グレード1およびブレード2のCPK高値、下痢、低アルブミン血症、アスパラギン酸アミノトランスフェラーゼ(AST)高値、および発疹であった。まれなグレード3およびグレード4の毒性作用として、CPK高値、発疹、好中球減少、嘔吐、および爪甲周囲炎が含まれた。
臨床評価段階にある治療法の選択肢
特定の患者には、初期段階の臨床試験が利用できる場合がある。これらの試験は、COG、Pediatric Brain Tumor Consortium、または他の団体を通して利用できる場合がある。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Nicolin G, Parkin P, Mabbott D, et al.: Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer 53 (7): 1231-7, 2009.[PUBMED Abstract]
- Kramm CM, Butenhoff S, Rausche U, et al.: Thalamic high-grade gliomas in children: a distinct clinical subset? Neuro Oncol 13 (6): 680-9, 2011.[PUBMED Abstract]
- Perilongo G, Carollo C, Salviati L, et al.: Diencephalic syndrome and disseminated juvenile pilocytic astrocytomas of the hypothalamic-optic chiasm region. Cancer 80 (1): 142-6, 1997.[PUBMED Abstract]
- Leibel SA, Sheline GE, Wara WM, et al.: The role of radiation therapy in the treatment of astrocytomas. Cancer 35 (6): 1551-7, 1975.[PUBMED Abstract]
- Udaka YT, Yeh-Nayre LA, Amene CS, et al.: Recurrent pediatric central nervous system low-grade gliomas: the role of surveillance neuroimaging in asymptomatic children. J Neurosurg Pediatr 11 (2): 119-26, 2013.[PUBMED Abstract]
- Austin EJ, Alvord EC: Recurrences of cerebellar astrocytomas: a violation of Collins' law. J Neurosurg 68 (1): 41-7, 1988.[PUBMED Abstract]
- Bowers DC, Krause TP, Aronson LJ, et al.: Second surgery for recurrent pilocytic astrocytoma in children. Pediatr Neurosurg 34 (5): 229-34, 2001.[PUBMED Abstract]
- Scheinemann K, Bartels U, Tsangaris E, et al.: Feasibility and efficacy of repeated chemotherapy for progressive pediatric low-grade gliomas. Pediatr Blood Cancer 57 (1): 84-8, 2011.[PUBMED Abstract]
- de Haas V, Grill J, Raquin MA, et al.: Relapses of optic pathway tumors after first-line chemotherapy. Pediatr Blood Cancer 52 (5): 575-80, 2009.[PUBMED Abstract]
- Merchant TE, Conklin HM, Wu S, et al.: Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol 27 (22): 3691-7, 2009.[PUBMED Abstract]
- Marcus KJ, Goumnerova L, Billett AL, et al.: Stereotactic radiotherapy for localized low-grade gliomas in children: final results of a prospective trial. Int J Radiat Oncol Biol Phys 61 (2): 374-9, 2005.[PUBMED Abstract]
- Bitterman DS, MacDonald SM, Yock TI, et al.: Revisiting the Role of Radiation Therapy for Pediatric Low-Grade Glioma. J Clin Oncol 37 (35): 3335-3339, 2019.[PUBMED Abstract]
- Cherlow JM, Shaw DWW, Margraf LR, et al.: Conformal Radiation Therapy for Pediatric Patients with Low-Grade Glioma: Results from the Children's Oncology Group Phase 2 Study ACNS0221. Int J Radiat Oncol Biol Phys 103 (4): 861-868, 2019.[PUBMED Abstract]
- Packer RJ, Lange B, Ater J, et al.: Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol 11 (5): 850-6, 1993.[PUBMED Abstract]
- Gnekow AK, Falkenstein F, von Hornstein S, et al.: Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol 14 (10): 1265-84, 2012.[PUBMED Abstract]
- Gururangan S, Fisher MJ, Allen JC, et al.: Temozolomide in children with progressive low-grade glioma. Neuro Oncol 9 (2): 161-8, 2007.[PUBMED Abstract]
- Lassaletta A, Scheinemann K, Zelcer SM, et al.: Phase II Weekly Vinblastine for Chemotherapy-Naïve Children With Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. J Clin Oncol 34 (29): 3537-3543, 2016.[PUBMED Abstract]
