

ご利用について
医療専門家向けの本PDQがん情報要約では、小児急性リンパ芽球性白血病の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児急性リンパ芽球性白血病(ALL)に関する一般情報
-
小児および青年におけるがんはまれであるが、ALLを含む小児がん全体の発生率は、1975年以降徐々に増加している。[ 1 ]小児および青年のがん患者の生存においては、劇的な改善が達成されている。[ 1 ][ 2 ][ 3 ]1975年から2010年の小児がんによる死亡率の減少は50%を超えている。[ 1 ][ 2 ][ 3 ]同じ期間に、ALLの5年生存率は、15歳未満の小児で60%から約90%に、15~19歳の青年で28%から75%超に上昇している。[ 4 ]小児および青年のがん生存者には、治療から数ヵ月または数年経過後もがん治療の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。(小児および青年のがん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)のPDQ要約を参照のこと。)
発生率
ALLは小児において診断されるがんの中で最も多く、15歳未満の小児で診断されるがんの約25%を占める。[ 2 ][ 3 ]米国におけるALLの年間発生率は、0~14歳で100万人当たり約41症例、および15~19歳で100万人当たり約17症例である。[ 4 ]米国では毎年約3,100人の小児および20歳未満の青年がALLの診断を受ける。[ 5 ]1975年以降、ALLの発生率に緩やかな増加が認められている。[ 4 ][ 6 ]
ALL発生率の明確なピークは2~3歳の小児に認められ(年間100万人当たり90症例を超える)、8歳までに100万人当たり30症例未満に発生率が低下する。[ 2 ][ 3 ]2~3歳のALL発生率は乳児の約4倍であり、同様に10歳以上の小児の4~5倍である。[ 2 ][ 3 ]
ALLの発生率はヒスパニック系の小児において最も高いと考えられている(100万人当たり43症例)。[ 2 ][ 3 ][ 7 ][ 8 ]白人小児におけるALLの発生率は黒人小児より大幅に高く、2~3歳の白人小児におけるALLの発生率は黒人小児より3倍近く高くなっている。[ 2 ][ 3 ][ 7 ]
解剖学
小児ALLは、骨髄に存在するTリンパ芽球およびBリンパ芽球を起源とする(図1を参照のこと)。

図1.血液細胞の分化。Tリンパ球およびBリンパ球を含むさまざまな血液細胞系列および免疫細胞系列が共通の血液幹細胞から分化する。 急性白血病は光学顕微鏡で確認された骨髄病変により、以下のように定義される:
急性白血病患者のほぼすべての患者で、M3の骨髄所見が得られる。
形態学的特徴
これまで、ALLリンパ芽球は、French-American-British(FAB)分類の基準を用いて、L1型、L2型、またはL3型に分類されていた。[ 9 ]しかしながら、この分類体系は、独立した予後的意義に欠け、本質的に主観的な分類であるため、もはや使用されていない。
形態学的にL3型を示すALL症例のほとんどが細胞表面免疫グロブリン(Ig)を発現し、バーキットリンパ腫にみられるものと同じMYC遺伝子転座(すなわち、t(8;14)(q24;q32)、t(2;8))が認められ、これはMYCがIg遺伝子のいずれかと結合したものである。この特異的でまれな形態の白血病(成熟B細胞またはバーキット白血病)の患者は、バーキットリンパ腫に対するプロトコルに従って治療すべきである。(成熟B細胞リンパ腫/白血病およびバーキットリンパ腫/白血病の治療に関する詳しい情報については、小児非ホジキンリンパ腫の治療に関するPDQ要約を参照のこと。)まれにL1/L2(L3ではない)の形態を有する芽球が表面Igを発現する。[ 10 ]これらの患者では、B-ALL患者と同じ治療法を使用すべきである。[ 10 ]
ALL発生の危険因子
ALL発生のリスク増大と関連している因子はほとんど同定されていない。ALLで主に受け入れられている危険因子および関連する遺伝子(重要な場合)には以下のものがある:
ダウン症候群
ダウン症候群の小児は、ALLおよびAMLの発症リスクがいずれも高く[ 22 ][ 23 ]、白血病の累積発症リスクは、5歳までが約2.1%、30歳までが約2.7%である。[ 22 ][ 23 ]
ダウン症候群の小児における急性白血病症例の約2分の1から3分の2がALLであり、小児ALL症例の約2~3%がダウン症候群の小児に発生する(小児期のダウン症候群の有病率は0.1%であることに注意)。[ 24 ][ 25 ][ 26 ][ 27 ]ダウン症候群の小児におけるALLは、ダウン症候群でない小児におけるALLとほぼ同じ年齢分布を示し、年齢中央値は3~4歳である。[ 24 ][ 25 ]対照的に、ダウン症候群の小児におけるAML症例の大多数は4歳前に発生する(年齢中央値、1歳)。[ 28 ]
ALLとダウン症候群を合併した患者では、予後良好(t(12;21)(p13;q22)/ETV6-RUNX1[TEL-AML1]および高二倍体[染色体数51~65])および予後不良(t(9;22)(q34;q11.2)またはt(4;11)(q21;q23)および低二倍体[染色体数が44未満])の細胞遺伝学的所見の発生率がいずれも低く、T細胞表現型はほとんど認められない。[ 24 ][ 25 ][ 26 ][ 28 ][ 29 ]
ダウン症候群患児におけるALL症例の約50~60%では、CRLF2に影響を及ぼすゲノム変化がみられ、この遺伝子により産生される蛋白が一般的に過剰発現しており、インターロイキン-7受容体αと二量体を形成してサイトカイン胸腺間質性リンパ球新生因子に対する受容体となる。[ 30 ][ 31 ][ 32 ]ダウン症候群を合併していないB-ALLの小児において、CRLF2のゲノム変化ははるかに低い頻度(10%未満)で観察される。[ 32 ][ 33 ][ 34 ]相対的に数が少ない公表されたシリーズに基づくと、ダウン症候群とALLを合併した小児におけるゲノムCRLF2異常は、予後的に重要でないと考えられるが、この問題に取り組むためにはさらに研究が必要である。[ 29 ][ 31 ]
ダウン症候群患児に発生するALLの約20%は、体細胞JAK2変異によるものであり[ 30 ][ 31 ][ 35 ][ 36 ][ 37 ]、この所見はALLの若年児であまりみられないが、主に高リスクB-ALLの年長児および青年の一部で観察される。[ 38 ]ダウン症候群とALLを合併し、JAK2変異を認める症例のほとんどすべてにCRLF2ゲノム変化も認められる。[ 30 ][ 31 ][ 32 ]予備的な証拠によると、ダウン症候群とALLを合併した小児では、JAK2変異の状態と5年イベントフリー生存(EFS)率との間に関連性がみられないことが示唆されるが[ 31 ][ 36 ]、この問題に取り組むためにはさらに研究が必要である。
ゲノムワイド関連解析により、ダウン症候群ではない集団においてB-ALLと関連している4つの感受性遺伝子座(IKZF1、CDKN2A、ARID5B、およびGATA3)は、ダウン症候群の小児におけるALLへの感受性とも関連していることが明らかになった。[ 39 ]CDKN2Aのリスクアレルの浸透度は、ダウン症候群の小児で高いようであった。
ダウン症候群とALLを合併した小児の最大35%にIKZF1遺伝子欠失が認められており、この患者群における有意に不良な転帰との関連性が指摘されている。[ 31 ][ 40 ]
低浸透度および高浸透度の遺伝性遺伝子多様体
ALLに対する遺伝的素因は、以下のようにおおまかにいくつかのカテゴリーに分類できる:
小児ALLの出生前起源
ALLの発生は、ほとんどの症例で多段階プロセスであり、明らかな白血病が発生するには複数の遺伝子変化が必要である。少なくとも小児ALLの一部症例では、最初の遺ゲノム変化が子宮内で発生すると考えられている。これを支持する証拠は、出生時に得られた血液サンプルに、各患者の白血病細胞に特異的な免疫グロブリンまたはT細胞受容体抗原の再構成が検出できるという観察からもたらされている。[ 58 ][ 59 ]同様に、特定の染色体異常を特徴とするALLでは、出生時の血液細胞に白血病のゲノム異常が1つ以上あり、さらに共同して働くゲノム変化を生後に獲得する患者が一部にいる。[ 58 ][ 59 ][ 60 ]遺伝的に一致した白血病を有する一卵性双生児のゲノム研究により、一部の白血病における出生前起源がさらに裏付けられている。[ 58 ][ 61 ]
ALLを発症していない小児の一部には、ALLに関連するゲノム変化を有するまれな血液細胞が出生時にみられるという証拠もある。初期の研究では、ETV6-RUNX1転座が注目され、遺伝子融合の存在を示すRNA転写産物を同定する逆転写(RT)ポリメラーゼ連鎖反応(PCR)が使用された。例えば、ある研究によると、新生児の血液スポット(ガスリーカード)検査で、ETV6-RUNX1転座の陽性率は1%であった。[ 62 ]その後の報告で、一部の小児の出生時におけるETV6-RUNX1転座の存在がおおむね確認されたが、陽性率および陽性度は大幅に異なっていた。
この疑問により断定的に取り組むため、感度および特異度が高いDNAベースのアプローチ(genomic inverse PCR for exploration of ligated breakpoints [GIPFEL])が1,000の臍帯血標本から得られたDNAに適用され、標本の5%にETV6-RUNX1転座が認められた。[ 63 ]TCF3-PBX1融合を検出するために、同じ方法を340の臍帯血標本に適用した場合、2つの臍帯血標本(0.6%)における存在が陽性であった。[ 64 ]ETV6-RUNX1およびTCF3-PBX1の両方で、これらの転座の1つが陽性の臍帯血標本の割合は、いずれかの種類のALLを発症する小児の割合(0.01%未満)をはるかに超えていた。
全般的な予後
ALL小児のうち約98%が寛解に達する。1~18歳で新たにALLと診断され、現行の治療を受けた患者の約85%が長期のイベントフリー生存者となると予想され、5年生存者は90%を超える。[ 70 ][ 71 ][ 72 ][ 73 ]微小残存病変(MRD)の結果と組み合わせた細胞遺伝学的所見およびゲノム所見により、EFS率が95%を超えるALLのサブセットに加え、逆にEFS率が50%以下のサブセットが定義できる(詳しい情報については、小児ALLの細胞遺伝学/ゲノミクスおよびリスクに基づく治療に影響する予後因子のセクションを参照のこと)。
小児ALLでは治療法が進歩しているにもかかわらず、治療関連毒性を最小限に抑え、すべてのALL患児を治癒させる目標が達成できるまでには、解決しなければならない重要な生物学的・治療的課題が多く残されている。これらの問題を体系的に研究するためには大規模な臨床試験が必要であり、これらの試験に参加する機会がほとんどの患者および家族に提供される。
ALLの小児および青年を対象とした臨床試験では、治癒率改善および/または毒性低減を目指した研究的なレジメンに対して、その時点で標準として受け入れられている治療法を比較するようデザインされるのが一般的である。対象となる患者群で治癒率がきわめて高い特定の試験では、治療縮小の課題が求められることもある。小児ALLおよびその他の小児がんで確認されている治癒的療法で行われた進歩のほとんどは、研究者による発見を通して達成され、注意深いランダム化対照比較による多施設共同臨床試験において検証されてきた。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 28. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 29. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Howlader N, Noone AM, Krapcho M: SEER Cancer Statistics Review (CSR) 1975-2013. Bethesda, Md: National Cancer Institute, 2015. Available online. Last accessed February 13, 2020.[PUBMED Abstract]
- Special section: cancer in children and adolescents. In: American Cancer Society: Cancer Facts and Figures 2014. Atlanta, Ga: American Cancer Society, 2014, pp 25-42. Available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Shah A, Coleman MP: Increasing incidence of childhood leukaemia: a controversy re-examined. Br J Cancer 97 (7): 1009-12, 2007.[PUBMED Abstract]
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Barrington-Trimis JL, Cockburn M, Metayer C, et al.: Rising rates of acute lymphoblastic leukemia in Hispanic children: trends in incidence from 1992 to 2011. Blood 125 (19): 3033-4, 2015.[PUBMED Abstract]
- Bennett JM, Catovsky D, Daniel MT, et al.: The morphological classification of acute lymphoblastic leukaemia: concordance among observers and clinical correlations. Br J Haematol 47 (4): 553-61, 1981.[PUBMED Abstract]
- Koehler M, Behm FG, Shuster J, et al.: Transitional pre-B-cell acute lymphoblastic leukemia of childhood is associated with favorable prognostic clinical features and an excellent outcome: a Pediatric Oncology Group study. Leukemia 7 (12): 2064-8, 1993.[PUBMED Abstract]
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.[PUBMED Abstract]
- Passarge E: Bloom's syndrome: the German experience. Ann Genet 34 (3-4): 179-97, 1991.[PUBMED Abstract]
- Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.[PUBMED Abstract]
- Taylor AM, Metcalfe JA, Thick J, et al.: Leukemia and lymphoma in ataxia telangiectasia. Blood 87 (2): 423-38, 1996.[PUBMED Abstract]
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.[PUBMED Abstract]
- Powell BC, Jiang L, Muzny DM, et al.: Identification of TP53 as an acute lymphocytic leukemia susceptibility gene through exome sequencing. Pediatr Blood Cancer 60 (6): E1-3, 2013.[PUBMED Abstract]
- Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.[PUBMED Abstract]
- Ilencikova D, Sejnova D, Jindrova J, et al.: High-grade brain tumors in siblings with biallelic MSH6 mutations. Pediatr Blood Cancer 57 (6): 1067-70, 2011.[PUBMED Abstract]
- Ripperger T, Schlegelberger B: Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet 59 (3): 133-42, 2016.[PUBMED Abstract]
- Moriyama T, Relling MV, Yang JJ: Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 125 (26): 3988-95, 2015.[PUBMED Abstract]
- Li Y, Schwab C, Ryan SL, et al.: Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508 (7494): 98-102, 2014.[PUBMED Abstract]
- Hasle H: Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol 2 (7): 429-36, 2001.[PUBMED Abstract]
- Whitlock JA: Down syndrome and acute lymphoblastic leukaemia. Br J Haematol 135 (5): 595-602, 2006.[PUBMED Abstract]
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.[PUBMED Abstract]
- Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.[PUBMED Abstract]
- Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.[PUBMED Abstract]
- de Graaf G, Buckley F, Skotko BG: Estimation of the number of people with Down syndrome in the United States. Genet Med 19 (4): 439-447, 2017.[PUBMED Abstract]
- Chessells JM, Harrison G, Richards SM, et al.: Down's syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch Dis Child 85 (4): 321-5, 2001.[PUBMED Abstract]
- Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.[PUBMED Abstract]
- Hertzberg L, Vendramini E, Ganmore I, et al.: Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood 115 (5): 1006-17, 2010.[PUBMED Abstract]
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.[PUBMED Abstract]
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.[PUBMED Abstract]
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.[PUBMED Abstract]
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.[PUBMED Abstract]
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.[PUBMED Abstract]
- Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.[PUBMED Abstract]
- Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.[PUBMED Abstract]
- Mullighan CG, Zhang J, Harvey RC, et al.: JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106 (23): 9414-8, 2009.[PUBMED Abstract]
- Brown AL, de Smith AJ, Gant VU, et al.: Inherited genetic susceptibility to acute lymphoblastic leukemia in Down syndrome. Blood 134 (15): 1227-1237, 2019.[PUBMED Abstract]
- Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.[PUBMED Abstract]
- Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al.: Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1006-10, 2009.[PUBMED Abstract]
- Treviño LR, Yang W, French D, et al.: Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1001-5, 2009.[PUBMED Abstract]
- Migliorini G, Fiege B, Hosking FJ, et al.: Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 122 (19): 3298-307, 2013.[PUBMED Abstract]
- Hungate EA, Vora SR, Gamazon ER, et al.: A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun 7: 10635, 2016.[PUBMED Abstract]
- Sherborne AL, Hosking FJ, Prasad RB, et al.: Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42 (6): 492-4, 2010.[PUBMED Abstract]
- Xu H, Yang W, Perez-Andreu V, et al.: Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105 (10): 733-42, 2013.[PUBMED Abstract]
- Ellinghaus E, Stanulla M, Richter G, et al.: Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 26 (5): 902-9, 2012.[PUBMED Abstract]
- Qian M, Zhao X, Devidas M, et al.: Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J Natl Cancer Inst 111 (12): 1350-1357, 2019.[PUBMED Abstract]
- Shah S, Schrader KA, Waanders E, et al.: A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 45 (10): 1226-31, 2013.[PUBMED Abstract]
- Auer F, Rüschendorf F, Gombert M, et al.: Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 28 (5): 1136-8, 2014.[PUBMED Abstract]
- Zhang MY, Churpek JE, Keel SB, et al.: Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet 47 (2): 180-5, 2015.[PUBMED Abstract]
- Topka S, Vijai J, Walsh MF, et al.: Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genet 11 (6): e1005262, 2015.[PUBMED Abstract]
- Noetzli L, Lo RW, Lee-Sherick AB, et al.: Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 47 (5): 535-8, 2015.[PUBMED Abstract]
- Rampersaud E, Ziegler DS, Iacobucci I, et al.: Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv 3 (7): 1039-1046, 2019.[PUBMED Abstract]
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.[PUBMED Abstract]
- Churchman ML, Qian M, Te Kronnie G, et al.: Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 33 (5): 937-948.e8, 2018.[PUBMED Abstract]
- Kuehn HS, Boisson B, Cunningham-Rundles C, et al.: Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med 374 (11): 1032-1043, 2016.[PUBMED Abstract]
- Greaves MF, Wiemels J: Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 3 (9): 639-49, 2003.[PUBMED Abstract]
- Taub JW, Konrad MA, Ge Y, et al.: High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood 99 (8): 2992-6, 2002.[PUBMED Abstract]
- Bateman CM, Colman SM, Chaplin T, et al.: Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 115 (17): 3553-8, 2010.[PUBMED Abstract]
- Greaves MF, Maia AT, Wiemels JL, et al.: Leukemia in twins: lessons in natural history. Blood 102 (7): 2321-33, 2003.[PUBMED Abstract]
- Mori H, Colman SM, Xiao Z, et al.: Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 99 (12): 8242-7, 2002.[PUBMED Abstract]
- Schäfer D, Olsen M, Lähnemann D, et al.: Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 131 (7): 821-826, 2018.[PUBMED Abstract]
- Hein D, Dreisig K, Metzler M, et al.: The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in ≈0.6% of healthy newborns. Blood 134 (16): 1355-1358, 2019.[PUBMED Abstract]
- Rabin KR, Gramatges MM, Margolin JF, et al.: Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 463-97.[PUBMED Abstract]
- Chessells JM; haemostasis and thrombosis task force, British committee for standards in haematology: Pitfalls in the diagnosis of childhood leukaemia. Br J Haematol 114 (3): 506-11, 2001.[PUBMED Abstract]
- Onciu M: Acute lymphoblastic leukemia. Hematol Oncol Clin North Am 23 (4): 655-74, 2009.[PUBMED Abstract]
- Heerema-McKenney A, Cleary M, Arber D: Pathology and molecular diagnosis of leukemias and lymphomas. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 113-30.[PUBMED Abstract]
- Cheng J, Klairmont MM, Choi JK: Peripheral blood flow cytometry for the diagnosis of pediatric acute leukemia: Highly reliable with rare exceptions. Pediatr Blood Cancer 66 (1): e27453, 2019.[PUBMED Abstract]
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.[PUBMED Abstract]
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.[PUBMED Abstract]
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.[PUBMED Abstract]
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.[PUBMED Abstract]
- 小児ALLの世界保健機関(WHO)分類システム
-
造血組織およびリンパ組織の腫瘍の2016年版WHO分類では、急性リンパ性白血病について以下の疾患名が掲載されている:[ 1 ]
B細胞リンパ芽球性白血病/リンパ腫の2016年版WHO分類
T細胞リンパ芽球性白血病/リンパ腫の2016年版WHO分類
細胞系列があいまいな急性白血病の2016年版WHO分類
細胞系列があいまいな急性白血病は、急性骨髄性白血病(AML)と急性リンパ芽球性白血病(ALL)の両方の特徴を有する急性白血病のグループで、そのWHO分類を表1で要約している。[ 2 ][ 3 ]混合表現型急性白血病(MPAL)の診断に対して細胞系列を割り当てるための基準を表2に示している。[ 1 ]
表1.造血器およびリンパ組織腫瘍に関する世界保健機関(WHO)分類による細胞系列があいまいな急性白血病a 疾患 定義 NOS = 他に特定されない。 a 出典: Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias!Haematologica 94 (7): 891-3, 2009.[ 2 ]Obtained from Haematologica/the Hematology Journal website http://www.haematologica.org. 急性未分化型白血病 リンパ系または骨髄系のいずれかの細胞系列に対して特異的と考えられるいずれのマーカーも発現していない急性白血病 混合表現型急性白血病、t(9;22)(q34;q11.2);BCR-ABL1を伴う(BCR-ABL1を伴うMPAL)。 芽球に(9;22)転座またはBCR-ABL1再構成も認められる混合表現型急性白血病の診断基準を満たす急性白血病 混合表現型急性白血病、t(v;11q23);KMT2A(MLL)再構成を伴う(KMT2Aを伴うMPAL)。 芽球にKMT2A遺伝子を巻き込んだ転座も認められる混合表現型急性白血病の診断基準を満たす急性白血病 混合表現型急性白血病、B細胞性/骨髄性、NOS(B/M MPAL) 芽球にBCR-ABL1またはKMT2Aを巻き込んだ遺伝子異常がみられないB細胞および骨髄細胞の両系列へ割り当てる診断基準を満たす急性白血病 混合表現型急性白血病、T細胞性/骨髄性、NOS(T/M MPAL) 芽球にBCR-ABL1またはKMT2Aを巻き込んだ遺伝子異常がみられないT細胞および骨髄細胞の両系列へ割り当てる診断基準を満たす急性白血病 混合表現型急性白血病、B細胞性/骨髄性、NOS—まれな種類 B細胞およびT細胞の両系列へ割り当てる診断基準を満たす急性白血病 他の細胞系列があいまいな白血病 ナチュラルキラー細胞リンパ芽球性白血病/リンパ腫 表2.骨髄腫瘍と急性白血病の2016年版世界保健機関分類による混合表現型急性白血病に対する細胞系列の割り当て基準a 細胞系列 基準 a 出典:Arber et al.[ 1 ] b 「強い」とは、標本内の正常なBまたはT細胞と比較して同等か、より明るいものと定義された。 骨髄細胞系列 ミエロペルオキシダーゼ(フローサイトメトリー、免疫組織化学、または細胞化学);あるいは単球分化(次のうち少なくとも2つ:非特異的エステラーゼ細胞化学、CD11c、CD14、CD64、リゾチーム) T細胞系列 強いb細胞質CD3(CD3イプシロン鎖に対する抗体を伴う);または細胞表面のCD3 B細胞系列 強いbCD19と次のうち少なくとも1つが強く発現している:CD79a、細胞質CD22、またはCD10;あるいは弱いCD19と次のうち少なくとも2つが強く発現している:CD79a、細胞質CD22、またはCD10 混合表現型の白血病では、以下のようなさまざまな所見がみられることがある:
- 通常はリンパ系細胞と骨髄系細胞の2つの異なった細胞集団を認める二細胞系列白血病。
- 個々の芽球がリンパ系細胞と骨髄系細胞の両方の特徴を示す二重表現型白血病。
二重表現型の症例は混合表現型白血病の大多数を占める。[ 4 ] TEL-AML1融合がみられない骨髄B細胞二重表現型白血病の患者では、B-ALLの患者と比較して、完全寛解(CR)率が低く、イベントフリー生存(EFS)率が有意に不良である。[ 4 ]一部の研究によると、二重表現型白血病患者は、骨髄系と対照的にリンパ系の治療レジメンを用いることで予後がよい可能性が示唆される。[ 5 ][ 6 ][ 7 ][ 8 ];[ 9 ][証拠レベル:3iiiA]国際ベルリン-フランクフルト-ミュンスター(BFM)グループによる大規模なレトロスペクティブ研究で、ALL向けレジメンによる初回治療に伴い、AML向けレジメンまたはALL/AMLの併用レジメンと比較して優れた転帰が得られ、特にCD19陽性例または他のリンパ系抗原発現例で顕著であった。この研究で、初回CR期での造血幹細胞移植(HSCT)は有益でなかったが、治療1ヵ月後に骨髄病変が残存する形態学的証拠(芽球が5%以上)がある場合は例外であると考えられる。[ 8 ]
これらの疾患単位に対する予後的意義に加え、重要な臨床的および生物学的特性については、本要約の小児ALLの細胞遺伝学/ゲノミクスのセクションを参照のこと。
参考文献- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[PUBMED Abstract]
- Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 150-5.[PUBMED Abstract]
- Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.[PUBMED Abstract]
- Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.[PUBMED Abstract]
- Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.[PUBMED Abstract]
- Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.[PUBMED Abstract]
- Hrusak O, de Haas V, Stancikova J, et al.: International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood 132 (3): 264-276, 2018.[PUBMED Abstract]
- Orgel E, Alexander TB, Wood BL, et al.: Mixed-phenotype acute leukemia: A cohort and consensus research strategy from the Children's Oncology Group Acute Leukemia of Ambiguous Lineage Task Force. Cancer 126 (3): 593-601, 2020.[PUBMED Abstract]
- 小児ALLの細胞遺伝学/ゲノミクス
-
小児ALLのゲノミクス
小児ALLのゲノミクスは、広範にわたり研究されており、細胞遺伝学的および分子遺伝学的特性に基づいて代表的な亜型がいくつか同定されており、それぞれが独自の臨床的および予後的特性パターンを有している。[ 1 ]図2に細胞遺伝学/分子的亜型別のALL症例の分布を例示する。[ 1 ]
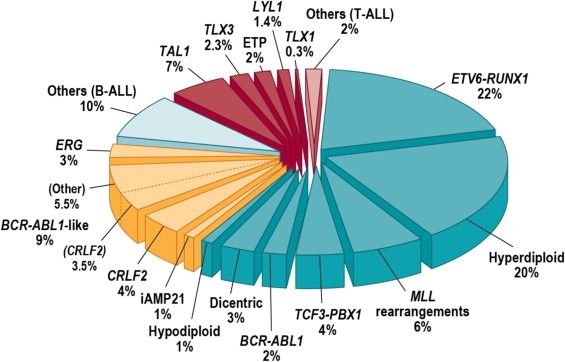
図2.小児ALLの亜分類。青色のウェッジは前駆B細胞型ALL、黄色のウェッジは最近同定されたB-ALLの亜型、赤色のウェッジはT細胞系列ALLを示す。Elsevierから許諾を得て転載:Seminars in Hematology, Volume 50, Charles G. Mullighan, Genomic Characterization of Childhood Acute Lymphoblastic Leukemia, Pages 314-324, Copyright (2013). B-ALLの細胞遺伝学/ゲノミクス
B-ALLにおけるゲノムの全体像は、正常なB細胞産生を妨げる一連のゲノム変化、また一部の例では増殖シグナルを発する遺伝子における変異(例、RASファミリー遺伝子における活性化変異またはキナーゼ経路シグナリングにつながる変異/転座)という特徴を示す。B細胞産生の阻止につながるゲノム変化には、転座(例、TCF3-PBX1およびETV6-RUNX1)、点変異(例、IKZF1およびPAX5)、および遺伝子内/遺伝子間欠失(例、IKZF1、PAX5、EBF、およびERG)がある。[ 2 ]
B-ALLにおけるゲノム変化がランダムに発生する傾向はみられないが、むしろその遺伝子発現プロファイルのような生物学的特徴により説明できる亜型内で群発する傾向がある。頻発性の染色体転座を認める症例(例、TCF3-PBX1、ETV6-RUNX1、およびKMT2A [MLL] 再構成ALL)は、独特な生物学的特徴を有し、この点を例示しており、特有な生物学的亜型内での特定のゲノム変化の以下の例も同様である:
キナーゼ遺伝子の活性化点変異は、高リスクB-ALLでまれである。変異していることが明らかになる第一のキナーゼ遺伝子がJAKである。これらの変異は、一般にCRLF2異常を伴うPh-like ALL患者で観察されるが、ダウン症候群のALL小児でも約15%にJAK2変異が観察される。[ 4 ][ 8 ][ 9 ]いくつかのキナーゼ遺伝子およびサイトカイン受容体遺伝子は、以下のPh+ ALLおよびPh-like ALLの考察で説明しているように、転座により活性化される。FLT3変異は、高二倍体ALLおよびKMT2A再構成ALLの少数例(約10%)で発生し、他の亜型ではまれである。[ 10 ]
再燃時のB-ALLのゲノミクスの解明は、診断時のALLのゲノミクスの解明より進展が遅れている。小児ALLは診断時にしばしば多クローン性であり、治療の選択的影響下で、一部のクローンが消滅し、特有なゲノムプロファイルを有する新たなクローンが発生することがある。[ 11 ]特に重要な点として、再燃時に特定の治療要素により選択されることがある新たな変異が発生する。1つの例として、NT5C2の変異は診断時に検出されないが、この変異について2件の研究で評価され、早期再燃を来したB-ALLの44例中7例(16%)および20例中9例(45%)にNT5C2の特異的変異が観察された。[ 11 ][ 12 ]NT5C2変異は、再燃が遅い患者でまれであり、メルカプトプリン(6-MP)およびthioguanineに対する抵抗性を誘導すると考えられている。[ 12 ]再燃時のみに変異が検出される他の遺伝子は、プリン生合成に関与する遺伝子のPRSP1である。[ 13 ]中国人コホートの13.0%およびドイツ人コホートの2.7%に変異が観察されており、治療中に再燃した患者で変異が観察された。再燃例で観察されたPRSP1変異は、白血病細胞株でチオプリン系薬物に対する抵抗性を誘導した。CREBBP変異も再燃時に豊富にみられ、グルココルチコイド系薬物に対する抵抗性増加に関係していると考えられている。[ 11 ][ 14 ]再燃のゲノミクスに関する理解が深まるにつれて、再燃を避けるように初期治療を修正すること、または抵抗性を誘導する変異を早期に検出して明らかな再燃前に介入することが可能になるかもしれない。
頻発する染色体異常の多くには予後的意義があり、特にB-ALLで顕著なことが明らかにされている。その中には、高度の高二倍体(染色体数が51~65)およびETV6-RUNX1融合のように、より予後良好な転帰と関連している染色体異常がある。他の変化は歴史的に予後不良に関連しており、Ph染色体(t(9;22)(q34;q11.2)、KMT2A遺伝子の再構成、低二倍体、およびAML1遺伝子の染色体内増幅(iAMP21)などがある。[ 15 ]
これらのゲノム変化の多くで臨床的意義が認識されたことから、造血組織およびリンパ組織の腫瘍の2016年版世界保健機関分類では、B-ALLに対して以下の病型が記載されている:[ 16 ]
小児ALLについて、これらおよびその他の染色体とゲノムの異常を以下に記載する。
- 染色体数。
- 染色体転座および染色体セグメントの増幅/欠失。
T-ALLの細胞遺伝学/ゲノミクス
T-ALLは、T細胞発生に関連する転写プログラムの活性化につながるゲノム変化、ならびにNOTCH1経路の活性化をもたらすNOTCH1および/またはFBXW7における変異を有する症例の頻度が高い(約60%)ことを特徴とする。[ 131 ]B-ALLと対照的に、T-ALLのゲノム変化の予後的意義はほとんど確定していない。B細胞系列ALLによくみられる細胞遺伝学的異常(例、染色体数が51~65の高二倍体)がT-ALLでみられるのはまれである。[ 132 ][ 133 ]
初期の前駆T細胞ALLの細胞遺伝学/ゲノミクス
初期の前駆T細胞ALLを対象とした分子的特徴の詳細な検索により、この疾患は分子レベルにおいて高度に不均一で、3分の1を超える症例では変異またはコピー数の変化により影響を受けた遺伝子が1つもみられないことが示された。[ 153 ]他のT-ALL症例と比較した場合、初期の前駆T細胞性グループでは、NOTCH1突然変異の発生率が低く、サイトカイン受容体およびRASの信号伝達、造血発生、およびヒストン修飾を調節している遺伝子の変異頻度が有意に高かった。初期の前駆T細胞ALLの転写プロファイルは、正常な造血幹細胞および骨髄性白血病幹細胞のものと類似性を示している。[ 153 ]
比較ゲノムハイブリダイゼーションおよび/または定量的DNA-PCRにより検出されるようなTCRγ遺伝子座の両アレル欠失の非検出(ABD)は、T-ALL患者の早期治療失敗に関連していることが研究により明らかになっている。[ 154 ][ 155 ]ABDは初期胸腺前駆細胞に特徴的な所見で、ABDを伴うT-ALL患者の多くが初期の前駆T細胞の表現型の診断と一致した免疫表現型を有する。
混合表現型急性白血病(MPAL)の細胞遺伝学/ゲノミクス
細胞系列があいまいな急性白血病について、WHO分類システムを表3に要約している。[ 156 ][ 157 ]MPALの診断に対して細胞系列を割り当てるための基準を表4に提示している。[ 16 ]
表3.造血器およびリンパ組織腫瘍に関する世界保健機関(WHO)分類による細胞系列があいまいな急性白血病a 疾患 定義 NOS = 他に特定されない。 a 出典: Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias!Haematologica 94 (7): 891-3, 2009.[ 156 ]Obtained from Haematologica/the Hematology Journal website http://www.haematologica.org. 急性未分化型白血病 リンパ系または骨髄系のいずれかの細胞系列に対して特異的と考えられるいずれのマーカーも発現していない急性白血病 混合表現型急性白血病、t(9;22)(q34;q11.2);BCR-ABL1を伴う(BCR-ABL1を伴うMPAL) 芽球に(9;22)転座またはBCR-ABL1再構成も認められる混合表現型急性白血病の診断基準を満たす急性白血病 混合表現型急性白血病、t(v;11q23);KMT2A(MLL)再構成を伴う(KMT2Aを伴うMPAL) 芽球にKMT2A遺伝子を巻き込んだ転座も認められる混合表現型急性白血病の診断基準を満たす急性白血病 混合表現型急性白血病、B細胞性/骨髄性、NOS(B/M MPAL) 芽球にBCR-ABL1またはKMT2Aを巻き込んだ遺伝子異常がみられないB細胞および骨髄細胞の両系列へ割り当てる診断基準を満たす急性白血病 混合表現型急性白血病、T細胞性/骨髄性、NOS(T/M MPAL) 芽球にBCR-ABL1またはKMT2Aを巻き込んだ遺伝子異常がみられないT細胞および骨髄細胞の両系列へ割り当てる診断基準を満たす急性白血病 混合表現型急性白血病、B細胞性/骨髄性、NOS—まれな種類 B細胞およびT細胞の両系列へ割り当てる診断基準を満たす急性白血病 他の細胞系列があいまいな白血病 ナチュラルキラー細胞リンパ芽球性白血病/リンパ腫 表4.骨髄腫瘍と急性白血病の2016年版世界保健機関分類による混合表現型急性白血病に対する細胞系列の割り当て基準a 細胞系列 基準 a 出典:Arber et al.[ 16 ] b 「強い」とは、標本内の正常なBまたはT細胞と比較して同等か、より明るいものと定義された。 骨髄細胞系列 ミエロペルオキシダーゼ(フローサイトメトリー、免疫組織化学、または細胞化学);あるいは単球分化(次のうち少なくとも2つ:非特異的エステラーゼ細胞化学、CD11c、CD14、CD64、リゾチーム) T細胞系列 強いb細胞質CD3(CD3イプシロン鎖に対する抗体を伴う);または細胞表面のCD3 B細胞系列 強いbCD19と次のうち少なくとも1つが強く発現している:CD79a、細胞質CD22、またはCD10;あるいは弱いCD19と次のうち少なくとも2つが強く発現している:CD79a、細胞質CD22、またはCD10 MPALの分類システムには、原発性分子変化により定義される2つの病型:BCR-ABL1転座を伴うMPALおよびKMT2A再構成を伴うMPALが含まれている。MPAL、B細胞性/骨髄性、NOS(B/M MPAL)およびMPAL, T細胞性/骨髄性、NOS(T/M MPAL)の病型と関連するゲノム変化は独特であり、以下に記載している:
参考文献- Mullighan CG: Genomic characterization of childhood acute lymphoblastic leukemia. Semin Hematol 50 (4): 314-24, 2013.[PUBMED Abstract]
- Mullighan CG, Goorha S, Radtke I, et al.: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 (7137): 758-64, 2007.[PUBMED Abstract]
- Mullighan CG, Miller CB, Radtke I, et al.: BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 (7191): 110-4, 2008.[PUBMED Abstract]
- Roberts KG, Li Y, Payne-Turner D, et al.: Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371 (11): 1005-15, 2014.[PUBMED Abstract]
- Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al.: Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7: 11790, 2016.[PUBMED Abstract]
- Zhang J, McCastlain K, Yoshihara H, et al.: Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48 (12): 1481-1489, 2016.[PUBMED Abstract]
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.[PUBMED Abstract]
- Loh ML, Zhang J, Harvey RC, et al.: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 121 (3): 485-8, 2013.[PUBMED Abstract]
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.[PUBMED Abstract]
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.[PUBMED Abstract]
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.[PUBMED Abstract]
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.[PUBMED Abstract]
- Li B, Li H, Bai Y, et al.: Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 21 (6): 563-71, 2015.[PUBMED Abstract]
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.[PUBMED Abstract]
- Moorman AV, Ensor HM, Richards SM, et al.: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11 (5): 429-38, 2010.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48 (8): 637-60, 2009.[PUBMED Abstract]
- Aricò M, Valsecchi MG, Rizzari C, et al.: Long-term results of the AIEOP-ALL-95 Trial for Childhood Acute Lymphoblastic Leukemia: insight on the prognostic value of DNA index in the framework of Berlin-Frankfurt-Muenster based chemotherapy. J Clin Oncol 26 (2): 283-9, 2008.[PUBMED Abstract]
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.[PUBMED Abstract]
- Synold TW, Relling MV, Boyett JM, et al.: Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest 94 (5): 1996-2001, 1994.[PUBMED Abstract]
- Moorman AV, Richards SM, Martineau M, et al.: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 102 (8): 2756-62, 2003.[PUBMED Abstract]
- Chilton L, Buck G, Harrison CJ, et al.: High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia 28 (7): 1511-8, 2014.[PUBMED Abstract]
- Sutcliffe MJ, Shuster JJ, Sather HN, et al.: High concordance from independent studies by the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children's Oncology Group (COG) initiative. Leukemia 19 (5): 734-40, 2005.[PUBMED Abstract]
- Harris MB, Shuster JJ, Carroll A, et al.: Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood 79 (12): 3316-24, 1992.[PUBMED Abstract]
- Heerema NA, Harbott J, Galimberti S, et al.: Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18 (4): 693-702, 2004.[PUBMED Abstract]
- Carroll AJ, Shago M, Mikhail FM, et al.: Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children's Oncology Group. Cancer Genet 238: 62-68, 2019.[PUBMED Abstract]
- Nachman JB, Heerema NA, Sather H, et al.: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 110 (4): 1112-5, 2007.[PUBMED Abstract]
- Raimondi SC, Zhou Y, Shurtleff SA, et al.: Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome. Cancer Genet Cytogenet 169 (1): 50-7, 2006.[PUBMED Abstract]
- Attarbaschi A, Mann G, König M, et al.: Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 18 (10): 1611-6, 2004.[PUBMED Abstract]
- Lemez P, Attarbaschi A, Béné MC, et al.: Childhood near-tetraploid acute lymphoblastic leukemia: an EGIL study on 36 cases. Eur J Haematol 85 (4): 300-8, 2010.[PUBMED Abstract]
- Paulsson K, Lilljebjörn H, Biloglav A, et al.: The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47 (6): 672-6, 2015.[PUBMED Abstract]
- Harrison CJ, Moorman AV, Broadfield ZJ, et al.: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125 (5): 552-9, 2004.[PUBMED Abstract]