- Avery RA, Hwang EI, Jakacki RI, et al.: Marked recovery of vision in children with optic pathway gliomas treated with bevacizumab. JAMA Ophthalmol 132 (1): 111-4, 2014.[PUBMED Abstract]
- Gururangan S, Fangusaro J, Poussaint TY, et al.: Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas--a Pediatric Brain Tumor Consortium study. Neuro Oncol 16 (2): 310-7, 2014.[PUBMED Abstract]
- Hwang EI, Jakacki RI, Fisher MJ, et al.: Long-term efficacy and toxicity of bevacizumab-based therapy in children with recurrent low-grade gliomas. Pediatr Blood Cancer 60 (5): 776-82, 2013.[PUBMED Abstract]
- Packer RJ, Jakacki R, Horn M, et al.: Objective response of multiply recurrent low-grade gliomas to bevacizumab and irinotecan. Pediatr Blood Cancer 52 (7): 791-5, 2009.[PUBMED Abstract]
- Foster KA, Ares WJ, Pollack IF, et al.: Bevacizumab for symptomatic radiation-induced tumor enlargement in pediatric low grade gliomas. Pediatr Blood Cancer 62 (2): 240-245, 2015.[PUBMED Abstract]
- Sievert AJ, Lang SS, Boucher KL, et al.: Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A 110 (15): 5957-62, 2013.[PUBMED Abstract]
- Karajannis MA, Legault G, Fisher MJ, et al.: Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol 16 (10): 1408-16, 2014.[PUBMED Abstract]
- Odogwu L, Mathieu L, Blumenthal G, et al.: FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist 23 (6): 740-745, 2018.[PUBMED Abstract]
- Kieran MW, Bouffet E, Tabori U, et al.: The first study of dabrafenib in pediatric patients with BRAF V600–mutant relapsed or refractory low-grade gliomas. [Abstract] Ann Oncol 27 (Suppl 6): A-LBA19 PR, 2016.[PUBMED Abstract]
- Rush S, Foreman N, Liu A: Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol 31 (10): e159-60, 2013.[PUBMED Abstract]
- del Bufalo F, Carai A, Figà-Talamanca L, et al.: Response of recurrent BRAFV600E mutated ganglioglioma to Vemurafenib as single agent. J Transl Med 12: 356, 2014.[PUBMED Abstract]
- Aguilera D, Janss A, Mazewski C, et al.: Successful Retreatment of a Child with a Refractory Brainstem Ganglioglioma with Vemurafenib. Pediatr Blood Cancer 63 (3): 541-3, 2016.[PUBMED Abstract]
- Marks AM, Bindra RS, DiLuna ML, et al.: Response to the BRAF/MEK inhibitors dabrafenib/trametinib in an adolescent with a BRAF V600E mutated anaplastic ganglioglioma intolerant to vemurafenib. Pediatr Blood Cancer 65 (5): e26969, 2018.[PUBMED Abstract]
- Touat M, Gratieux J, Condette Auliac S, et al.: Vemurafenib and cobimetinib overcome resistance to vemurafenib in BRAF-mutant ganglioglioma. Neurology 91 (11): 523-525, 2018.[PUBMED Abstract]
- Banerjee A, Jakacki RI, Onar-Thomas A, et al.: A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol 19 (8): 1135-1144, 2017.[PUBMED Abstract]
- Fangusaro JR, Onar-Thomas A, Young-Poussaint T, et al.: A phase II prospective study of selumetinib in children with recurrent or refractory low-grade glioma (LGG): a Pediatric Brain Tumor Consortium (PBTC) study. [Abstract] J Clin Oncol 35 (Suppl 15): A-10504, 2017. Also available online. Last accessed June 26, 2020.[PUBMED Abstract]
- 小児高悪性度星細胞腫の治療