- Mullighan CG, Jeha S, Pei D, et al.: Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 126 (26): 2896-9, 2015.[PUBMED Abstract]
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.[PUBMED Abstract]
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.[PUBMED Abstract]
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.[PUBMED Abstract]
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.[PUBMED Abstract]
- Rubnitz JE, Wichlan D, Devidas M, et al.: Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26 (13): 2186-91, 2008.[PUBMED Abstract]
- Kanerva J, Saarinen-Pihkala UM, Niini T, et al.: Favorable outcome in 20-year follow-up of children with very-low-risk ALL and minimal standard therapy, with special reference to TEL-AML1 fusion. Pediatr Blood Cancer 42 (1): 30-5, 2004.[PUBMED Abstract]
- Aldrich MC, Zhang L, Wiemels JL, et al.: Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev 15 (3): 578-81, 2006.[PUBMED Abstract]
- Loh ML, Goldwasser MA, Silverman LB, et al.: Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 107 (11): 4508-13, 2006.[PUBMED Abstract]
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.[PUBMED Abstract]
- Madzo J, Zuna J, Muzíková K, et al.: Slower molecular response to treatment predicts poor outcome in patients with TEL/AML1 positive acute lymphoblastic leukemia: prospective real-time quantitative reverse transcriptase-polymerase chain reaction study. Cancer 97 (1): 105-13, 2003.[PUBMED Abstract]
- Bhojwani D, Pei D, Sandlund JT, et al.: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26 (2): 265-70, 2012.[PUBMED Abstract]
- Enshaei A, Schwab CJ, Konn ZJ, et al.: Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 27 (11): 2256-9, 2013.[PUBMED Abstract]
- Barbany G, Andersen MK, Autio K, et al.: Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t(12;21)(p13;q22)-positive B-cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-ALL-2000 protocol. Leuk Res 36 (7): 936-8, 2012.[PUBMED Abstract]
- Forestier E, Heyman M, Andersen MK, et al.: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 140 (6): 665-72, 2008.[PUBMED Abstract]
- Seeger K, Stackelberg AV, Taube T, et al.: Relapse of TEL-AML1--positive acute lymphoblastic leukemia in childhood: a matched-pair analysis. J Clin Oncol 19 (13): 3188-93, 2001.[PUBMED Abstract]
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.[PUBMED Abstract]
- Zuna J, Ford AM, Peham M, et al.: TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10 (16): 5355-60, 2004.[PUBMED Abstract]
- van Delft FW, Horsley S, Colman S, et al.: Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 (23): 6247-54, 2011.[PUBMED Abstract]
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.[PUBMED Abstract]
- Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.[PUBMED Abstract]
- Ribeiro RC, Broniscer A, Rivera GK, et al.: Philadelphia chromosome-positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11 (9): 1493-6, 1997.[PUBMED Abstract]
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.[PUBMED Abstract]
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.[PUBMED Abstract]
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.[PUBMED Abstract]
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.[PUBMED Abstract]
- Johansson B, Moorman AV, Haas OA, et al.: Hematologic malignancies with t(4;11)(q21;q23)--a cytogenetic, morphologic, immunophenotypic and clinical study of 183 cases. European 11q23 Workshop participants. Leukemia 12 (5): 779-87, 1998.[PUBMED Abstract]
- Raimondi SC, Peiper SC, Kitchingman GR, et al.: Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 73 (6): 1627-34, 1989.[PUBMED Abstract]
- Harrison CJ, Moorman AV, Barber KE, et al.: Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol 129 (4): 520-30, 2005.[PUBMED Abstract]
- Pui CH, Pei D, Campana D, et al.: A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 28 (12): 2336-43, 2014.[PUBMED Abstract]
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.[PUBMED Abstract]
- Pui CH, Gaynon PS, Boyett JM, et al.: Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359 (9321): 1909-15, 2002.[PUBMED Abstract]
- Rubnitz JE, Camitta BM, Mahmoud H, et al.: Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol 17 (1): 191-6, 1999.[PUBMED Abstract]
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.[PUBMED Abstract]
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.[PUBMED Abstract]
- Fischer U, Forster M, Rinaldi A, et al.: Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47 (9): 1020-9, 2015.[PUBMED Abstract]
- Pui CH, Sandlund JT, Pei D, et al.: Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290 (15): 2001-7, 2003.[PUBMED Abstract]
- Crist WM, Carroll AJ, Shuster JJ, et al.: Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 76 (1): 117-22, 1990.[PUBMED Abstract]
- Andersen MK, Autio K, Barbany G, et al.: Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155 (2): 235-43, 2011.[PUBMED Abstract]
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.[PUBMED Abstract]
- Jeha S, Pei D, Raimondi SC, et al.: Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 23 (8): 1406-9, 2009.[PUBMED Abstract]
- Minson KA, Prasad P, Vear S, et al.: t(17;19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013: 563291, 2013.[PUBMED Abstract]
- Zaliova M, Potuckova E, Hovorkova L, et al.: ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 104 (7): 1407-1416, 2019.[PUBMED Abstract]
- Harvey RC, Mullighan CG, Wang X, et al.: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 (23): 4874-84, 2010.[PUBMED Abstract]
- Zaliova M, Zimmermannova O, Dörge P, et al.: ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28 (1): 182-5, 2014.[PUBMED Abstract]
- Clappier E, Auclerc MF, Rapion J, et al.: An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 28 (1): 70-7, 2014.[PUBMED Abstract]
- Gu Z, Churchman M, Roberts K, et al.: Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7: 13331, 2016.[PUBMED Abstract]
- Liu YF, Wang BY, Zhang WN, et al.: Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8: 173-83, 2016.[PUBMED Abstract]
- Suzuki K, Okuno Y, Kawashima N, et al.: MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol 34 (28): 3451-9, 2016.[PUBMED Abstract]
- Lilljebjörn H, Ågerstam H, Orsmark-Pietras C, et al.: RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia 28 (4): 977-9, 2014.[PUBMED Abstract]
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.[PUBMED Abstract]
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.[PUBMED Abstract]
- Shago M, Abla O, Hitzler J, et al.: Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63 (11): 1915-21, 2016.[PUBMED Abstract]
- Yao L, Cen J, Pan J, et al.: TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet 211: 1-4, 2017.[PUBMED Abstract]
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.[PUBMED Abstract]
- Hogan TF, Koss W, Murgo AJ, et al.: Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol 5 (3): 382-90, 1987.[PUBMED Abstract]
- Grimaldi JC, Meeker TC: The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 73 (8): 2081-5, 1989.[PUBMED Abstract]
- Meeker TC, Hardy D, Willman C, et al.: Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76 (2): 285-9, 1990.[PUBMED Abstract]
- Sutton R, Lonergan M, Tapp H, et al.: Two cases of hypereosinophilia and high-risk acute lymphoblastic leukemia. Leukemia 22 (7): 1463-5, 2008.[PUBMED Abstract]
- Heerema NA, Carroll AJ, Devidas M, et al.: Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. J Clin Oncol 31 (27): 3397-402, 2013.[PUBMED Abstract]
- Moorman AV, Robinson H, Schwab C, et al.: Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol 31 (27): 3389-96, 2013.[PUBMED Abstract]
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.[PUBMED Abstract]
- Gu Z, Churchman ML, Roberts KG, et al.: PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51 (2): 296-307, 2019.[PUBMED Abstract]
- Nebral K, Denk D, Attarbaschi A, et al.: Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia 23 (1): 134-43, 2009.[PUBMED Abstract]
- Strehl S, König M, Dworzak MN, et al.: PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17 (6): 1121-3, 2003.[PUBMED Abstract]
- Schwab C, Nebral K, Chilton L, et al.: Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv 1 (19): 1473-7, 2017.[PUBMED Abstract]
- Den Boer ML, van Slegtenhorst M, De Menezes RX, et al.: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10 (2): 125-34, 2009.[PUBMED Abstract]
- Mullighan CG, Su X, Zhang J, et al.: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360 (5): 470-80, 2009.[PUBMED Abstract]
- Reshmi SC, Harvey RC, Roberts KG, et al.: Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood 129 (25): 3352-3361, 2017.[PUBMED Abstract]
- Roberts KG, Morin RD, Zhang J, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22 (2): 153-66, 2012.[PUBMED Abstract]
- van der Veer A, Waanders E, Pieters R, et al.: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 122 (15): 2622-9, 2013.[PUBMED Abstract]
- Roberts KG, Reshmi SC, Harvey RC, et al.: Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children's Oncology Group. Blood 132 (8): 815-824, 2018.[PUBMED Abstract]
- Roberts KG, Pei D, Campana D, et al.: Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 32 (27): 3012-20, 2014.[PUBMED Abstract]
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.[PUBMED Abstract]
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.[PUBMED Abstract]
- Iacobucci I, Li Y, Roberts KG, et al.: Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29 (2): 186-200, 2016.[PUBMED Abstract]
- Cario G, Zimmermann M, Romey R, et al.: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 115 (26): 5393-7, 2010.[PUBMED Abstract]
- Ensor HM, Schwab C, Russell LJ, et al.: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 117 (7): 2129-36, 2011.[PUBMED Abstract]
- Schmäh J, Fedders B, Panzer-Grümayer R, et al.: Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr Blood Cancer 64 (10): , 2017.[PUBMED Abstract]
- Vesely C, Frech C, Eckert C, et al.: Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia 31 (7): 1491-1501, 2017.[PUBMED Abstract]
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.[PUBMED Abstract]
- Potter N, Jones L, Blair H, et al.: Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia 33 (4): 893-904, 2019.[PUBMED Abstract]
- Morak M, Attarbaschi A, Fischer S, et al.: Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood 120 (26): 5134-42, 2012.[PUBMED Abstract]
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.[PUBMED Abstract]
- Chen IM, Harvey RC, Mullighan CG, et al.: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512-22, 2012.[PUBMED Abstract]
- Palmi C, Vendramini E, Silvestri D, et al.: Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 26 (10): 2245-53, 2012.[PUBMED Abstract]
- Clappier E, Grardel N, Bakkus M, et al.: IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29 (11): 2154-61, 2015.[PUBMED Abstract]
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.[PUBMED Abstract]
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.[PUBMED Abstract]
- Feng J, Tang Y: Prognostic significance of IKZF1 alteration status in pediatric B-lineage acute lymphoblastic leukemia: a meta-analysis. Leuk Lymphoma 54 (4): 889-91, 2013.[PUBMED Abstract]
- Dörge P, Meissner B, Zimmermann M, et al.: IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98 (3): 428-32, 2013.[PUBMED Abstract]
- Olsson L, Castor A, Behrendtz M, et al.: Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28 (2): 302-10, 2014.[PUBMED Abstract]
- Boer JM, van der Veer A, Rizopoulos D, et al.: Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia 30 (1): 32-8, 2016.[PUBMED Abstract]
- Tran TH, Harris MH, Nguyen JV, et al.: Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2 (5): 529-533, 2018.[PUBMED Abstract]
- Vrooman LM, Blonquist TM, Harris MH, et al.: Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2 (12): 1449-1458, 2018.[PUBMED Abstract]
- van der Veer A, Zaliova M, Mottadelli F, et al.: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123 (11): 1691-8, 2014.[PUBMED Abstract]
- Stanulla M, Dagdan E, Zaliova M, et al.: IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 36 (12): 1240-1249, 2018.[PUBMED Abstract]
- Yeoh AEJ, Lu Y, Chin WHN, et al.: Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J Clin Oncol 36 (26): 2726-2735, 2018.[PUBMED Abstract]
- Liu Y, Easton J, Shao Y, et al.: The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49 (8): 1211-1218, 2017.[PUBMED Abstract]
- Armstrong SA, Look AT: Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 23 (26): 6306-15, 2005.[PUBMED Abstract]
- Karrman K, Forestier E, Heyman M, et al.: Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosomes Cancer 48 (9): 795-805, 2009.[PUBMED Abstract]
- Weng AP, Ferrando AA, Lee W, et al.: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306 (5694): 269-71, 2004.[PUBMED Abstract]
- Gallo Llorente L, Luther H, Schneppenheim R, et al.: Identification of novel NOTCH1 mutations: increasing our knowledge of the NOTCH signaling pathway. Pediatr Blood Cancer 61 (5): 788-96, 2014.[PUBMED Abstract]
- Petit A, Trinquand A, Chevret S, et al.: Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131 (3): 289-300, 2018.[PUBMED Abstract]
- Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al.: Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol 31 (34): 4333-42, 2013.[PUBMED Abstract]
- Paganin M, Grillo MF, Silvestri D, et al.: The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br J Haematol 182 (5): 705-711, 2018.[PUBMED Abstract]
- Bergeron J, Clappier E, Radford I, et al.: Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110 (7): 2324-30, 2007.[PUBMED Abstract]
- van Grotel M, Meijerink JP, Beverloo HB, et al.: The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91 (9): 1212-21, 2006.[PUBMED Abstract]
- Cavé H, Suciu S, Preudhomme C, et al.: Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: results of EORTC studies 58881 and 58951. Blood 103 (2): 442-50, 2004.[PUBMED Abstract]
- Baak U, Gökbuget N, Orawa H, et al.: Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22 (6): 1154-60, 2008.[PUBMED Abstract]
- Ferrando AA, Neuberg DS, Dodge RK, et al.: Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 363 (9408): 535-6, 2004.[PUBMED Abstract]
- Mansour MR, Abraham BJ, Anders L, et al.: Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346 (6215): 1373-7, 2014.[PUBMED Abstract]
- Burmeister T, Gökbuget N, Reinhardt R, et al.: NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood 108 (10): 3556-9, 2006.[PUBMED Abstract]
- Graux C, Stevens-Kroef M, Lafage M, et al.: Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 23 (1): 125-33, 2009.[PUBMED Abstract]
- Hagemeijer A, Graux C: ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49 (4): 299-308, 2010.[PUBMED Abstract]
- Quintás-Cardama A, Tong W, Manshouri T, et al.: Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22 (6): 1117-24, 2008.[PUBMED Abstract]
- Clarke S, O'Reilly J, Romeo G, et al.: NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res 35 (7): e131-3, 2011.[PUBMED Abstract]
- Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al.: Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 23 (3): 627-9, 2009.[PUBMED Abstract]
- Crombet O, Lastrapes K, Zieske A, et al.: Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59 (2): 333-4, 2012.[PUBMED Abstract]
- Seki M, Kimura S, Isobe T, et al.: Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet 49 (8): 1274-1281, 2017.[PUBMED Abstract]
- Zhang J, Ding L, Holmfeldt L, et al.: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380): 157-63, 2012.[PUBMED Abstract]
- Gutierrez A, Dahlberg SE, Neuberg DS, et al.: Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28 (24): 3816-23, 2010.[PUBMED Abstract]
- Yang YL, Hsiao CC, Chen HY, et al.: Absence of biallelic TCRγ deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (6): 846-51, 2012.[PUBMED Abstract]
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[PUBMED Abstract]
- Borowitz MJ, Béné MC, Harris NL, et al.: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. Lyon, France: International Agency for Research on Cancer, 2017, pp 179-87.[PUBMED Abstract]
- Davies SM, Bhatia S, Ross JA, et al.: Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100 (1): 67-71, 2002.[PUBMED Abstract]
- Krajinovic M, Costea I, Chiasson S: Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 359 (9311): 1033-4, 2002.[PUBMED Abstract]
- Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al.: Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J 4 (1): 66-72, 2004.[PUBMED Abstract]
- Schmiegelow K, Forestier E, Kristinsson J, et al.: Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23 (3): 557-64, 2009.[PUBMED Abstract]
- Relling MV, Hancock ML, Boyett JM, et al.: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93 (9): 2817-23, 1999.[PUBMED Abstract]
- Stanulla M, Schaeffeler E, Flohr T, et al.: Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293 (12): 1485-9, 2005.[PUBMED Abstract]
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.[PUBMED Abstract]
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.[PUBMED Abstract]
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.[PUBMED Abstract]
- Tanaka Y, Kato M, Hasegawa D, et al.: Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol 171 (1): 109-15, 2015.[PUBMED Abstract]
- Diouf B, Crews KR, Lew G, et al.: Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313 (8): 815-23, 2015.[PUBMED Abstract]
- Yang JJ, Cheng C, Yang W, et al.: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301 (4): 393-403, 2009.[PUBMED Abstract]
- Gregers J, Christensen IJ, Dalhoff K, et al.: The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 115 (23): 4671-7, 2010.[PUBMED Abstract]
- Radtke S, Zolk O, Renner B, et al.: Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121 (26): 5145-53, 2013.[PUBMED Abstract]
- リスクに基づく治療割り付け
-
リスクに基づく治療概論
急性リンパ芽球性白血病(ALL)の小児は、通常、臨床像および検査所見によって定義されたリスクグループに従って治療される。治癒を得るために必要な治療強度は、ALL患児のサブセット間で実質的に異なる。リスクに基づく治療割り付けがALLの小児において利用されており、それにより、臨床的および生物学的に予後良好な特徴があり、控え目の治療できわめて良好な転帰が得られる可能性が高い患者が強力で毒性の強い治療を避けることができる一方で、長期生存の可能性が乏しい患者に対して積極的で毒性の可能性が高い治療アプローチを提供できる。[ 1 ][ 2 ]
小児腫瘍学グループ(COG)のような特定のALL研究グループでは、一部の治療前因子に基づいて、程度の差はあるものの強力な寛解導入レジメンを用いているが、すべての患者に対して同様な寛解導入レジメンを実施しているグループもある。
COGで導入療法の強度を判断するために用いられた因子には、以下が含まれる:
B-ALLに対するNCIリスクグループ分類では、次のように年齢および白血球(WBC)数に従ってリスクを層別化している:[ 3 ]
NCIリスクグループ、免疫表現型、早期反応判定、ならびに細胞遺伝学的およびゲノム的変化など、さまざまな予後因子に基づいて、すべての研究グループが寛解導入後療法の強度を修正している。[ 4 ]フィラデルフィア染色体の検出(すなわち、フィラデルフィア染色体陽性[Ph+]ALL)は、導入療法の即時の変更につながる。[ 5 ]
リスクに基づく治療割り付けには、転帰を予測する信頼性の高い予後因子を得ることが必要である。ALLの小児では、多数の因子が予後的価値を示しており、その一部を以下に記載する。[ 6 ]予後に影響する因子は、以下の3つのカテゴリーに分けられる:
予後因子のどんな議論でも同様であるが、その重要性の相対的順序および変数相互の関係は治療法に左右されることが多く、どの因子が予後変数として独立して働くかを明らかにするには多変量解析が必要である。予後因子は治療に左右されるため、治療の改善により、これらの推定されている予後因子の重要性が低下したり、なくなったりすることがある。
以下に記述する予後因子および臨床的因子の一部は、治療割り付けを目的としたALL小児の初期層別化にも使用される。(米国で進行中の臨床試験に現在適用されている予後分類の概要については、本要約の臨床評価段階にある予後[リスク]グループのセクションを参照のこと。)
(再燃時に重要な予後因子に関する情報については、本要約の小児ALLの初回再燃後の予後因子のセクションを参照のこと。)
リスクに基づく治療に影響する予後因子
患者特性と臨床的疾患特性
予後に影響する患者特性と臨床的疾患特性には以下のものがある:
診断時年齢
診断時年齢には強い予後的意義があり、年齢層が異なれば基礎にあるALLの生物学的特徴に差があることを反映している。[ 7 ]
-
乳児(1歳未満)。
ALLの乳児は治療失敗のリスクが特に高い。治療失敗は、以下のグループに最も多くみられる:
ALLの乳児の最大80%には11q23の転座がみられ、さまざまな染色体パートナーによりKMT2A遺伝子再構成が生じる。[ 9 ][ 11 ][ 13 ][ 14 ]最も多くみられる再構成はKMT2A-AFF1(t(4;11)(q21;q23))であるが、他に多くの転座パートナーとのKMT2A再構成も観察される。
生後6ヵ月未満の乳児では、KMT2A遺伝子転座の発生率が極端に高い;生後6ヵ月から1歳までは、KMT2A再構成の発生率は低下するが、これを上回る年齢の小児にみられる発生率よりは依然として高い。[ 9 ][ 15 ]ALLの黒人の乳児は、KMT2A再構成を有する可能性が白人の乳児より有意に低い。[ 15 ]
KMT2A再構成を認める白血病の乳児では、典型的にWBC数がきわめて多く、CNS浸潤の発生率が高い。KMT2A再構成を認めるALLの乳児では、イベントフリー生存(EFS)および全生存(OS)が不良で、5年EFS率およびOS率はわずか35~40%である。[ 9 ][ 10 ][ 11 ]KMT2A再構成を認めるALL乳児と小児における体細胞変異の全体像を比較すると、2つ年齢層間で有意な差が明らかになり、KMT2A再構成を認めるALLに特有な年齢関連の生物学的挙動がみられ、乳児で有意に不良な転帰に関連している可能性が示唆された。[ 16 ][ 17 ]
KMT2A再構成を認める乳児の芽球は、しばしばCD10陰性で、FLT3の発現レベルが高い。[ 9 ][ 10 ][ 14 ][ 18 ]対照的に、白血病細胞に生殖細胞KMT2A遺伝子配置がみられる乳児はCD10陽性の前駆B細胞免疫表現型を示す頻度が高い。これらの乳児は、KMT2A再構成を特徴とするALLの乳児より、転帰が有意に良好である。[ 9 ][ 10 ][ 14 ][ 19 ]
(ALLの乳児に関する詳しい情報については、本要約の特定のALLサブグループに対する寛解導入後療法のセクションのALLの乳児のサブセクションを参照のこと。)
-
年少児(1歳から10歳未満)。
年少児(1歳から10歳未満)は、年長児、青年、および乳児よりも無病生存が良好である。[ 3 ][ 7 ][ 20 ][ 21 ][ 22 ]年少児における予後の改善は、白血病芽球において染色体数が51~65の高二倍体および/または予後良好な染色体トリソミー、またはETV6-RUNX1融合(TEL-AML1転座としても知られるt(12;21)(p13;q22))を含む予後良好な細胞遺伝学的特徴がより高頻度に認められることから、少なくともある程度は説明できる。[ 7 ][ 23 ][ 24 ]
-
青年および若年成人(10歳以上)。
一般に10歳以上の患者の転帰は、1歳から10歳未満の患者より不良である。ただし、年齢が高い小児、特に青年の転帰は、時間の経過とともに著しく改善している。[ 25 ][ 26 ][ 27 ]15~19歳の青年では、5年生存率が36%(1975年~1984年)から72%(2003年~2009年)に増加した。[ 28 ][ 29 ][ 30 ]
複数のレトロスペクティブ研究により、16~21歳の青年は成人用プロトコルと比べて小児用プロトコルによる治療を受けた方が転帰良好であることが確立されている。[ 31 ][ 32 ][ 33 ](ALLの青年に関する詳しい情報については、本要約の特定のALLサブグループに対する寛解導入後療法のセクションを参照のこと。)
診断時のWBC数
WBC数と予後との関係は、段階的というより連続的に機能するが、予後が良好か不良かの実務上の分岐ポイントとして、一般にはWBC数の50,000/μLが用いられている。[ 3 ]B-ALLで診断時のWBC数が多い患者は、最初のWBC数が少ない患者と比較して治療失敗のリスクが高い。[ 34 ]
診断時のWBC数の中央値は、T-ALL(50,000/μL超過)の方がB-ALL(10,000/μL未満)よりはるかに多いが、診断時のWBC数によるT-ALLの予後への一貫した影響は認められていない。[ 34 ][ 35 ][ 36 ][ 37 ][ 38 ][ 39 ][ 40 ][ 41 ]
診断時のCNSへの浸潤
診断時のCNS白血病の有無は予後的に重要である。診断時に非外傷性腰椎穿刺であった患者は、WBC数/μLおよび遠沈での芽球の有無に応じて、以下のような3つのカテゴリーのいずれかになる:
診断時にCNS病態(CNS3)を呈するALL小児は、CNS1またはCNS2に分類される患者より、治療(CNS内治療および全身的治療の両方)が失敗するリスクが高い。[ 42 ][ 43 ]CNS1患者と比較して、CNS2患者ではCNS再燃のリスクが高く、かつ/またはEFSが劣っていることを報告している研究がいくつかあるが[ 44 ][ 45 ]、これらを認めていない報告もある。[ 42 ][ 46 ][ 47 ][ 48 ]
診断時の芽球を含む外傷性腰椎穿刺(赤血球数が10個/μL以上)は、CNS再燃のリスクが高いこと、および全体的により不良な転帰と関連していることが一部の研究で認められているが[ 42 ][ 47 ][ 49 ]、これらを認めていない研究もある。[ 45 ][ 46 ][ 50 ]CNS2、CNS3、または外傷性腰椎穿刺に分類される患者は、診断時のWBC数が有意に多い、診断時年齢が高い、T-ALL表現型を示す頻度が高い、およびKMT2A遺伝子再構成が認められるといった不良な予後的特徴を示す頻度がCNS1に分類される患者より高い。[ 42 ][ 46 ][ 47 ]
ほとんどの臨床試験グループでは、主に寛解導入療法中に髄腔内療法を追加するといったより強力な治療法を使用することで、CNS2および外傷性腰椎穿刺患者の治療に対処している。[ 42 ][ 51 ][ 52 ];[ 46 ][証拠レベル:2A];[ 53 ][証拠レベル:1iiA]
外傷性腰椎穿刺(芽球を伴う)が認められた患者をCNS3として治療すべきかどうかを判断するために、COGは、脊髄液および末梢血中のWBCおよび赤血球の数と関連付けたアルゴリズムを用いている。[ 54 ]
診断時の精巣浸潤
診断時に顕性の精巣浸潤が約2%の男児に認められ[ 55 ][ 56 ]、T-ALL患者における頻度がB-ALL患者より高い。[ 56 ]
初期のALL試験では、診断時の精巣浸潤は不良な予後因子であった。しかしながら、より積極的な初期治療を行った場合、診断時の精巣浸潤に予後的意義はないと考えられる。[ 55 ][ 56 ]例えば、European Organization for Research and Treatment of Cancer(EORTC [EORTC-58881])では、診断時の顕性の精巣浸潤に不良な予後的意義はないことを報告している。[ 56 ]
精巣浸潤に対する放射線療法の役割は不明である。St. Jude Children's Research Hospital(SJCRH)の研究は、放射線を用いない積極的な従来の化学療法により良好な転帰が達成可能であることを示唆している。[ 55 ]COGも精巣病変を認める男児に対してこの戦略を採用し、精巣病変は導入療法を終えるまでに完全に消失する。COGは、他に呈する特徴とは無関係に精巣病変を認める患者を高リスクとみなしているが、他の米国および欧州の大規模な臨床試験グループのほとんどが精巣病変を高リスクの特徴としてみなしていない。
ダウン症候群(21トリソミー)
ダウン症候群とALLを合併した小児の転帰は、一般的にダウン症候群でない小児にみられる転帰よりもいくぶん劣ると報告されている。[ 57 ][ 58 ][ 59 ][ 60 ][ 61 ][ 62 ]一部の研究でダウン症候群の小児のEFSおよびOSが短いことは、導入療法失敗および再燃のリスクが高いことに加え、治療関連死亡率が高いことも関係していると考えられている。[ 57 ][ 58 ][ 59 ][ 60 ][ 63 ][ 64 ]ダウン症候群のALL患者における白血病に対する治療転帰が劣る理由の1つは、ETV6-RUNX1または4番および10番染色体のトリソミーを伴う高二倍体(染色体数が51~65)などの予後良好な生物学的特徴の保有率低下である可能性がある。[ 63 ][ 64 ]
性別
数件の研究において、ALLの女児の予後は、ALLの男児の予後よりもわずかに良好である。[ 71 ][ 72 ][ 73 ]女児の予後が優位である理由の1つは、男児の精巣再燃であるが、男児は骨髄およびCNS再燃のリスクも高いと考えられている(その理由は十分に解明されているとはいえない)。[ 71 ][ 72 ][ 73 ]男児の転帰は女児の転帰ときわめて近いと報告している研究も一部にはあるが[ 22 ][ 51 ][ 74 ]、大規模な臨床試験の経験および国内データによると、男児の生存率がやや低いことが依然として示されている。[ 21 ][ 28 ][ 29 ][ 75 ]
人種と民族
過去数十年間、米国における黒人およびヒスパニック系のALL小児の生存率は、白人のALL小児の生存率よりもいくぶん低くなっている。[ 76 ][ 77 ][ 78 ][ 79 ]
以下の人種と民族に関連する因子は生存に影響を及ぼす:
診断時および治療中の体重
ALLの転帰に対する肥満の影響に関する研究の結果は多様である。これらの研究のほとんどで、肥満は、年齢および身長別の第95パーセンタイルを超える体重と定義される。
ALL小児患者762人(年齢2~17歳)を対象にした研究において、Dutch Childhood Oncology Groupは、診断時体重が標準以下の小児患者(集団の8%)の再燃リスクが標準以上の体重の患者(リスクグループおよび年齢で調整後)と比較してほぼ2倍高かったことを明らかにしたが、このことはEFSまたはOSにおける差につながらなかった。治療開始から最初の32週間以内にBMIが低下した患者は他の患者と同程度の再燃率を示したが、主として再燃後の救助率が不良であったために、OSは有意に不良であった。[ 92 ]
白血病細胞の特徴
予後に影響する白血病細胞の特徴には以下のものがある:
免疫表現型
骨髄腫瘍と急性白血病の2016年版世界保健機関(WHO)分類では、ALLをB細胞リンパ芽球性白血病またはT細胞リンパ芽球性白血病のいずれかに分類しており、分子的特徴に基づいてさらに細分している。[ 93 ][ 94 ](詳しい情報については、本要約の診断のセクションを参照のこと。)
B細胞またはT細胞リンパ芽球性白血病は、いずれも骨髄系抗原を共発現している可能性がある。このような症例は、細胞系列があいまいな白血病と区別する必要がある。
-
B-ALL(WHO分類でB細胞リンパ芽球性白血病)。
2008年より前にWHOは、B細胞リンパ芽球性白血病を前駆B細胞リンパ芽球性白血病として分類していたが、この用語は小児ALLの文献で成熟B細胞ALLと区別するために依然として頻繁に使用されている。成熟B細胞ALLは、現在バーキット白血病と呼ばれており、B-ALL(前駆B細胞ALL)に対して実施されているものと異なる治療が必要である。
B-ALLは、CD19、HLA-DR、細胞質CD79a、およびその他のB細胞関連抗原の発現により定義され、小児ALLの80~85%を占める。B-ALL症例の約90%は、CD10表面抗原(以前は共通ALL抗原[cALLa]として知られていた)を発現している。CD10陰性は通常、KMT2A再構成、特にt(4;11)(q21;q23)および不良な転帰と関連している。[ 9 ][ 95 ]KMT2A遺伝子再構成が認められない場合に、CD10陰性が何らかの独立した予後的意義を有するかどうかは不明である。[ 96 ]
免疫表現型による主なB-ALL亜型は、以下の通りである:
-
T-ALL。
T-ALLは、白血病芽球上にT細胞関連抗原(細胞質CD3およびCD7に加え、CD2またはCD5)の発現によって定義される。T-ALLでは、以下を含むさまざまな臨床像との関連が頻繁に認められる:[ 20 ][ 36 ][ 74 ]
歴史的には当てはまらないが、今では適切な強化療法により、T-ALLの小児はB細胞系列ALLの小児に近い転帰が得られる。[ 20 ][ 36 ][ 39 ][ 40 ][ 74 ][ 101 ]
T-ALLの患者で一般に認められた予後因子はほとんどない。T-ALLで認められる白血球数の予後的意義に関して、矛盾したデータが存在する。[ 35 ][ 36 ][ 37 ][ 38 ][ 39 ][ 40 ][ 41 ][ 102 ]診断時に縦隔腫瘤が存在するかどうかには予後的意義がない。縦隔腫瘤がある患者で、腫瘤の退縮速度に予後的意義はない。[ 103 ]
初期の前駆T細胞ALL
初期の前駆T細胞ALLは小児T-ALLの別のサブセットであり、正常な初期の前駆T細胞の発現プロファイルに強く相関している遺伝子発現プロファイルを示すT-ALLを特定することによって初めて定義された。[ 104 ]このような解析によって特定されたT-ALL症例のサブセットは、全症例の13%を占め、特有な免疫表現型(CD1aおよびCD8陰性で、CD5の弱い発現と幹細胞または骨髄系細胞マーカーの共発現を伴う)を示す特徴がみられた。
初期前駆T細胞ALLについて記述した初期の報告で、このサブセットは他のT-ALL患者より予後が不良なことが示唆された。[ 104 ][ 105 ][ 106 ]しかしながら、別の研究では、初期前駆T細胞ALLのサブグループにおける5年EFS率が初期前駆T細胞以外の患者と比較して劣っていた(76% vs 84%)ものの有意ではなかったことが示された。[ 107 ]同様に、COG-AALL0434試験では、初期前駆T細胞の患者と初期前駆T細胞以外の患者で同程度の5年EFS率が観察され、いずれも約87%であった。[ 108 ]初期前駆T細胞ALLの予後的意義をしっかりと確定するには、患者コホートを追加した研究がさらに必要であるが、ほとんどのALL治療グループは、初期前駆T細胞の状態に基づいて患者の治療法を変更していない。
-
骨髄細胞系抗原の発現。
小児ALL患者の最大で3分の1には、骨髄系関連の表面抗原を発現する白血病細胞が認められる。骨髄系関連抗原の発現は、ALLの特定のサブグループ、特にKMT2A再構成、ETV6-RUNX1、およびBCR-ABL1を認めるサブグループと関連しているとみられる。[ 109 ][ 110 ][ 111 ]ZNF384を巻き込んだ遺伝子再構成を認めるB-ALL患者も骨髄系抗原発現を示すことが多い。[ 112 ][ 113 ]骨髄系表面抗原の発現に関する独立した不良な予後的意義は示されていない。[ 109 ][ 110 ]
(細胞系列があいまいな白血病に関する情報については、本要約の細胞系列があいまいな急性白血病の2016年版WHO分類のセクションを参照のこと。)
細胞遺伝学的/ゲノム変化
(B-ALLおよびT-ALLの細胞遺伝学/ゲノミクスおよび薬物代謝経路における遺伝子多型に関する情報については、本要約の小児ALLの細胞遺伝学/ゲノミクスのセクションを参照のこと。)
初回治療に対する反応