-
至適管理を決定しこれを実施するために、小児高悪性度星細胞腫の治療は、小児脳腫瘍の治療経験があるがん専門医の集学的チームによって導かれるべきである。
小児期に発生する高悪性度グリオーマの転帰は、しばしば成人における転帰よりも良好である。この差が腫瘍の特徴における生物学的差、使用した治療法、腫瘍の切除可能性、または他の因子のいずれによって生じているのか明らかではない。
成人でも小児でも、高悪性度テント上星細胞腫の治療には、手術、放射線療法、および化学療法が挙げられる。
新規診断小児高悪性度星細胞腫に対する標準治療法の選択肢には以下のものがある:
手術
完全切除が可能であれば、より良好な予後が期待できる。[ 1 ][ 2 ]手術、放射線療法、およびニトロソウレア(ロムスチン)ベースの化学療法により治療された患者の5年無増悪生存率は、19%(±3%)であった;全摘出患者の生存率は40%であった。[ 3 ]同様に、多剤化学放射線療法およびバルプロ酸に加えた補助化学療法の試験で、全体の5年イベントフリー生存(EFS)率は13%であったが、腫瘍を完全切除した小児では、EFS率が48%であった。[ 4 ][証拠レベル:2A]
補助療法
放射線療法
放射線療法では、腫瘍全体を大きく囲む領域にルーチンで照射される。通常、腫瘍床への放射線量は54Gy以上である。このような治療法にもかかわらず、全生存(OS)率は依然として不良である。放射線療法で治療された脊髄が原発部位の小児および視床の高悪性度グリオーマ(すなわち、びまん性正中グリオーマ、H3 K27M変異型の腫瘍)の小児も同様に生存は不良である。[ 5 ][ 6 ];[ 7 ][ 8 ][証拠レベル:3iiiA]
化学療法
1件の試験では、補助薬としてロムスチン、ビンクリスチン、およびプレドニゾンを用いたプロスペクティブ・ランダム化試験で治療を受けた膠芽腫の小児は、放射線療法単独で治療された小児よりも経過が良好であった。[ 9 ]さらに、亜全切除された腫瘍、特にメチル化O6-メチルグアニン-DNA-メチルトランスフェラーゼ(MGMT)過剰発現を伴う膠芽腫に対してテモゾロミドに加えてロムスチンを投与された小児では、わずかに治療成績の改善がみられた。[ 10 ]IDH1変異を有する患者の1年OS率(100%)は、IDH1野生型腫瘍の患者(1年OS率、81%)と比較して良好であり、基礎にある生物学的特徴の潜在的な重要性が強調されている。[ 11 ]
膠芽腫を治療するためのテモゾロミドの使用は、最初は成人で研究された。この集団では、放射線療法中およびその後のテモゾロミドの追加により、放射線療法単独による治療と比較して2年EFS率の改善がもたらされた。MGMTプロモーターを認める膠芽腫の成人患者がテモゾロミドから利益を得たのに対し、メチル化MGMTプロモーターを認めない患者はテモゾロミドから利益を得られなかった。[ 12 ][ 13 ]高悪性度テント上グリオーマの患児に対して放射線療法と同時にテモゾロミドを投与した場合の成績は、ニトロソウレアをベースとした治療に匹敵するようであり[ 14 ]、MGMTの過剰発現が認められない患児におけるEFSの優位性が再度実証された。
放射線療法後のベバシズマブによる補助療法は、新たに高悪性度グリオーマと診断された小児患者のOSまたは無増悪生存期間を延長させなかった。[ 15 ]
より若年の小児は化学療法または地固めの大量化学療法から有益性が得られ、放射線療法の開始を遅らせ、照射内容を変更することができ、選択された症例によっては照射の必要がなくなることもある。[ 16 ][ 17 ][ 18 ]
臨床評価段階にある治療法の選択肢
特定の患者には、初期段階の臨床試験が利用できる場合がある。これらの試験は、小児腫瘍学グループ(COG)、Pediatric Brain Tumor Consortium、または他の団体を通して利用できる場合がある。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Wisoff JH, Boyett JM, Berger MS, et al.: Current neurosurgical management and the impact of the extent of resection in the treatment of malignant gliomas of childhood: a report of the Children's Cancer Group trial no. CCG-945. J Neurosurg 89 (1): 52-9, 1998.[PUBMED Abstract]
- Yang T, Temkin N, Barber J, et al.: Gross total resection correlates with long-term survival in pediatric patients with glioblastoma. World Neurosurg 79 (3-4): 537-44, 2013 Mar-Apr.[PUBMED Abstract]
- Fouladi M, Hunt DL, Pollack IF, et al.: Outcome of children with centrally reviewed low-grade gliomas treated with chemotherapy with or without radiotherapy on Children's Cancer Group high-grade glioma study CCG-945. Cancer 98 (6): 1243-52, 2003.[PUBMED Abstract]
- Wolff JE, Driever PH, Erdlenbruch B, et al.: Intensive chemotherapy improves survival in pediatric high-grade glioma after gross total resection: results of the HIT-GBM-C protocol. Cancer 116 (3): 705-12, 2010.[PUBMED Abstract]
- Kramm CM, Butenhoff S, Rausche U, et al.: Thalamic high-grade gliomas in children: a distinct clinical subset? Neuro Oncol 13 (6): 680-9, 2011.[PUBMED Abstract]
- Tendulkar RD, Pai Panandiker AS, Wu S, et al.: Irradiation of pediatric high-grade spinal cord tumors. Int J Radiat Oncol Biol Phys 78 (5): 1451-6, 2010.[PUBMED Abstract]
- Wolff B, Ng A, Roth D, et al.: Pediatric high grade glioma of the spinal cord: results of the HIT-GBM database. J Neurooncol 107 (1): 139-46, 2012.[PUBMED Abstract]