治療開始後に白血病細胞が除去される速度および寛解導入療法終了時点における残存病変のレベルは、長期的な転帰と関連している。治療に対する反応は、白血病細胞の薬物感受性ならびに宿主の薬力学および薬理ゲノミクスによる影響を受けるため[ 114 ]、早期反応は強い予後的意義を有する。治療に対する白血病細胞の反応を評価するために、以下のようなさまざまな方法が用いられている:
- MRD測定。
- 治療7日目および14日目の骨髄反応。
- ステロイドによる前治療に対する末梢血反応。
- 多剤併用寛解導入療法に対する末梢血反応。
- 寛解導入療法終了前(8日目、15日目)の末梢血MRD。
- 寛解導入療法終了時の白血病残存(寛解導入失敗)。
MRD測定
血液または骨髄中に残存している白血病細胞を形態的に評価することは困難なことが多く、相対的に感度が低い。伝統的に、骨髄中の芽球5%(光学顕微鏡による判定)がカットオフ値として寛解状態の判定に使用されてきた。これは20個中1個の悪性細胞レベルに相当する。血液または骨髄のいずれかで低レベルの白血病細胞を検出するためには、特異的なIg/T細胞受容体遺伝子再構成および染色体転座により産生される融合転写産物を測定するポリメラーゼ連鎖反応(PCR)分析法、または白血病に特異的な免疫表現型を検出するフローサイトメトリー分析法のような専門技術が必要である。これらの技術を用いると、100,000個の正常細胞の中からわずか1個の白血病細胞の検出が可能であり、細胞10,000個中1個のレベルでMRDをルーチンで検出できる。[ 115 ]Ig/T細胞受容体遺伝子再構成のハイスループット配列決定法(HTS)などの比較的新しい技術により、MRDの検出感度を細胞100万個中の1個(10-6、つまり0.0001%)にまで高めることができる。[ 116 ]
複数の研究で、寛解導入療法終了時のMRDは、B細胞系ALLの小児および青年における転帰の独立した予測因子として重要であることが明らかにされている。[ 117 ][ 118 ][ 119 ]年齢、白血球数、および細胞遺伝学的異常によって定義された患者サブグループでは、MRDの結果により転帰が分かれる。[ 120 ]一般に、寛解導入療法終了時のMRDレベルが高い患者は、それが低いレベルであるかまたは検出できない患者よりも予後が不良である。[ 115 ][ 117 ][ 118 ][ 119 ]しかしながら、特定のMRDレベルと関係する再燃の全体リスクは、遺伝的サブタイプにより異なる。例えば、ある所定のレベルで導入療法終了時のMRDが検出可能とすると、ETV6-RUNX1または高度の高二倍体などの細胞遺伝学的に予後良好な患者は、その後の再燃の絶対リスクが他の患者より低いが、細胞遺伝学的に高リスクの患者は、その後の再燃の絶対リスクが他の患者より高い。[ 121 ]この観察は、MRDを用いてリスク分類計画を策定する際に重要な見識となる可能性がある。
寛解導入療法終了時のMRDは、寛解導入後療法の強度を決定する因子としてほとんどすべてのグループが使用している;MRDレベルがより高い(典型的に0.1%を超え0.01%まで)ことが確認された患者は、より強力な治療法に割り当てられる。[ 115 ][ 118 ][ 122 ];[ 123 ][証拠レベル:2A]
ALLの小児619人を対象とした研究で、フローサイトメトリーによるMRDの予後的有用性がより感度の高いHTS分析法と比較された。0.01%の寛解導入療法終了時のMRDカットポイント値を用いたところ、ハイスループット配列決定法では、約30%多くの症例が陽性(すなわち、>0.01%)であると判定された。HTSにより陽性と判定されたが、フローサイトメトリーでは陰性であった患者の予後は、両方の方法で陽性または陰性のいずれかに分類された患者と比較して中間であった。標準リスクALLの基準を満たす患者でHTSによりMRDを検出できなかった場合は、予後が特に良好であった(5年EFS率、98.1%)。[ 116 ]
治療開始後10~12週間経過時(地固め療法終了時)に得られるMRDレベルも予後において重要であることが示されている;この時点でMRDレベルが高い患者は他の患者と比較してEFSが有意に不良である。[ 119 ][ 120 ]
MRDの測定は、患者にみられるその他の特徴とともに、再燃のリスクがきわめて低い患者のサブセットを同定するためにも使用されてきた。COGの報告によると、前駆B細胞表現型、NCI標準リスクの年齢/白血球数、CNS1状態、および予後良好な細胞遺伝学的異常(予後良好なトリソミーを含む高度の高二倍体またはETV6-RUNX1融合のいずれか)を有しており、8日目(末梢血にて)および寛解導入療法終了時(骨髄にて)のいずれもMRDレベルが0.01%未満であった患者は、予後がきわめて良好であった(5年EFS率、97%±1%)。[ 118 ]寛解導入療法終了時のMRDレベルが低い患者における優れた転帰は、診断から10年以上維持された。[ 125 ]
MRD測定に基づいて治療法を修正することで転帰が改善することが示されている。
治療7日目および14日目の骨髄反応
多剤併用化学療法開始から7日または14日以内に、骨髄中の白血病細胞が5%未満に急速に減少する患者は、骨髄からの白血病細胞の除去が遅い患者よりも予後良好である。[ 128 ]寛解導入療法終了時のMRD評価は、治療への反応の予後指標として、7日目および14日目の形態学的評価からおおむね置き換えられているが、これは、多変量解析でMRDを解析に含めた場合に、後者では予後的意義が失われるためである。[ 118 ][ 129 ]
ステロイドによる前治療に対する末梢血反応
寛解導入前にプレドニゾンによる治療を7日間実施し、メトトレキサート髄注を1回行うことにより末梢血芽球数が1,000/μL未満に減少した(プレドニゾンに対する反応が良好な)患者は、末梢血芽球数が1,000/μLを超えて残存している(プレドニゾンに対する反応が不良な)患者よりも予後良好である。[ 20 ]プレドニゾンに対する反応不良は、10%未満の患者に認められている。[ 20 ][ 130 ]ベルリン-フランクフルト-ミュンスター(BFM)臨床試験グループのプロトコルで行っている治療層別化の一部は、7日間のプレドニゾンによる前治療(多剤併用寛解導入療法開始直前に投与)に対する早期反応に基づいている。
多剤併用寛解導入療法に対する末梢血反応
多剤併用化学療法の開始から7~10日経過しても白血病細胞が持続的に循環血中にみられる患者は、治療開始後1週間以内に末梢血芽球の除去がみられる患者よりも再燃するリスクが高い。[ 131 ]末梢血芽球の除去率は、T細胞ALLとB細胞系列ALLの両方で予後に重要であることが明らかにされている。[ 131 ]
寛解導入療法終了前(8日目、15日目)の末梢血MRD
多剤併用寛解導入化学療法の開始から1週間後に採取した末梢血を用いたMRDは、早期の治療への反応の予後因子としても評価されている。
両研究で、多剤併用寛解導入療法から1週間後に低いMRDレベルを達成し、その後の治療失敗率が低かった患者群が特定された。
寛解導入療法終了時の白血病残存(寛解導入失敗)
ALL小児の大多数が、治療後最初の1ヵ月以内に形態学的完全寛解を達成する。寛解導入相の終了時点で形態学的評価による5%を超えるリンパ芽球の存在がALL小児の1~2%に認められる。[ 21 ][ 22 ][ 133 ][ 134 ][ 135 ]
寛解導入失敗リスクが高いことに関連する特徴には以下がある:[ 135 ][ 136 ][ 137 ]
大規模なレトロスペクティブ研究で、寛解導入に失敗した患者のOS率はわずか32%であった。[ 133 ]しかしながら、臨床的にも生物学的にも顕著な不均一性が認められた。有害な細胞遺伝学的所見(KMT2A再構成またはBCR-ABL1)がみられない年齢が1~5歳のB-ALL患者では、比較的良好な転帰が観察された。このグループでは10年生存率が50%を超えており、このサブセットに対して第一寛解期におけるHSCTを化学療法単独と比較した場合、生存の優位性との関連はみられなかった。転帰が最も不良な(10年生存率が20%未満の)患者には、年齢が14~18歳の患者またはPh染色体またはKMT2A再構成を認める患者が含まれていた。B-ALLの6歳未満の患者およびT-ALLの患者(年齢は問わない)では、CRに達した後に同種HSCTによる治療を受けた場合、その後も化学療法単独による治療を受けた患者より転帰が良好であるとみられた。
フローサイトメトリー vs 形態学
形態学的完全寛解にもかかわらずMRDレベルが5%以上の患者は形態学的に寛解導入に失敗した患者と同程度の転帰を有することが示された研究に基づいて、MRDは現在、寛解導入療法に対する反応の形態学的評価と統合されている。
- UKALL2003(NCT00222612)研究では、患者3,113人中59人(1.9%)が形態学的に寛解導入失敗であった。[ 135 ]
- 2004年から2014年の間にCOG臨床試験に登録された患者9,350人の研究では、形態学(M1 vs M2/M3)およびフローサイトメトリーで評価したMRD状態(5%未満 vs 5%以上)で分類された患者の特徴と転帰が比較された。形態学的寛解(M1状態)は寛解導入療法終了時にB-ALL患者の98.6%およびT-ALL患者の93.8%で達成された。[ 139 ]
表5.寛解導入療法終了時の骨髄について寛解状態で一致している、一致していない、非寛解状態で一致している患者における5年イベントフリー生存率および全生存率a 転帰 M1/MRDが5%未満 値 M1/MRDが5%以上 値 M2/MRDが5%以上 HR = 高リスク;MRD = 微小残存病変;SR = 標準リスク。 a出典:Gupta et al.[ ] bP値は、M1/MRDが5%未満とM1/MRDが5%以上を比較している。 cP値は、M1/MRDが5%以上とM2/MRDが5%以上を比較している。 B-ALL、全体 87.1% ± 0.4%(n = 7,682) <.0001 59.1% ± 6.5%(n = 66) .009 39.1% ± 7.9%(n = 40) B-ALL、SR 90.8% ± 0.4%(n = 5,000) .25 85.9% ± 7.6%(n = 22) .45 76.2% ± 15.2%(n = 9) B-ALL、HR 80% ± 0.9%(n = 2,682) <.0001 44.9% ± 8.3%(n = 44) .05 29% ± 8.2%(n = 31) T-ALL 87.6% ± 1.5%(n = 1,303) .01 80.3% ± 7.3%(n = 97) .13 62.7% ± 13.5%(n = 40) B-ALL、全体 93.8% ± 0.3%(n = 7,682) <.0001 77.2% ± 5.6%(n = 66) .01 59% ± 8.9%(n = 40) B-ALL、SR 96.6% ± 0.3%(n = 5,000) .24 95.5% ± 4.6%(n = 22) .75 88.9% ± 12.1%(n = 9) B-ALL、HR 88.4% ± 0.7%(n = 2,682) <.0001 66.9% ± 8.3%(n = 44) .06 51.4% ± 10.4%(n = 31) T-ALL 91.9% ± 1.3%(n = 1,303) .005 83.4% ± 6.8%(n = 97) .34 76.7% ± 12.3%(n = 40) 予後(リスク)グループ
数十年にわたって小児ALLについて研究している臨床試験グループは、リスク分類スキームを利用して治療失敗の推定リスクに基づいて患者に治療レジメンを割り当てるようになってきた。初期のリスク分類システムでは、年齢および初診時のWBC数のような臨床的因子を利用していた。その後、治療への反応の判定が追加されたが、早期の形態学的骨髄反応(例えば、8日目または15日目)を利用しているグループもあれば、プレドニゾン単剤に対する循環白血病細胞の反応を利用しているグループもある。最新のリスク分類システムでは、年齢および初診時のWBC数のような臨床的因子を依然として利用しており、さらに診断時の白血病細胞における細胞遺伝学的およびゲノム的損傷(例えば、予後良好な転座および予後不良な転座)ならびに寛解導入療法終了時(および一部の症例では、その後の時点)のMRD検出に基づいた治療への反応を採用している。[ 124 ]COGおよびBFMグループのリスク分類システムについて、以下に簡潔に示す。
小児腫瘍学グループ(COG)によるリスクグループ
COGプロトコルでは、最初に以下のような予後因子のサブセットに基づいてALL患児が各治療群(治療失敗リスクの程度は異なる)に層別化される:
良好リスク基準(年齢が1歳から10歳未満、WBC数が50,000/μL未満、および前駆B細胞免疫表現型)に該当する小児では、EFS率が85%を超える;高リスク基準に該当する小児では、EFS率が約75%である。[ 4 ][ 51 ][ 130 ][ 140 ][ 141 ]発症年齢、WBC数、免疫表現型、髄外性病変の存在、およびステロイド前治療と合わせて、細胞遺伝学的異常および治療に対する早期反応判定(例えば、8日目の末梢血および寛解導入療法終了時点の骨髄サンプルにおけるMRDレベル)などの追加因子を考慮することで、寛解導入後療法を行う患者集団を識別することができ、EFS率は40%未満から95%を超える範囲と推定される。[ 4 ][ 118 ]
ベルリン-フランクフルト-ミュンスター(BFM)によるリスクグループ
2000年以降、BFMプロトコルでのリスク層別化分類は、ほぼ治療に対する反応の基準だけに基づいている。プレドニゾンを用いた前治療への反応に加えて、治療に対する反応は2つの時点、つまり寛解導入療法終了時(5週間経過時)および地固め療法終了時(12週間経過時)にMRDを測定して評価される。
BFMリスクグループには以下のものがある:[ 120 ]
表現型、白血病細胞の推定腫瘍量(BFM危険因子としても知られる)、および診断時のCNS病変の状態は、現在のリスク分類スキームの要素として考えられていない。t(9;22)(q34;q11.2)またはt(4;11)(q21;q23)のいずれかを認める患者は、早期反応の基準に関係なく高リスクと考えられる。
臨床評価段階にある予後(リスク)グループ
- COGのAALL1731(NCT03914625)による標準リスクおよびAALL1732(NCT03959085)による高リスクの臨床試験:COGでは、以下の基準に基づいて、B-ALL患者を6つのリスクグループ(予後良好な標準リスク、平均的な標準リスク、高度の標準リスク、予後良好な高リスク、高リスク、超高リスク)に分類している:
リスク層別化の一部として、骨髄における初期反応の形態学的評価が寛解導入療法の8日目および15日目に実施されることはない。T細胞表現型を示す患者は、別個の研究で治療を受けており、このような方法ではリスク分類されていない。
B-ALL患者で、予後良好、予後不良、および中間予後の細胞遺伝学的所見の定義を以下に示す:
NCI標準リスクの患者は、きわめて予後良好なグループ(予後良好な標準リスク;5年DFS率、95%超過)、予後良好な転帰を有するグループ(平均的な標準リスク;5年DFS率、90%-95%)、および5年DFS率が90%未満のグループ(高度の標準リスク)に細分される。高度の標準リスクに分類される患者では、強化地固め療法、中間維持療法、および再寛解導入療法による高リスクB-ALLレジメンのような基本骨格の化学療法を施行する。これらの3つのグループに対する基準は、以下の表6、表7、および表8に示している。
表6.予後良好な標準リスク(SR)B-ALL(非ダウン症候群およびダウン症候群) NCIリスクグループ CNS病期 ステロイド前治療 予後良好な遺伝学的所見( 8日目のPBのMRD 29日目のBMのMRD BM = 骨髄;CNS = 中枢神経系;DT = ダブルトリソミー;MRD = 微小残存病変;NCI = 米国国立がん研究所;PB = 末梢血。 a診断前1ヵ月以内。 SR 1, 2 なし あり <1% <0.01% 表7.平均的な標準リスク(SR)B-ALL(非ダウン症候群およびダウン症候群) NCIリスクグループ CNS病期 DT 中間予後の細胞遺伝学的所見 8日目のPBのMRD 29日目のBMのMRD BM = 骨髄;CNS = 中枢神経系;DT = ダブルトリソミー;MRD = 微小残存病変;NCI = 米国国立がん研究所;PB = 末梢血。 SR 1, 2 いずれかあり なし ≥1% <0.01% SR 1, 2 なし あり なし 任意 ≥0.01%~<0.1% SR 1 なし なし あり 任意 <0.01% 表8.高度の標準リスク(SR)B-ALL NCIリスクグループ CNS病期 DT 中間予後の細胞遺伝学的所見 予後不良な細胞遺伝学的所見 8日目のPBのMRD 29日目のBMのMRD BM = 骨髄;CNS = 中枢神経系;DT = ダブルトリソミー;MRD = 微小残存病変;NCI = 米国国立がん研究所;PB = 末梢血。 SR 1, 2 あり なし なし なし 任意 ≥0.01% SR 1, 2 なし あり なし なし 任意 ≥0.1% SR 1 なし なし あり なし 任意 ≥0.01% SR 2 なし なし あり なし 任意 任意 SR 1, 2 なし なし なし あり 任意 任意 予後良好な高リスクB-ALLは、表9に示す特徴により定義される。これらの患者では、過去の高リスク患者のCOG臨床試験でEFS率が90%を超えている。
表9.予後良好な高リスク(HR)B-ALL患者の特徴 NCIリスクグループ 年齢(歳) CNS状態 精巣白血病 ステロイド前治療 予後良好な遺伝学的所見( EOI時の骨髄のMRD HR <10 1 なし 24時間以下a あり <0.01% 高リスクB-ALLは、表10に示す特徴により定義される。NCI標準リスクの患者は、ステロイド前治療ならびにCNSおよび/または精巣病変に基づき高リスク状態に移行する。
表10.高リスク(HR)B-ALL患者の特徴 NCIリスクグループ 年齢(歳) CNSおよび/または精巣白血病 ステロイド前治療 細胞遺伝学的所見 EOI時の骨髄のMRD EOC時の骨髄のMRD CNS = 中枢神経系;EOC = 地固め療法終了;EOI = 寛解導入療法終了;MRD = 微小残存病変;N/A = 適用されない;NCI = 米国国立がん研究所;SR = 標準リスク。 aCNS3。 bフィラデルフィア染色体陽性(Ph+)ALLは除外する。 cEOI時の骨髄のMRDが0.01%以上の対象者のみ、EOC時に骨髄のMRD評価を実施する。 d診断の2週間以内。 eCNS2またはCNS3。 SR <10 ありa 任意 任意b 任意 <1%c SR <10 なし 24時間超過d 任意b 任意 <1%c HR ≥10 任意 任意 任意b <0.01% N/A HR <10 ありe 任意 任意b <0.01% N/A HR <10 なし 24時間超過d 任意b <0.01% N/A HR <10 なし 24時間以下d 中間予後/予後不良b <0.01% N/A HR 任意 任意 任意 任意b ≥0.01% <0.01% 地固め療法終了時の骨髄MRDが0.01%以上のNCI高リスク患者は、超高リスクに分類され、第一寛解時にキメラ抗原受容体(CAR)T細胞の臨床試験(NCT03792633)に適格である。
ダウン症候群のB-ALL患者は、他の小児に類似したリスクグループに分類されるが、高リスクに分類されたダウン症候群患者では、毒性を弱くするように修正された治療レジメンを使用する。
- NCI-2014-00712;AALL1231(NCT02112916)(新たにT-ALLまたはII~IV期T細胞リンパ芽球性リンパ腫と診断された若年患者の治療においてボルテゾミブを併用するまたは併用しない併用化学療法):T-ALL患者のリスクカテゴリーへの割り付けに、COGは以下の基準を用いている:
- SJCRH Total 17研究(NCT03117751)(新たにALLおよびリンパ腫と診断された患者に対するTotal Therapy XVII):本研究の包括的目標は、ALLおよび急性リンパ芽球性リンパ腫の小児の治癒率および生活の質を改善するために、遺伝性および白血病特異的なゲノムの特徴に基づいた新規の正確な医薬(precision medicine)戦略および標的療法アプローチを用いることである。
-
DFCI ALL 16-001(NCT03020030)(ALLの小児および成人に対してより優れた治療法選択肢を特定するリスク分類スキーム):患者は初診時の特徴および白血病の生物学的所見に基づいて治療10日目までに以下の初期リスク群に割り当てられる:
BCR-ABL1を認める患者は、15日目でプロトコルに従う治療から除外された。以下の最終リスク群は、初期リスク群および寛解導入療法終了時点(32日目;第1時点)および治療10週目(第2時点)でのMRD(次世代の塩基配列決定法により評価)に基づいている:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Hunger SP, Loh ML, Whitlock JA, et al.: Children's Oncology Group's 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer 60 (6): 957-63, 2013.[PUBMED Abstract]
- Hunger SP, Mullighan CG: Acute Lymphoblastic Leukemia in Children. N Engl J Med 373 (16): 1541-52, 2015.[PUBMED Abstract]
- Smith M, Arthur D, Camitta B, et al.: Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol 14 (1): 18-24, 1996.[PUBMED Abstract]
- Schultz KR, Pullen DJ, Sather HN, et al.: Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109 (3): 926-35, 2007.[PUBMED Abstract]
- Jeha S, Coustan-Smith E, Pei D, et al.: Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 120 (10): 1514-9, 2014.[PUBMED Abstract]
- Vrooman LM, Silverman LB: Childhood acute lymphoblastic leukemia: update on prognostic factors. Curr Opin Pediatr 21 (1): 1-8, 2009.[PUBMED Abstract]
- Möricke A, Zimmermann M, Reiter A, et al.: Prognostic impact of age in children and adolescents with acute lymphoblastic leukemia: data from the trials ALL-BFM 86, 90, and 95. Klin Padiatr 217 (6): 310-20, 2005 Nov-Dec.[PUBMED Abstract]
- Reaman GH, Sposto R, Sensel MG, et al.: Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children's Cancer Group. J Clin Oncol 17 (2): 445-55, 1999.[PUBMED Abstract]
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.[PUBMED Abstract]
- Hilden JM, Dinndorf PA, Meerbaum SO, et al.: Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108 (2): 441-51, 2006.[PUBMED Abstract]
- Dreyer ZE, Hilden JM, Jones TL, et al.: Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer 62 (3): 419-26, 2015.[PUBMED Abstract]
- Chessells JM, Harrison CJ, Watson SL, et al.: Treatment of infants with lymphoblastic leukaemia: results of the UK Infant Protocols 1987-1999. Br J Haematol 117 (2): 306-14, 2002.[PUBMED Abstract]
- Isoyama K, Eguchi M, Hibi S, et al.: Risk-directed treatment of infant acute lymphoblastic leukaemia based on early assessment of MLL gene status: results of the Japan Infant Leukaemia Study (MLL96). Br J Haematol 118 (4): 999-1010, 2002.[PUBMED Abstract]
- Nagayama J, Tomizawa D, Koh K, et al.: Infants with acute lymphoblastic leukemia and a germline MLL gene are highly curable with use of chemotherapy alone: results from the Japan Infant Leukemia Study Group. Blood 107 (12): 4663-5, 2006.[PUBMED Abstract]
- Sam TN, Kersey JH, Linabery AM, et al.: MLL gene rearrangements in infant leukemia vary with age at diagnosis and selected demographic factors: a Children's Oncology Group (COG) study. Pediatr Blood Cancer 58 (6): 836-9, 2012.[PUBMED Abstract]
- Kang H, Wilson CS, Harvey RC, et al.: Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (8): 1872-81, 2012.[PUBMED Abstract]
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.[PUBMED Abstract]
- Stam RW, Schneider P, de Lorenzo P, et al.: Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood 110 (7): 2774-5, 2007.[PUBMED Abstract]
- De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.[PUBMED Abstract]
- Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.[PUBMED Abstract]
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.[PUBMED Abstract]
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.[PUBMED Abstract]
- Forestier E, Schmiegelow K; on behalf of the Nordic Society of Paediatric Haematology and Oncology NOPHO: The incidence peaks of the childhood acute leukemias reflect specific cytogenetic aberrations. J Pediatr Hematol Oncol 28 (8): 486-95, 2006.[PUBMED Abstract]
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.[PUBMED Abstract]
- Nachman JB, La MK, Hunger SP, et al.: Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children's oncology group. J Clin Oncol 27 (31): 5189-94, 2009.[PUBMED Abstract]
- Pulte D, Gondos A, Brenner H: Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st century. Blood 113 (7): 1408-11, 2009.[PUBMED Abstract]
- Pui CH, Pei D, Campana D, et al.: Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29 (4): 386-91, 2011.[PUBMED Abstract]
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 28. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 29. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- de Bont JM, Holt B, Dekker AW, et al.: Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia 18 (12): 2032-5, 2004.[PUBMED Abstract]
- Boissel N, Auclerc MF, Lhéritier V, et al.: Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 21 (5): 774-80, 2003.[PUBMED Abstract]
- Stock W, La M, Sanford B, et al.: What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112 (5): 1646-54, 2008.[PUBMED Abstract]
- Hastings C, Gaynon PS, Nachman JB, et al.: Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (≥200 × 10(9) /l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol 168 (4): 533-46, 2015.[PUBMED Abstract]
- Pullen J, Shuster JJ, Link M, et al.: Significance of commonly used prognostic factors differs for children with T cell acute lymphocytic leukemia (ALL), as compared to those with B-precursor ALL. A Pediatric Oncology Group (POG) study. Leukemia 13 (11): 1696-707, 1999.[PUBMED Abstract]
- Goldberg JM, Silverman LB, Levy DE, et al.: Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol 21 (19): 3616-22, 2003.[PUBMED Abstract]
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.[PUBMED Abstract]
- Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.[PUBMED Abstract]
- Gaynon PS, Angiolillo AL, Carroll WL, et al.: Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children's Oncology Group Report. Leukemia 24 (2): 285-97, 2010.[PUBMED Abstract]
- Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.[PUBMED Abstract]
- Vaitkevičienė G, Forestier E, Hellebostad M, et al.: High white blood cell count at diagnosis of childhood acute lymphoblastic leukaemia: biological background and prognostic impact. Results from the NOPHO ALL-92 and ALL-2000 studies. Eur J Haematol 86 (1): 38-46, 2011.[PUBMED Abstract]
- Bürger B, Zimmermann M, Mann G, et al.: Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 21 (2): 184-8, 2003.[PUBMED Abstract]
- Vora A, Andreano A, Pui CH, et al.: Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 34 (9): 919-26, 2016.[PUBMED Abstract]
- Mahmoud HH, Rivera GK, Hancock ML, et al.: Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 329 (5): 314-9, 1993.[PUBMED Abstract]
- Winick N, Devidas M, Chen S, et al.: Impact of Initial CSF Findings on Outcome Among Patients With National Cancer Institute Standard- and High-Risk B-Cell Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group. J Clin Oncol 35 (22): 2527-2534, 2017.[PUBMED Abstract]
- Sirvent N, Suciu S, Rialland X, et al.: Prognostic significance of the initial cerebro-spinal fluid (CSF) involvement of children with acute lymphoblastic leukaemia (ALL) treated without cranial irradiation: results of European Organization for Research and Treatment of Cancer (EORTC) Children Leukemia Group study 58881. Eur J Cancer 47 (2): 239-47, 2011.[PUBMED Abstract]
- te Loo DM, Kamps WA, van der Does-van den Berg A, et al.: Prognostic significance of blasts in the cerebrospinal fluid without pleiocytosis or a traumatic lumbar puncture in children with acute lymphoblastic leukemia: experience of the Dutch Childhood Oncology Group. J Clin Oncol 24 (15): 2332-6, 2006.[PUBMED Abstract]
- Gilchrist GS, Tubergen DG, Sather HN, et al.: Low numbers of CSF blasts at diagnosis do not predict for the development of CNS leukemia in children with intermediate-risk acute lymphoblastic leukemia: a Childrens Cancer Group report. J Clin Oncol 12 (12): 2594-600, 1994.[PUBMED Abstract]
- Gajjar A, Harrison PL, Sandlund JT, et al.: Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 96 (10): 3381-4, 2000.[PUBMED Abstract]
- Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.[PUBMED Abstract]
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.[PUBMED Abstract]
- Levinsen M, Taskinen M, Abrahamsson J, et al.: Clinical features and early treatment response of central nervous system involvement in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 61 (8): 1416-21, 2014.[PUBMED Abstract]
- Jeha S, Pei D, Choi J, et al.: Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol 37 (35): 3377-3391, 2019.[PUBMED Abstract]
- Cherlow JM, Sather H, Steinherz P, et al.: Craniospinal irradiation for acute lymphoblastic leukemia with central nervous system disease at diagnosis: a report from the Children's Cancer Group. Int J Radiat Oncol Biol Phys 36 (1): 19-27, 1996.[PUBMED Abstract]
- Hijiya N, Liu W, Sandlund JT, et al.: Overt testicular disease at diagnosis of childhood acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia 19 (8): 1399-403, 2005.[PUBMED Abstract]
- Sirvent N, Suciu S, Bertrand Y, et al.: Overt testicular disease (OTD) at diagnosis is not associated with a poor prognosis in childhood acute lymphoblastic leukemia: results of the EORTC CLG Study 58881. Pediatr Blood Cancer 49 (3): 344-8, 2007.[PUBMED Abstract]
- Bassal M, La MK, Whitlock JA, et al.: Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer 44 (1): 21-8, 2005.[PUBMED Abstract]
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.[PUBMED Abstract]
- Whitlock JA, Sather HN, Gaynon P, et al.: Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 106 (13): 4043-9, 2005.[PUBMED Abstract]
- Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.[PUBMED Abstract]
- Lundin C, Forestier E, Klarskov Andersen M, et al.: Clinical and genetic features of pediatric acute lymphoblastic leukemia in Down syndrome in the Nordic countries. J Hematol Oncol 7 (1): 32, 2014.[PUBMED Abstract]
- Athale UH, Puligandla M, Stevenson KE, et al.: Outcome of children and adolescents with Down syndrome treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium protocols 00-001 and 05-001. Pediatr Blood Cancer 65 (10): e27256, 2018.[PUBMED Abstract]
- Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.[PUBMED Abstract]
- Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.[PUBMED Abstract]
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.[PUBMED Abstract]
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.[PUBMED Abstract]
- Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.[PUBMED Abstract]
- Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.[PUBMED Abstract]
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.[PUBMED Abstract]
- Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.[PUBMED Abstract]
- Pui CH, Boyett JM, Relling MV, et al.: Sex differences in prognosis for children with acute lymphoblastic leukemia. J Clin Oncol 17 (3): 818-24, 1999.[PUBMED Abstract]
- Shuster JJ, Wacker P, Pullen J, et al.: Prognostic significance of sex in childhood B-precursor acute lymphoblastic leukemia: a Pediatric Oncology Group Study. J Clin Oncol 16 (8): 2854-63, 1998.[PUBMED Abstract]
- Chessells JM, Richards SM, Bailey CC, et al.: Gender and treatment outcome in childhood lymphoblastic leukaemia: report from the MRC UKALL trials. Br J Haematol 89 (2): 364-72, 1995.[PUBMED Abstract]
- Silverman LB, Gelber RD, Dalton VK, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97 (5): 1211-8, 2001.[PUBMED Abstract]
- Hunger SP, Lu X, Devidas M, et al.: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 30 (14): 1663-9, 2012.[PUBMED Abstract]
- Bhatia S: Influence of race and socioeconomic status on outcome of children treated for childhood acute lymphoblastic leukemia. Curr Opin Pediatr 16 (1): 9-14, 2004.[PUBMED Abstract]
- Kadan-Lottick NS, Ness KK, Bhatia S, et al.: Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA 290 (15): 2008-14, 2003.[PUBMED Abstract]
- Tai EW, Ward KC, Bonaventure A, et al.: Survival among children diagnosed with acute lymphoblastic leukemia in the United States, by race and age, 2001 to 2009: Findings from the CONCORD-2 study. Cancer 123 (Suppl 24): 5178-5189, 2017.[PUBMED Abstract]
- Kahn JM, Cole PD, Blonquist TM, et al.: An investigation of toxicities and survival in Hispanic children and adolescents with ALL: Results from the Dana-Farber Cancer Institute ALL Consortium protocol 05-001. Pediatr Blood Cancer 65 (3): , 2018.[PUBMED Abstract]
- Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.[PUBMED Abstract]
- Bhatia S, Landier W, Hageman L, et al.: 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 124 (15): 2345-53, 2014.[PUBMED Abstract]
- Yang JJ, Cheng C, Devidas M, et al.: Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 43 (3): 237-41, 2011.[PUBMED Abstract]
- Xu H, Cheng C, Devidas M, et al.: ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol 30 (7): 751-7, 2012.[PUBMED Abstract]
- Aldhafiri FK, McColl JH, Reilly JJ: Prognostic significance of being overweight and obese at diagnosis in children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 36 (3): 234-6, 2014.[PUBMED Abstract]
- Baillargeon J, Langevin AM, Lewis M, et al.: Obesity and survival in a cohort of predominantly Hispanic children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 28 (9): 575-8, 2006.[PUBMED Abstract]
- Hijiya N, Panetta JC, Zhou Y, et al.: Body mass index does not influence pharmacokinetics or outcome of treatment in children with acute lymphoblastic leukemia. Blood 108 (13): 3997-4002, 2006.[PUBMED Abstract]
- Butturini AM, Dorey FJ, Lange BJ, et al.: Obesity and outcome in pediatric acute lymphoblastic leukemia. J Clin Oncol 25 (15): 2063-9, 2007.[PUBMED Abstract]
- Gelelete CB, Pereira SH, Azevedo AM, et al.: Overweight as a prognostic factor in children with acute lymphoblastic leukemia. Obesity (Silver Spring) 19 (9): 1908-11, 2011.[PUBMED Abstract]
- Orgel E, Sposto R, Malvar J, et al.: Impact on survival and toxicity by duration of weight extremes during treatment for pediatric acute lymphoblastic leukemia: A report from the Children's Oncology Group. J Clin Oncol 32 (13): 1331-7, 2014.[PUBMED Abstract]
- Orgel E, Tucci J, Alhushki W, et al.: Obesity is associated with residual leukemia following induction therapy for childhood B-precursor acute lymphoblastic leukemia. Blood 124 (26): 3932-8, 2014.[PUBMED Abstract]
- Eissa HM, Zhou Y, Panetta JC, et al.: The effect of body mass index at diagnosis on clinical outcome in children with newly diagnosed acute lymphoblastic leukemia. Blood Cancer J 7 (2): e531, 2017.[PUBMED Abstract]
- den Hoed MA, Pluijm SM, de Groot-Kruseman HA, et al.: The negative impact of being underweight and weight loss on survival of children with acute lymphoblastic leukemia. Haematologica 100 (1): 62-9, 2015.[PUBMED Abstract]
- Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. Lyon, France: International Agency for Research on Cancer, 2017.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.[PUBMED Abstract]
- Möricke A, Ratei R, Ludwig WD, et al.: Prognostic factors in CD10 negative precursor b-cell acute lymphoblastic leukemia in children: data from three consecutive trials ALL-BFM 86, 90, and 95. [Abstract] Blood 104 (11): A-1957, 540a, 2004.[PUBMED Abstract]
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.[PUBMED Abstract]
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.[PUBMED Abstract]
- Koehler M, Behm FG, Shuster J, et al.: Transitional pre-B-cell acute lymphoblastic leukemia of childhood is associated with favorable prognostic clinical features and an excellent outcome: a Pediatric Oncology Group study. Leukemia 7 (12): 2064-8, 1993.[PUBMED Abstract]
- Wagener R, López C, Kleinheinz K, et al.: IG-MYC+ neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood 132 (21): 2280-2285, 2018.[PUBMED Abstract]
- Winter SS, Dunsmore KP, Devidas M, et al.: Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children's Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol 36 (29): 2926-2934, 2018.[PUBMED Abstract]
- Slack JL, Arthur DC, Lawrence D, et al.: Secondary cytogenetic changes in acute promyelocytic leukemia--prognostic importance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol 15 (5): 1786-95, 1997.[PUBMED Abstract]
- Attarbaschi A, Mann G, Dworzak M, et al.: Mediastinal mass in childhood T-cell acute lymphoblastic leukemia: significance and therapy response. Med Pediatr Oncol 39 (6): 558-65, 2002.[PUBMED Abstract]
- Coustan-Smith E, Mullighan CG, Onciu M, et al.: Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 10 (2): 147-56, 2009.[PUBMED Abstract]
- Ma M, Wang X, Tang J, et al.: Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med 6 (4): 416-20, 2012.[PUBMED Abstract]
- Inukai T, Kiyokawa N, Campana D, et al.: Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children's Cancer Study Group Study L99-15. Br J Haematol 156 (3): 358-65, 2012.[PUBMED Abstract]
- Patrick K, Wade R, Goulden N, et al.: Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 166 (3): 421-4, 2014.[PUBMED Abstract]