- Ononiwu C, Mehta V, Bettegowda C, et al.: Pediatric spinal glioblastoma multiforme: current treatment strategies and possible predictors of survival. Childs Nerv Syst 28 (5): 715-20, 2012.[PUBMED Abstract]
- Sposto R, Ertel IJ, Jenkin RD, et al.: The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Childrens Cancer Study Group. J Neurooncol 7 (2): 165-77, 1989.[PUBMED Abstract]
- Jakacki RI, Cohen KJ, Buxton A, et al.: Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: a report of the Children's Oncology Group ACNS0423 study. Neuro Oncol 18 (10): 1442-50, 2016.[PUBMED Abstract]
- Pollack IF, Hamilton RL, Sobol RW, et al.: IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27 (1): 87-94, 2011.[PUBMED Abstract]
- Stupp R, Mason WP, van den Bent MJ, et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352 (10): 987-96, 2005.[PUBMED Abstract]
- Hegi ME, Diserens AC, Gorlia T, et al.: MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352 (10): 997-1003, 2005.[PUBMED Abstract]
- Cohen KJ, Pollack IF, Zhou T, et al.: Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol 13 (3): 317-23, 2011.[PUBMED Abstract]
- Grill J, Massimino M, Bouffet E, et al.: Phase II, Open-Label, Randomized, Multicenter Trial (HERBY) of Bevacizumab in Pediatric Patients With Newly Diagnosed High-Grade Glioma. J Clin Oncol 36 (10): 951-958, 2018.[PUBMED Abstract]
- Duffner PK, Horowitz ME, Krischer JP, et al.: Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328 (24): 1725-31, 1993.[PUBMED Abstract]
- Dufour C, Grill J, Lellouch-Tubiana A, et al.: High-grade glioma in children under 5 years of age: a chemotherapy only approach with the BBSFOP protocol. Eur J Cancer 42 (17): 2939-45, 2006.[PUBMED Abstract]
- Espinoza JC, Haley K, Patel N, et al.: Outcome of young children with high-grade glioma treated with irradiation-avoiding intensive chemotherapy regimens: Final report of the Head Start II and III trials. Pediatr Blood Cancer 63 (10): 1806-13, 2016.[PUBMED Abstract]
- 再発小児高悪性度星細胞腫の治療
-
至適管理を決定しこれを実施するために、小児高悪性度星細胞腫の治療は、小児脳腫瘍の治療経験があるがん専門医の集学的チームによって導かれるべきである。
高悪性度星細胞腫またはグリオーマ患者のほとんどでは、最終的に腫瘍が再発する。再発は通常、最初の診断から3年以内であるが、初期治療後何年も経過してから再発する患者もいる。疾患は原発腫瘍部位、切除断端/放射線照射部、または遠隔の中枢神経系部位に再発することもある。全身に再燃をみることはまれである。
再発時には、悪性腫瘍すべてに対して再燃の範囲を確認するための完全な評価が適応とされる。二次腫瘍および治療に関連する脳壊死などの実体は、臨床的には腫瘍再発と鑑別できないため、再燃の確認には生検または外科的切除術が必要とされる。
再発小児高悪性度星細胞腫に対する治療法の選択肢には以下のものがある:
- 手術。
- 幹細胞移植(SCT)を伴う大量化学療法。[ 1 ]
- 放射線療法。
- BRAF V600E変異を有する患者に対してBRAF阻害薬を用いた標的療法。
手術
外科的介入の有用性は、以下の点を基に個別に決定しなければならない:
SCTを伴う大量化学療法
造血幹細胞移植を伴う高用量骨髄破壊的化学療法は、再発時に微小残存病変を有する患者の厳選したサブセットにおいて有効な場合がある。[ 1 ][証拠レベル:3iiiA]しかしながら、さまざまな標的化学療法および多剤併用化学療法を検証したこれまでの臨床試験の結果のほとんどは、登録した患者に対して説得力のある有益性を示すことができていない。[ 2 ][ 3 ][ 4 ]
放射線療法
以前に放射線を照射されていない患者には、放射線療法が適切である。放射線量および放射線照射容積は、新たに診断された患者に使用する場合と同等である。一般に、放射線療法は、最初は放射線を回避する戦略で治療される幼児に対しては制限される。
以前に放射線照射を受けた患者に対して再照射が用いられているが、利益を示すデータは乏しい。少分割放射線療法または標準分割照射法のいずれかを用いる定位放射線手術(SRS)または定位放射線療法(SRT)技術を検討してもよい。容積の小さい明瞭な病変については、SRSにより正常組織を最大限に温存できる。より浸潤している病変には、分割放射線療法が正常組織の温存に比較的優れている。[ 5 ]
標的療法
再発高悪性度グリオーマに対する分子標的には制限がある。BRAF V600E変異はこれらの患者の小規模なサブセットにしかみられず、BRAF阻害薬に対する反応は少数の症例でしか認められていない。
BRAF V600変異が認められる再発膠芽腫患者においてBRAF阻害剤のベムラフェニブにより完全奏効が得られたことを示した症例報告がある。[ 6 ]1件の第I相試験では、BRAF V600E変異を認める進行性の高悪性度グリオーマを有する患児8人がダブラフェニブによる治療を受け、3人で完全奏効、3人で部分奏効が得られ、2人に病勢進行を認めたことが抄録にて報告された。[ 7 ]
臨床評価段階にある治療法の選択肢
再発高悪性度星細胞腫を有する小児の治療における免疫チェックポイント阻害の役割が現在研究段階にある。両アレル性ミスマッチ修復遺伝子異常が認められる小児は、変異による負荷が非常に強く、ネオアンチゲン発現が認められ、血液悪性腫瘍、消化管がん、脳腫瘍などさまざまながんを発症するリスクが高い。変異およびネオアンチゲンの強い負荷は免疫チェックポイント阻害への良好な反応と相関している。初期の症例報告により、抗プログラム死1(抗PD-1)阻害薬により治療された小児において臨床的およびX線画像での反応が実証されている。[ 8 ]
初期治療が失敗した患者には、新たな治療アプローチの臨床試験への参加など、他の治療法が有益である可能性がある。[ 9 ]特定の患者には、初期段階の臨床試験が利用できる場合がある。これらの試験は、小児腫瘍学グループ(COG)、Pediatric Brain Tumor Consortium、または他の団体を通して利用できる場合がある。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Finlay JL, Dhall G, Boyett JM, et al.: Myeloablative chemotherapy with autologous bone marrow rescue in children and adolescents with recurrent malignant astrocytoma: outcome compared with conventional chemotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 51 (6): 806-11, 2008.[PUBMED Abstract]
- Fouladi M, Nicholson HS, Zhou T, et al.: A phase II study of the farnesyl transferase inhibitor, tipifarnib, in children with recurrent or progressive high-grade glioma, medulloblastoma/primitive neuroectodermal tumor, or brainstem glioma: a Children's Oncology Group study. Cancer 110 (11): 2535-41, 2007.[PUBMED Abstract]
- Nicholson HS, Kretschmar CS, Krailo M, et al.: Phase 2 study of temozolomide in children and adolescents with recurrent central nervous system tumors: a report from the Children's Oncology Group. Cancer 110 (7): 1542-50, 2007.[PUBMED Abstract]
- Wetmore C, Daryani VM, Billups CA, et al.: Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: a children's Oncology Group Study ACNS1021. Cancer Med 5 (7): 1416-24, 2016.[PUBMED Abstract]
- Tsang DS, Oliveira C, Bouffet E, et al.: Repeat irradiation for children with supratentorial high-grade glioma. Pediatr Blood Cancer 66 (9): e27881, 2019.[PUBMED Abstract]
- Robinson GW, Orr BA, Gajjar A: Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 14: 258, 2014.[PUBMED Abstract]
- Kieran MW, Hargrave DR, Cohen KJ, et al.: Phase 1 study of dabrafenib in pediatric patients (pts) with relapsed or refractory BRAF V600E high- and low-grade gliomas (HGG, LGG), Langerhans cell histiocytosis (LCH), and other solid tumors (OST). [Abstract] J Clin Oncol 33 (15 Suppl): A-10004, 2015.[PUBMED Abstract]
- Bouffet E, Larouche V, Campbell BB, et al.: Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 34 (19): 2206-11, 2016.[PUBMED Abstract]
- Warren KE, Gururangan S, Geyer JR, et al.: A phase II study of O6-benzylguanine and temozolomide in pediatric patients with recurrent or progressive high-grade gliomas and brainstem gliomas: a Pediatric Brain Tumor Consortium study. J Neurooncol 106 (3): 643-9, 2012.[PUBMED Abstract]
- 本要約の変更点(07/06/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
本要約は再編集された。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児星細胞腫の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、ウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Childhood Astrocytomas Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/brain/hp/child-astrocytoma-treament-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389382]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。