- Wood BL, Winter SS, Dunsmore KP, et al.: T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children's Oncology Group (COG) study AALL0434. [Abstract] Blood 124 (21): A-1, 2014. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Pui CH, Rubnitz JE, Hancock ML, et al.: Reappraisal of the clinical and biologic significance of myeloid-associated antigen expression in childhood acute lymphoblastic leukemia. J Clin Oncol 16 (12): 3768-73, 1998.[PUBMED Abstract]
- Uckun FM, Sather HN, Gaynon PS, et al.: Clinical features and treatment outcome of children with myeloid antigen positive acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 90 (1): 28-35, 1997.[PUBMED Abstract]
- Corrente F, Bellesi S, Metafuni E, et al.: Role of flow-cytometric immunophenotyping in prediction of BCR/ABL1 gene rearrangement in adult B-cell acute lymphoblastic leukemia. Cytometry B Clin Cytom 94 (3): 468-476, 2018.[PUBMED Abstract]
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.[PUBMED Abstract]
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.[PUBMED Abstract]
- Relling MV, Dervieux T: Pharmacogenetics and cancer therapy. Nat Rev Cancer 1 (2): 99-108, 2001.[PUBMED Abstract]
- van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.[PUBMED Abstract]
- Wood B, Wu D, Crossley B, et al.: Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 131 (12): 1350-1359, 2018.[PUBMED Abstract]
- Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.[PUBMED Abstract]
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.[PUBMED Abstract]
- Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.[PUBMED Abstract]
- Conter V, Bartram CR, Valsecchi MG, et al.: Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 115 (16): 3206-14, 2010.[PUBMED Abstract]
- O'Connor D, Enshaei A, Bartram J, et al.: Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol 36 (1): 34-43, 2018.[PUBMED Abstract]
- Basso G, Veltroni M, Valsecchi MG, et al.: Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol 27 (31): 5168-74, 2009.[PUBMED Abstract]
- Pui CH, Pei D, Coustan-Smith E, et al.: Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: a prospective study. Lancet Oncol 16 (4): 465-74, 2015.[PUBMED Abstract]
- Schrappe M, Valsecchi MG, Bartram CR, et al.: Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118 (8): 2077-84, 2011.[PUBMED Abstract]
- Bartram J, Wade R, Vora A, et al.: Excellent outcome of minimal residual disease-defined low-risk patients is sustained with more than 10 years follow-up: results of UK paediatric acute lymphoblastic leukaemia trials 1997-2003. Arch Dis Child 101 (5): 449-54, 2016.[PUBMED Abstract]
- Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.[PUBMED Abstract]
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.[PUBMED Abstract]
- Gaynon PS, Desai AA, Bostrom BC, et al.: Early response to therapy and outcome in childhood acute lymphoblastic leukemia: a review. Cancer 80 (9): 1717-26, 1997.[PUBMED Abstract]
- Borowitz MJ, Wood BL, Devidas M, et al.: Assessment of end induction minimal residual disease (MRD) in childhood B precursor acute lymphoblastic leukemia (ALL) to eliminate the need for day 14 marrow examination: A Children's Oncology Group study. [Abstract] J Clin Oncol 31 (Suppl 15): A-10001, 2013. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.[PUBMED Abstract]
- Griffin TC, Shuster JJ, Buchanan GR, et al.: Slow disappearance of peripheral blood blasts is an adverse prognostic factor in childhood T cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. Leukemia 14 (5): 792-5, 2000.[PUBMED Abstract]
- Volejnikova J, Mejstrikova E, Valova T, et al.: Minimal residual disease in peripheral blood at day 15 identifies a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with superior prognosis. Haematologica 96 (12): 1815-21, 2011.[PUBMED Abstract]
- Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.[PUBMED Abstract]
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.[PUBMED Abstract]
- O'Connor D, Moorman AV, Wade R, et al.: Use of Minimal Residual Disease Assessment to Redefine Induction Failure in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol 35 (6): 660-667, 2017.[PUBMED Abstract]
- Silverman LB, Gelber RD, Young ML, et al.: Induction failure in acute lymphoblastic leukemia of childhood. Cancer 85 (6): 1395-404, 1999.[PUBMED Abstract]
- Oudot C, Auclerc MF, Levy V, et al.: Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol 26 (9): 1496-503, 2008.[PUBMED Abstract]
- Schwab C, Ryan SL, Chilton L, et al.: EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood 127 (18): 2214-8, 2016.[PUBMED Abstract]
- Gupta S, Devidas M, Loh ML, et al.: Flow-cytometric vs. -morphologic assessment of remission in childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group (COG). Leukemia 32 (6): 1370-1379, 2018.[PUBMED Abstract]
- Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.[PUBMED Abstract]
- Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.[PUBMED Abstract]
- Kosaka Y, Koh K, Kinukawa N, et al.: Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 104 (12): 3527-34, 2004.[PUBMED Abstract]
- Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.[PUBMED Abstract]
- Schrauder A, Reiter A, Gadner H, et al.: Superiority of allogeneic hematopoietic stem-cell transplantation compared with chemotherapy alone in high-risk childhood T-cell acute lymphoblastic leukemia: results from ALL-BFM 90 and 95. J Clin Oncol 24 (36): 5742-9, 2006.[PUBMED Abstract]
- Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007.[PUBMED Abstract]
-
乳児(1歳未満)。
- 小児ALLに対する治療法選択肢の概要
-
小児がん治療に関する特別な考慮事項
急性リンパ芽球性白血病(ALL)の小児の治療では、リスク分類および治療が複雑化し、集中的な支持療法(例えば、輸血;感染性合併症の管理;ならびに情緒的、財政的、および発育支援)が必要になるため、小児の支持療法に必要な設備をすべて備えたがんセンターまたは病院の集学的チームが評価および治療の調整を行うのが最も良い。[ 1 ]集学的チームのアプローチは、至適生存期間および至適QOLを得られるような治療、支持療法、およびリハビリテーションを小児が必ず受けられるようにするために、以下に示す医療専門家などのスキルを集結したものである:
がん施設とこれらの施設が小児がん患者の治療において担う役割に関するガイドラインが米国小児科学会により概説されている。[ 1 ]小児ALLの治療では、一般に2~3年にわたって化学療法が実施される。白血病および化学療法による治療の結果として骨髄抑制および全身の免疫抑制が予想されるため、全治療期間を通じて、血液学的サポートおよび感染症や他の合併症の治療を実施するための十分な設備を直ちに利用できなければならない。患者の約1~3%が寛解導入相で死亡し、さらに1~3%が完全寛解に達した後に治療関連合併症により死亡する。[ 2 ][ 3 ][ 4 ][ 5 ]患者のケアを指示する医療センターおよび専門医は地域の委託医師との連絡を保つことが重要である。コミュニケーションの強い絆によって、患児が在宅中に必要なすべての緊急的または暫定的ケアが最適に保たれる。
ALL患児では臨床試験が一般的に利用可能であり、治療失敗のリスクが標準(低い)の小児、および治療失敗のリスクがより高い小児に特化してデザインされたプロトコルがある。一般に、ALL患児を対象とした臨床試験は、特定のリスク群に対する標準として現在受け入れられている治療法に対して、生存転帰を改善できる、および/または標準の治療レジメンに関連する毒性を低減できる可能性のあるより良い治療アプローチを比較するためにデザインされる。ALL患児の生存率の増大をもたらした治療法の革新の多くは、臨床試験により達成されており、ALLの小児および青年に臨床試験への参加を提案することが適切である。
リスクに基づく治療法の選定は、ALL小児に用いられる重要な治療戦略である。このアプローチにより、歴史的にみて転帰が非常に良好な小児は、控え目な治療を受け、より毒性の強い治療を避けることができると同時に、歴史的にみて長期生存の可能性が低い小児は、治癒の可能性が広がる可能性のあるより強力な治療を受けられるようになる。(予後的価値が実証されている多くの臨床的特徴および臨床検査所見に関する詳しい情報については、本要約のリスクに基づく治療割り付けのセクションを参照のこと。)
聖域部位
歴史的に、特定の髄外部位は聖域部位(すなわち、典型的にALLの治療に用いられ、経口および静脈内に投与される化学療法薬の多くが浸透しにくい解剖学的空間)と考えられてきた。小児ALLで最も重要な聖域部位の2つは、中枢神経系(CNS)および精巣である。ALLの治療を成功させるためには、これらの髄外聖域部位における症候性または無症候性の白血病病変に対して効果的に対処する治療法が必要である。
中枢神経系(CNS)
診断時に患者の約3%にCNS病変がみられる(脳脊髄液検体にリンパ芽球を含む白血球が5個/μL以上認められ、かつ/または脳神経麻痺が存在することで定義される)。しかしながら、CNSに対して特異的な治療を実施しない限り、初回診断時の脊髄液にリンパ芽球が検出されるかどうかにかかわらず、最終的にほとんどの小児が顕性のCNS白血病を発症する。CNSに向けた治療法には、髄腔内化学療法、CNSに向けた全身化学療法、および頭蓋照射療法がある;これらの一部またはすべてが現行のALLレジメンに含まれている。(詳しい情報については、本要約の小児ALLに対するCNSに向けた治療のセクションを参照のこと。)
参考文献- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.[PUBMED Abstract]
- Rubnitz JE, Lensing S, Zhou Y, et al.: Death during induction therapy and first remission of acute leukemia in childhood: the St. Jude experience. Cancer 101 (7): 1677-84, 2004.[PUBMED Abstract]
- Christensen MS, Heyman M, Möttönen M, et al.: Treatment-related death in childhood acute lymphoblastic leukaemia in the Nordic countries: 1992-2001. Br J Haematol 131 (1): 50-8, 2005.[PUBMED Abstract]
- Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.[PUBMED Abstract]
- Lund B, Åsberg A, Heyman M, et al.: Risk factors for treatment related mortality in childhood acute lymphoblastic leukaemia. Pediatr Blood Cancer 56 (4): 551-9, 2011.[PUBMED Abstract]
- Hijiya N, Liu W, Sandlund JT, et al.: Overt testicular disease at diagnosis of childhood acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia 19 (8): 1399-403, 2005.[PUBMED Abstract]
- Sirvent N, Suciu S, Bertrand Y, et al.: Overt testicular disease (OTD) at diagnosis is not associated with a poor prognosis in childhood acute lymphoblastic leukemia: results of the EORTC CLG Study 58881. Pediatr Blood Cancer 49 (3): 344-8, 2007.[PUBMED Abstract]
- 新たに診断された小児ALLの治療
-
新規診断ALLに対する標準寛解導入療法の選択肢
新規診断小児急性リンパ芽球性白血病(ALL)に対する標準治療法の選択肢には以下のものがある:
- 化学療法。
寛解導入化学療法
第1段階の治療(寛解導入療法)の目標は、完全寛解(CR)に誘導することである。この寛解導入段階は、典型的に4週間継続される。全体として、この段階の終了までに、新たにB-ALLと診断された患者の約98%がCRに達するが、T-ALLまたは初診時の白血球数が多い乳児および乳児以外の患者では達成率がやや低い。[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ]
寛解導入化学療法には、アントラサイクリン系薬剤(ドキソルビシンまたはダウノルビシンのいずれか)を含む場合も含まない場合もあるが、典型的に以下の薬剤で構成される:
小児腫瘍学グループ(COG)のプロトコルでは、米国国立がん研究所(NCI)標準リスクのB-ALL患者に対して、3剤併用の寛解導入療法(ビンクリスチン、コルチコステロイド、およびpegaspargase)を施行し、NCI高リスクB-ALLおよびすべてのT-ALL患者に対して、4剤併用の寛解導入療法(ビンクリスチン、コルチコステロイド、およびpegaspargaseにアントラサイクリン系薬剤を追加)を施行する。他のグループはすべての患者に対して4剤併用の寛解導入療法を使用している、[ 1 ][ 2 ][ 3 ]
コルチコステロイド療法
現在のレジメンの多くで寛解導入中およびその後の治療段階にプレドニゾンの代わりにデキサメタゾンが用いられているが、患者のすべてのサブセットでデキサメタゾンが有益であるかどうかに関しては異論がある。数件の試験でも、寛解導入中のデキサメタゾンは、感染、ミオパチー、および行動の変化など、プレドニゾンより大きな毒性に関連している可能性が示唆されている。[ 1 ][ 6 ][ 7 ][ 8 ]寛解導入中のデキサメタゾンは、年長の(10歳を超える)患者で骨壊死のリスクが高いことに関連していることがCOGから報告されたが[ 8 ]、他のランダム化研究でこの知見は確認されていない。[ 1 ][ 7 ]
証拠(寛解導入中のデキサメタゾン vs プレドニゾン):
- Children's Cancer Groupは、アントラサイクリン系薬剤を含まない3剤併用の寛解導入療法を受けている標準リスクのB-ALL患者を対象にデキサメタゾンとプレドニゾンを比較するランダム化試験を実施した。[ 6 ]
- 標準リスクおよび高リスクのいずれの患者も含まれていた別のランダム化試験がUnited Kingdom Medical Research Councilによって実施された。[ 7 ]
- Associazione Italiana di Ematologia e Oncologia Pediatrica(AIEOP)のALL-BFM-2000(NCT00430118)試験では、7日間のプレドニゾン前治療に続く多剤併用寛解導入療法(すべての患者でアントラサイクリン系薬剤が含まれた)中にデキサメタゾン(10mg/m2/日)またはプレドニゾン(60mg/m2/日)のいずれかを受ける群に3,720人の患者がランダムに割り付けられた。[ 9 ]
- COGは、NCI高リスクのB-ALL患者を対象にデキサメタゾンとプレドニゾンのランダム化試験を実施した。[ 8 ]4剤併用の寛解導入療法(アントラサイクリン系薬剤を含む)中、デキサメタゾンの14日間投与またはプレドニゾンの28日間投与に患者がランダムに割り付けられた。この試験には、中間維持相の大量メトトレキサートと漸増用量メトトレキサートのランダム化比較も含まれていた。
使用されるプレドニゾン対デキサメタゾンの用量比が転帰に影響する可能性がある。デキサメタゾンとプレドニゾンの比率が1:5~1:7であった研究では、デキサメタゾンの方が良好な結果が示されているが、1:10の比率を用いた研究でも同様な結果が示されている。[ 10 ]
L-アスパラギナーゼ
ALL患児の治療には、以下のようないくつかの種類のL-アスパラギナーゼが使用されている:
pegaspargase(PEGアスパラギナーゼ)
pegaspargaseは、L-アスパラギナーゼの1形態であり、大腸菌(E. coli)酵素がポリエチレングリコールとの共有結合によって修飾されている。米国および西洋で治療を受ける新規診断患者における治療の寛解導入段階および寛解導入後の段階の両方で最も多く使用される製剤である。
pegaspargaseは、筋肉内(IM)注射または静脈内(IV)注射のいずれかで投与される。[ 11 ]pegaspargaseのIM投与とIV投与では、薬物動態および毒性プロファイルがほぼ同じである。[ 11 ]pegaspargaseのIV投与の方がIM投与より毒性が強いという証拠はない。[ 11 ][ 12 ][ 13 ]
pegaspargaseは、天然の大腸菌(E. coli)由来L-アスパラギナーゼよりも血清中半減期がはるかに長く、1回の注射でアスパラギンを長期に枯渇させることができる。[ 14 ]
血清中アスパラギナーゼ酵素活性レベルが0.1IU/mLを超えると、血清中アスパラギンの枯渇につながることが示されている。多剤併用寛解導入療法の一部としてpegaspargaseをIMまたはIVのいずれかで単回投与すると、ほぼすべての患者で2~3週間以上にわたって0.1IU/mLを超える血清中酵素活性が得られることが複数の研究で示されている。[ 11 ][ 12 ][ 15 ][ 16 ]1件のランダム化研究で、より高いpegaspargaseの用量(3,500U/m2)でも、標準の用量(2,500U/m2)と比較して転帰は改善しなかった。[ 17 ][証拠レベル:1iiA]
証拠(pegaspargase vs 天然の大腸菌[E. coli]由来L-アスパラギナーゼ):
- pegaspargaseのIV投与と天然の大腸菌(E. coli)由来アスパラギナーゼのIM投与のランダム化比較が実施された。各薬剤がCR達成後の30週間にわたって投与された。[ 13 ][証拠レベル:1iiC]
- 別の標準リスクのALL患者を対象としたランダム化試験では、寛解導入療法中および2つの遅延強化療法コースのそれぞれの期間中に、pegaspargaseまたは天然の大腸菌(E. coli)由来アスパラギナーゼのいずれかを投与する群に患者が割り付けられた。[ 15 ]
pegaspargaseに対するアレルギー反応がみられる患者では、典型的にErwinia菌由来L-アスパラギナーゼに切り替えられる。pegaspargaseに対する反応が軽度または疑わしい場合のSAAレベルの測定は、Erwiniaへの切り替えが適応となる(SAAが不十分なため)患者と製剤変更が必要ないであろう患者との区別に役立つ可能性がある。[ 18 ][ 19 ]
いくつかの研究で、顕性のアレルギーなしに、SAAの治療濃度に達しないことで定義されるアスパラギナーゼのサイレント不活性化を認める患者サブセットが特定されている。[ 20 ][ 21 ]Dana-Farber Cancer Institute(DFCI)Consortiumが実施した試験で、天然の大腸菌(E. coli)由来L-アスパラギナーゼによる初回治療を受けた患者の12%でサイレント不活化が実証された;これらの患者は、アスパラギナーゼ製剤を変更した場合にEFSが優れていた。[ 21 ]pegaspargaseによる初回治療を受けた患者では、サイレント不活化の頻度が低い(10%未満)ようである。[ 13 ][ 20 ]pegaspargaseによる治療を受けた患者に対する薬物動態モニタリングの至適頻度の決定、およびそのようなスクリーニングにより転帰に影響がみられるかについては、今後の研究が待たれる。
ペグ化アスパラギナーゼの別の製剤であるcalaspargase pegolも、ALLの小児および青年の治療に使用できる。[ 22 ]この製剤は、pegaspargaseと構造が類似しているが、L-アスパラギナーゼ酵素とPEG部分の間にあるリンカーが異なるため、半減期が長い。[ 23 ][ 24 ]
asparaginase Erwinia chrysanthemi(Erwinia菌由来L-アスパラギナーゼ)
天然の大腸菌(E. coli)由来L-アスパラギナーゼまたはpegaspargaseに対するアレルギーが確認されている患者には、典型的にErwinia菌由来L-アスパラギナーゼが使用される。
Erwinia菌由来L-アスパラギナーゼの半減期(0.65日)は、天然の大腸菌(E. coli)由来アスパラギナーゼ(1.2日)またはpegaspargase(5.7日)よりもはるかに短い。[ 14 ]Erwinia菌由来L-アスパラギナーゼを使用する場合、Erwinia製剤の半減期が短いため、アスパラギンの十分な除去を達成するためには、より頻回に投与する必要がある。
証拠(Erwinia菌由来L-アスパラギナーゼの投与頻度を多くして目標の治療効果を達成する必要性):
- COG試験で、pegaspargaseに対するアレルギーが認められた患者に対してErwinia菌由来L-アスパラギナーゼを週3回IM投与したところ、治療効果がある血清中アスパラギナーゼ酵素活性レベル(0.1IU/mL以上として定義)につながったことが実証された。[ 25 ]
- pegaspargaseに対するアレルギーが確認された患者にErwinia菌由来L-アスパラギナーゼを月-水-金スケジュールでIV投与した試験によると、治療効果がある血清中アスパラギナーゼ酵素活性(0.1IU/mL以上として定義)が示されたのは、投与48時間後が患者の83%であったが、投与72時間後では患者のわずか43%であった。[ 26 ]
寛解導入中に使用するアントラサイクリン系薬剤
COGプロトコルでは、NCI標準リスクのB-ALL患者に対して、3剤併用の寛解導入療法(ビンクリスチン、コルチコステロイド、およびpegaspargase)を施行し、NCI高リスクB-ALLおよびすべてのT-ALL患者に対して、4剤併用の寛解導入療法(ビンクリスチン、コルチコステロイド、およびpegaspargaseにアントラサイクリン系薬剤を追加)を施行する。他のグループはすべての患者に対して4剤併用の寛解導入療法を使用している、[ 1 ][ 2 ][ 3 ]
アントラサイクリン系薬剤を含む寛解導入レジメンでは、典型的にダウノルビシンまたはドキソルビシンが用いられる。寛解導入中に2つの薬剤を比較する1件のランダム化試験において、治療1週目の末梢血芽球数の減少、15日目の骨髄形態、寛解導入療法終了後の微小残存病変(MRD)レベルなどの早期反応の測定値に差は認められなかった。[ 27 ][証拠レベル:1iiDiv]
寛解導入療法に対する反応
新たに診断されたALL患児の95%以上が治療開始4週間以内に完全寛解(CR)を達成する。最初の4週間以内にCRに至らなかった患者では、約半数が寛解導入相で毒性により死亡し(通常は感染に起因する)、残りの半数が抵抗性の病態(形態学的に白血病が残存)を示す。[ 28 ][ 29 ][ 30 ];[ 31 ][証拠レベル:3iA]
4週間の寛解導入相終了時点で形態学的に検出可能な白血病が残存している患者のほとんどは予後不良であるが、CRに達した時点で同種造血幹細胞移植(HSCT)を実施することにより恩恵が得られる可能性がある。[ 4 ][ 32 ][ 33 ]大規模なレトロスペクティブ・シリーズで、このような患者の10年OS率は32%であった。[ 34 ]T細胞表現型の患者(年齢は不問)およびB-ALLの6歳を超える患者では、化学療法単独と比較して同種HSCTによる転帰が優れている傾向が観察された。診断時年齢が1~5歳で、有害な細胞遺伝学的異常(KMT2A[MLL]再構成、BCR-ABL1)がいずれも認められないB-ALLの患者では、予後が比較的良好であり、化学療法単独と比較してHSCTの実施による転帰の優位性は認められなかった。[ 34 ]
CRに達した患者では、芽球のクリアランスの迅速性の測定およびMRDの判定には、特に以下に示す重要な予後的意義がある:
(詳しい情報については、本要約の初回治療に対する反応のセクションを参照のこと。)
(新たにALLと診断された小児におけるCNS再燃を予防するCNS療法に関する具体的な情報については、本要約の小児ALLに対するCNSに向けた治療のセクションを参照のこと。)
小児ALLに対する標準寛解導入後療法の選択肢
地固め/強化療法および維持療法(寛解導入後療法)に対する標準治療法の選択肢には以下のものがある:
- 化学療法。
すべてのグループが、維持化学療法前に中枢神経系(CNS)向けの治療を実施している。維持療法中に継続して髄腔内化学療法を行うプロトコル(小児腫瘍学グループ[COG]、St. Jude Children's Research Hospital [SJCRH]、およびDana-Farber Cancer Institute[DFCI])もあれば、行わないプロトコル(ベルリン-フランクフルト-ミュンスター[BFM])もある。(寛解導入後療法を受けている急性リンパ芽球性白血病[ALL]患児におけるCNS再燃を予防するCNS療法に関する具体的な情報については、本要約の小児ALLに対するCNSに向けた治療のセクションを参照のこと。)
地固め/強化療法
完全寛解(CR)が得られた時点で、CNSに向けた治療と合わせて全身療法を続ける。寛解導入後化学療法の強度は、リスクグループの割り付けに応じて大幅に異なるが、すべての患者は、CRを得た後に維持療法を開始する前に、何らかの強化療法を受ける。
最も多く使用されている強化スキームは、BFMの基本骨格である。この治療基本骨格は、最初にBFM臨床試験グループにより導入されたもので、以下を含んでいる:[ 1 ]
- 初回の寛解導入相の直後に実施する最初の地固め相(寛解導入IBと呼ばれる)。この地固め相では、シクロホスファミド、低量シタラビン、およびメルカプトプリンが使用される。
ロイコボリン救援を伴う高用量のメトトレキサート(典型的に5g/m2)の4回投与を含む中間維持相。
- 典型的に寛解導入相および初回地固め相と同様な薬剤およびスケジュールを使用する再寛解導入相(または遅延強化相)。
- 典型的に髄腔内療法を継続しながら、メルカプトプリン(6-MP)1日1回投与および低量メトトレキサート週1回投与を行い、ときにビンクリスチンおよびコルチコステロイドを投与する維持相。
この基本骨格は、COGを含む多くのグループが採用している。この基本骨格の変法には以下のものがある:
他の臨床試験グループは、寛解導入後療法期間に以下のように異なった治療基本骨格を用いている:
標準リスクのALL
標準リスクのB-ALL患児では、アントラサイクリン系薬剤およびアルキル化剤のように晩期毒性作用のリスク増加と関連している可能性がある薬物への曝露を制限する試みがなされている。[ 50 ][ 51 ][ 52 ]BFM基本骨格を使用しているレジメン(COGなど)では、漸増用量メトトレキサート(ロイコボリン救援を伴わない)およびビンクリスチンからなる中間維持相と合わせて単回の再寛解導入/遅延強化相を実施することで、良好な転帰が得られている。[ 53 ]地固め療法とその後の維持療法(再寛解導入相はなし)として、コース数を制限した中等量または大量メトトレキサートを使用した試験でも、標準リスクの患者で予後良好な転帰が報告された。[ 51 ][ 54 ][ 55 ]DFCI ALL Consortium研究では、地固め療法としてpegaspargaseの反復投与(30週間)を用いて、寛解導入後療法でアルキル化剤またはアントラサイクリン系薬剤への曝露がなかった。[ 56 ][ 57 ]
しかしながら、導入療法および/または地固め療法終了時の微小残存病変(MRD)の予後的影響は、最初に米国国立がん研究所(NCI)の標準リスクALLと診断された患者の治療に影響を及ぼしている。多数の研究で、導入療法終了時のMRDレベルが高い場合、予後不良に関連することが実証されている。[ 36 ][ 38 ][ 39 ][ 58 ][ 59 ]導入療法終了時のMRDレベルが高い標準リスク患者において、治療の強化により転帰が改善されることが示されている。[ 42 ]したがって、導入療法終了時のMRDレベルが高い標準リスク患者は、導入療法終了時のMRDレベルが低い標準リスク患者に対して記述されているアプローチでは治療されず、通常は高リスクレジメンで治療される。
証拠(標準リスクALLに対する治療強度):
- 1980年代および1990年代初期に実施された臨床試験により、BFM基本骨格を用いたレジメンによる治療を受けた標準リスクのALL患児では、遅延強化相を用いることで転帰が改善することが実証された。[ 60 ][ 61 ][ 62 ]このようなレジメンでCOGのレジメンを含めた遅延強化相は、8週間の再寛解導入相(アントラサイクリン系薬剤を含む)、および寛解に至ってから約4~6ヵ月間投与するシクロホスファミド、シタラビン、および6-thioguanineを含む再地固め相から構成される。[ 29 ][ 60 ][ 63 ]
- 標準リスクのALLを対象とした旧Children's Cancer Group(CCG)の研究(CCG-1991/COG-1991)では、3剤併用の導入療法期にデキサメタゾンを用い、2回目の遅延強化療法相の利用を検証した。この研究では、2回の中間維持相に投与する経口メトトレキサートを含む標準的な維持併用療法に対して、ビンクリスチンと併用した漸増用量の静注(IV)メトトレキサート(ロイコボリン救援を伴わない)も比較された。[ 53 ][証拠レベル:1iiDi]
- COG AALL0331(NCT00103285)研究では、生物学的特徴および初期反応に基づいてNCI標準リスクの患者に対する治療強度を層別化した。迅速な初期反応は、施設内での形態学的解釈に基づく15日目までの骨髄芽球が5%未満であり、29日目でMRDレベルが0.1%未満のM1骨髄として定義される。低度の標準リスクの患者は、予後良好な生物学的特徴(ETV6-RUNX1またはトリプルトリソミーを伴う高度の高二倍体)、CNS1状態、および迅速な初期反応を示す患者である。平均的な標準リスクの患者は、予後良好または予後不良な生物学的特徴がみられず、同様に迅速な初期反応を示す患者である。高度の標準リスクの患者は、緩徐な初期反応を示し、かつ/またはCNS3状態の患者、または迅速な初期反応を示すKMT2A再構成を有する患者である。すべての患者が3剤併用寛解導入療法(アントラサイクリン系薬剤を含まない)を受けた。平均的な標準リスクの患者は、強化された地固め療法(強化BFM)または標準の地固め療法のいずれかにランダムに割り当てられた。高度の標準リスクの患者は、2つの遅延強化相を含むNCI高リスク患者に使用される完全な強化BFM療法に非ランダム的に割り当てられた。[ 64 ]
- 英国で実施された1件のランダム化研究では、高リスクの特徴(有害な細胞遺伝学的異常、および/または寛解導入療法8日目または15日目のM3の骨髄形態所見など)がみられないALLの小児および若年成人を対象に、寛解導入療法終了時(4週目)および治療11週目のMRDレベルに基づくリスク層別化が行われた。4週目にMRDが検出不能であった(または4週目のMRDレベルが低く、11週目までに検出不能になった)患者は低リスクとみなされ、1または2コースの遅延強化相による治療へのランダム化割り付けに適格とされた。[ 65 ][証拠レベル:1iiDi]
- Associazione Italiana di Ematologia e Oncologia Pediatrica(AIEOP)ALL-BFM-2000(NCT00430118)試験で、標準リスクの患者(33日目および78日目でMRDが検出されず、高リスクの細胞遺伝学的所見がみられない患者と定義)が標準強度または強度縮小(デキサメタゾン、ビンクリスチン、ドキソルビシン、シクロホスファミドの投与期間を短縮して総投与量を減量)のいずれかの単回遅延強化相による治療を受ける群にランダムに割り付けられた。[ 66 ]
- 診断時に標準リスクまたは中リスクであっても導入療法終了時のMRDレベルが高い患者は、比較的予後不良であることが示されており、高リスク患者として治療すべきである。UKALL2003(NCT00222612)試験では、導入療法終了時のMRDレベルが高い標準リスクまたは中リスクの患者に対する治療に、強化した導入後療法(過剰用量のpegaspargaseとビンクリスチンおよびロイコボリン救援を伴わない漸増用量のIVメトトレキサート)が使用された。[ 42 ][証拠レベル:1iiDi]
高リスクのALL
高リスク群の患者では、多数のさまざまなアプローチが用いられており、同程度の効力が認められている。[ 56 ][ 67 ];[ 63 ][証拠レベル:2Di]高リスクの患者に対する治療は、一般に標準リスクの患者に対する治療よりも強力であり、通常はアントラサイクリン系薬剤および/またはアルキル化剤を含む複数の薬剤の累積投与量が大きい。これらの薬剤の用量が高くなるほど短期および長期毒性のリスクがいずれも増加するため、多くの臨床試験では、これらの強化レジメンによる副作用を抑えることを中心に検討している。
証拠(高リスクALLに対する治療強度):
- 旧CCGは、2回目の中間維持相および遅延強化相を加えた増強BFM治療レジメンを開発した。このレジメンは、中間維持相でビンクリスチンとpegaspargaseを併用投与する漸増用量のIVメトトレキサート(ロイコボリン救援なし)のコースを反復し、初回地固め相および遅延強化相にビンクリスチンとpegaspargaseの律動的投与を追加することを特徴としている。CCG-1882試験では、早期反応が緩慢な(寛解導入療法から7日目でM3の骨髄所見)NCI高リスク患者を、標準BFM療法施行群または増強BFM療法施行群のいずれかにランダムに割り付けた。[ 47 ]
- イタリアのある研究では、遅延強化療法の2回の適用(プロトコルII)が、プレドニゾンによる前治療に対する反応が鈍い患児の転帰を有意に向上させたことが、研究者により示された。[ 69 ]
- CCG-1961研究では、初期反応が早期に認められたNCI高リスク患者を対象に、2×2要因デザインを用いて、標準強化療法 vs 増強強化療法の比較に加え、標準治療期間(中間維持相および遅延強化相を1回) vs 延長治療期間(中間維持相および遅延強化相を2回)についても比較した。この試験では、遅延強化相におけるデキサメタゾンの連日投与と隔週投与で、骨壊死の発生率に影響がみられるかどうかについても検証が行われた。
- COGのAALL0232(NCT00075725)研究(2004年~2011年)で、高リスクのB-ALL患者は、1回の中間維持相と遅延強化相とともに、強化されたBFMの基本骨格による治療を受けた;寛解導入療法終了時のMRDが0.1%を超えるか、5日目でM2/M3骨髄所見であった患者のみが2回の中間維持相/遅延強化相による治療を受けた。中間維持相(これらの相を2回受ける患者のみが最初の相)に大量メトトレキサートまたは漸増用量のIVメトトレキサート(Capizzi方式のメトトレキサート)のいずれかを投与する群に患者がランダムに割り付けられた。[ 8 ][ 37 ]
高リスクALLに対する治療にはより強力な治療が使用されるため、急性および長期毒性の高いリスクにつながり、EFSに有害な影響を与えないで副作用を抑える介入が多くの臨床試験で検討されている。検討されている介入には、アントラサイクリン系薬剤関連の心毒性作用の防止を目的とした心筋保護薬デクスラゾキサンの使用および骨壊死のリスク低減を目的としたコルチコステロイドの代替投与スケジュールがある。
証拠(デクスラゾキサンの心筋保護作用):
- DFCI ALL Consortium試験では、高リスクALL患児を対象に、多剤併用化学療法による寛解導入相および強化相で、ドキソルビシン(1回当たり30mg/m2で、累積用量300mg/m2まで)単独群、またはデクスラゾキサンとの併用群のいずれかにランダムに割り付けた。[ 72 ][ 73 ]
- POG-9404試験で、T-ALLの患者がドキソルビシン(累積用量、360mg/m2)の各投与前にデクスラゾキサンを投与する群または投与しない群にランダムに割り付けられた。[ 74 ]
証拠(骨壊死のリスク低減):
- CCG-1961研究で、骨壊死の頻度低減を目的として、遅延強化相でデキサメタゾンの隔週投与が検討された。[ 71 ]寛解導入療法に対する早期の形態学的反応が速やかな高リスクのB-ALL患者が1または2コースの遅延強化相による治療を受ける群にランダムに割り付けられた。1コースの遅延強化相にランダムに割り付けられた患者は、デキサメタゾンの1日1回投与(連続21日間)を受け、2コースの遅延強化相にランダムに割り付けられた患者は、各遅延強化相でデキサメタゾンの隔週投与(0~6日目と14~21日)を受けた。
(詳しい情報については、本要約の骨壊死のセクションを参照のこと。)
超高リスクのALL
ALL患者の約10~20%が超高リスクとして分類され、その中には以下の患者が含まれる:[ 63 ][ 75 ]
超高リスクの特徴を示す患者は、(通常、典型的なBFM基本骨格の強化相に加えて)地固め相で強化化学療法サイクルを複数回繰り返す治療を受けている。これらの追加サイクルには、標準リスクおよび高リスクの患者に対する初期ALLレジメンでは一般に使用されていない高用量シタラビン、イホスファミド、およびエトポシドといった薬剤が含まれることが多い。[ 63 ]しかし、この強化療法を併用しても、この患者サブセットで報告された長期EFS率は30~50%の範囲である。[ 32 ][ 63 ]
数件の臨床試験で、超高リスクの患者も第一CR期での同種造血幹細胞移植(HSCT)の候補とみなされている。[ 32 ][ 76 ][ 77 ][ 78 ]しかしながら、第一CR期での同種HSCTによる治療を受けた超高リスクの患者の転帰に関するデータは限られている。HSCTから利益が得られる可能性のある亜集団に関しては意見の相違がある。
証拠(超高リスク患者に対する第一寛解期における同種HSCT):
- 1995年から2000年に実施された欧州の共同研究グループによる研究では、超高リスクが次のいずれかで定義された:4剤併用寛解導入療法後に形態学的に病変が残存、t(9;22)(q34;q11.2)もしくはt(4;11)(q21;q23)、またはT細胞表現型もしくは初診時の白血球(WBC)が100,000/μLを超えている患者でプレドニゾン前治療に対する反応が不良。これらの患者が第一CR期における同種HSCT(ヒト白血球抗原適合血縁ドナーが得られることが前提)施行群または強化化学療法施行群のいずれかに割り付けられた。[ 32 ]
- 最初の寛解導入に失敗した患者を対象とした大規模なレトロスペクティブ・シリーズによると、白血病が残存していた患者の10年OS率は32%であった。[ 34 ]
- AIEOPのALL-BFM-2000(NCT00430118)研究(2000年~2006年)では、以下の基準のいずれかを満たす場合に高リスクとして患者を分類した:プレドニゾン前治療に対する不良な反応、治療1ヵ月目終了時にCRに達しない場合、寛解導入IB(治療78日目)後のMRDレベルが高い場合、およびt(4;11)(q21;q23)。これらの患者は、ドナーの入手性および研究者の判断を基にプロトコルに従い第一CR期での同種HSCTに割り付けられた。[ 79 ][証拠レベル:2Dii]
- 2件のレトロスペクティブ解析により、低二倍体ALL患者に対する第一CR期のHSCTの役割が調査された。研究では、1)低二倍体ALLのすべての患者に移植を行う場合、または2)寛解導入療法後にMRDが多いために高リスクと考えられる低二倍体の患者に移植を行う場合、HSCTにより転帰が改善するという明確な証拠は示されなかった。研究では、地固め療法後に持続するMRDに対するHSCTの戦略について調査されず、HSCT時のMRDの状態についても解析されなかった。
維持療法
維持療法の基本骨格
ほとんどのプロトコルにおける維持療法の基本骨格は、メルカプトプリン連日経口投与および週1回のメトトレキサート経口または非経口投与である。多くのプロトコルで、CNS聖域療法としての髄腔内化学療法は維持療法期間を通して継続される。維持療法を受けている小児は、薬物関連毒性および維持療法中に使用される経口化学療法薬の服薬遵守について注意深くモニターすることが肝要である。[ 82 ]COGが行った研究により、さまざまな人種および社会経済的状態の集団間でメルカプトプリンの服薬遵守について有意差が実証された。重要なことに、維持相でのメルカプトプリンによる治療に対する不遵守に伴って、再燃リスクに有意な増加が認められている。[ 82 ][ 83 ]
過去における臨床診療では、メルカプトプリンの経口投与を夕方行うよう広く求められたが、その根拠は、これを実践することでEFSが改善する可能性があるという過去の研究からの証拠であった。[ 84 ]しかしながら、Nordic Society for Pediatric Hematology and Oncology(NOPHO)グループが実施した研究では、経口摂取の詳細がプロスペクティブに収集され、メルカプトプリンの投与時間(夕方 vs 他の時間)は予後的に重要ではなかった。[ 85 ]COG研究によると、常に夕方ではなく、1日のさまざまな時間にメルカプトプリンを投与すると、非遵守の割合が高いという関連がみられた;しかしながら、遵守患者(すなわち、処方された用量の摂取量が95%を超える患者)で、メルカプトプリンの摂取時間と再燃リスクの間に関連は認められなかった。[ 86 ]
一部の患者では、メルカプトプリンを不活性化させる酵素であるチオプリンS-メチルトランスフェラーゼが先天的に欠損(ホモ接合性変異)しているために、従来の用量でメルカプトプリンを投与すると重度の造血毒性を来すことがある。[ 87 ][ 88 ]このような患者は、従来の投与量よりはるかに低い用量を投与した場合にのみ、メルカプトプリンに耐えることができる。[ 87 ][ 88 ]この変異がヘテロ接合体である患者は、一般に重篤な毒性なしにメルカプトプリンに忍容性を示すが、実際には正常アレルがホモ接合体である患者よりも造血毒性に対する用量減量が頻繁に必要となる。[ 87 ]東アジアおよびヒスパニック系の患者で高頻度に観察されるNUDT15遺伝子の多型は、メルカプトプリンの骨髄抑制作用に対する過度の感受性にもつながっている。[ 89 ][ 90 ][ 91 ]
証拠(維持療法):
- ランダム化試験のメタアナリシスでチオプリン系薬剤が比較され、以下が明らかになった:
- シクロホスファミドおよびエピポドフィロトキシンとともに、これより標準的な維持療法薬剤も含めて、2剤を順次入れ替えて使用する強化維持療法レジメンがSJCRHおよび他のグループにより実施された数件の臨床試験で評価されている。[ 2 ]
ビンクリスチン/コルチコステロイドの律動的投与
ビンクリスチンおよびコルチコステロイドの律動的投与は標準維持療法の基本骨格に加えられることが多いが、現代的な多剤併用化学療法レジメンが使用されている状況下で、こうした律動的投与の有益性については依然として異論がある。
証拠(ビンクリスチン/コルチコステロイドの律動的投与):
- 1980年代に実施されたCCGのランダム化試験では、月1回のビンクリスチン/プレドニゾンの律動的投与を受けた患者の転帰が改善したことが実証された。[ 101 ]
- 治療法が同じ時代の6件の臨床試験から得られたデータをまとめたメタアナリシスでは、ビンクリスチン/プレドニゾンの律動的投与でEFSが優れていることが示された。[ 102 ][ 103 ]しかしながら、これらの試験による全体的なEFSは、より現代的なレジメンで観察されるものより低かった。
- つい最近の臨床試験から得られたビンクリスチンとステロイドの律動的投与の効果に関する系統的レビューでは、より強力な早期の治療ならびに早期反応(MRD)および生物学的因子を組み込んだリスク層別化を含む現行のALL治療において、このような律動的投与に価値があるのかという疑問が提起された。[ 103 ]
- 中リスクのALL患児を対象にBFMレジメンによる治療を行った多施設ランダム化試験では、治療継続期間にビンクリスチン/デキサメタゾンの律動的投与を6回追加したが、有益性が実証されている他の試験よりも律動的投与の頻度が少なかったとはいえ、関連した有益性は認められなかった。[ 104 ]
- 平均リスクの患者を対象とした小規模の多施設共同試験では、ビンクリスチンとコルチコステロイドの律動的投与を受けた患者でEFSが優れていることが示された。この研究では、ステロイドの種類(プレドニゾン vs デキサメタゾン)に基づいた転帰に違いは認められなかった。[ 105 ][証拠レベル:1iiA]
ビンクリスチン/コルチコステロイドの律動的投与を含むレジメンでは、どちらのステロイド(デキサメタゾンまたはプレドニゾン)を使用すべきかという問題に多くの研究が取り組んでいる。これらの研究から、デキサメタゾンではEFSが優れているという関係がみられるが、特に年長の小児および青年では骨毒性および感染などのステロイド関連合併症の頻度が高まる原因となる可能性もある。[ 6 ][ 7 ][ 21 ][ 60 ][ 106 ]プレドニゾンとの比較で、デキサメタゾンでは、行動障害の高い頻度との関係も認められている。[ 7 ]維持療法を受けた3~16歳の患者50人を対象としたランダム化研究では、デキサメタゾンのパルス療法中のヒドロコルチゾン(生理的用量で)の併用投与により、行動障害、情緒不安定、および睡眠障害の頻度が減少した。[ 107 ]
証拠(デキサメタゾン vs プレドニゾン):
- CCG研究で、1歳から10歳未満の低リスクALL患児を対象に、寛解導入相および維持相でのデキサメタゾンがプレドニゾンと比較された。[ 6 ][ 60 ]
- Medical Research Council(MRC)のUnited Kingdom Acute Lymphoblastic Leukaemia(UKALL)試験では、標準リスクおよび高リスクの両患者を対象に、寛解導入相および維持相でのデキサメタゾンがプレドニゾロンと比較された。[ 7 ]
- DFCI ALL Consortium試験では、すべての寛解導入後治療相でデキサメタゾン投与群またはプレドニゾン投与群のいずれかに患者をランダムに割り付けた。[ 21 ]
10~18歳の小児集団ではステロイド誘発性骨壊死のリスクが増大するため、この年齢の小児に対してデキサメタゾンを使用する有益性についてはさらに調査が必要である。[ 68 ][ 106 ]
維持療法の期間
維持化学療法は、一般にCRが持続する限り、2~3年にわたり継続する。一部の研究では、男児は女児よりも治療期間が長かったが[ 60 ]、別の研究では性別に基づく治療期間の差はみられなかった。[ 56 ][ 63 ]特に現在の治療状況で、男児において維持治療期間を長くするほど再燃が少なくなるかどうかは明らかではない。[ 63 ][証拠レベル:2Di]維持療法期間を延長して3年を超えても転帰の改善は認められない。[ 102 ]
維持療法中の経口投与に対する遵守
維持療法中のメルカプトプリンによる治療に対する不遵守は、有意な再燃リスクと関連している。[ 82 ]
証拠(治療に対する遵守):
- COGは、小児および青年327人(ヒスパニック系169人および非ヒスパニック系白人158人)を対象に、維持療法中のメルカプトプリンに対する不遵守の影響について検討した。[ 82 ]
- 遵守に関する2つ目の研究がALLの小児298人(アジア系米国人71人、アフリカ系米国人68人、および非ヒスパニック系白人159人)を対象に実施された。[ 83 ]
- 小児742人を対象とした3番目の研究で、以下の主要な観察がみられた:[ 108 ]
- 上記の研究の著者らは、自己報告が治療遵守の信頼できる測定法ではないことも明らかにしており、患者の84%が少なくとも時々はメルカプトプリン服薬遵守を過剰に報告している。[ 109 ]このデータは、自己報告以外の別の遵守測定法が必要なことを示唆している。
- 追跡研究において、上述の著者らは、メルカプトプリン摂取習慣、赤血球thioguanine nucleotide(TGN)値、遵守、および再燃リスクについて調査した。[ 86 ][証拠レベル:2Diii]
臨床評価段階にある治療法の選択肢
リスクをベースとした治療割り付けは、ALL患児に用いられる重要な治療戦略であり、治療失敗リスクの程度が異なる特定の患者集団ごとにプロトコルがデザインされる。本要約のリスクに基づく治療割り付けのセクションでは、ALL患児をリスクに基づく治療グループに初期層別化するために用いられる臨床的特徴および臨床検査所見について記載している。
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
B-ALLに対するCOGの研究
標準リスクのALL
-
COG-AALL1731(NCT03914625)(限局性B細胞リンパ芽球性リンパ腫患者を対象に標準リスクB-ALL向け治療を施行した場合における転帰を判定する研究):このプロトコルは、初診時のWBCにかかわらず、非ダウン症候群のNCI標準リスクのB-ALL患者およびダウン症候群のすべてのB-ALL患者(年齢が31歳未満)を対象としている。このプロトコルでは、ブリナツモマブ抗体を結合させた二重特異性T細胞の追加により転帰を改善できるか、および男児における治療期間短縮(中間維持相1の開始後3年から同相の開始後2年へ)がDFSに悪影響を与えないかについて検証される。
すべての患者は、3剤併用寛解導入療法(アントラサイクリン系薬剤を含まない)を受ける。寛解導入療法完了後の患者は、生物学的測定値および早期反応測定値に基づいて3つのグループのいずれかに分類される:
予後良好な標準リスクの患者は標準の治療法による治療を受ける。
平均的な標準リスクのすべての患者は、ハイスループット配列決定法(HTS)-MRD分析法を用いて寛解導入療法の29日目にMRDの評価を受ける。HTS-MRDで病変が検出されない患者は、標準の治療法による治療を受けるが、HTS-MRDで病変が検出された(またはHTS-MRDで判定できないか、HTS-MRDが利用できない)患者は、ダブルトリソミーで29日目の骨髄MRDが0.01%以上から0.1%未満の患者と同様に、標準の治療法または標準の治療法 + ブリナツモマブの2サイクル追加のランダム化への参加に適格である。
高度の標準リスクの患者は、強化BFM(NCI高リスク向け)の基本骨格による治療を受ける。地固め療法終了時のMRDが1%を超えるすべての患者は、プロトコルに従う治療から除外される。地固め療法終了時のMRDが0.1%未満の患者は、NCI高リスク向け基本骨格単独または同治療法 + ブリナツモマブの2サイクルのランダム化への参加に適格である。地固め療法終了時のMRDが0.1%以上から1%未満の患者は、NCI高リスク向け基本骨格 + ブリナツモマブの2サイクルを受けるように直接割り付けられる。
NCI標準リスクのダウン症候群患者で、平均的な標準リスクの定義を満たす場合は、詳細を上に示したように非ダウン症候群の平均的な標準リスクの患者と同じ方法による治療を受ける。NCI高リスクのダウン症候群患者、予後不良な生物学的所見を認める患者、および29日目のMRDが多い患者を含む他のすべてのダウン症候群患者は、高度のダウン症候群とみなし、強化BFM療法の基本骨格から強力な要素を省略した緩和化学療法レジメンに加えてブリナツモマブの2サイクルを受けるように非ランダム的に割り付けられる。省略される要素には、寛解導入期のアントラサイクリン系薬剤および遅延強化期の後半でのシクロホスファミド/シタラビンをベースとした化学療法がある。
すべての患者は、リスクグループにかかわりなく、同じ期間(中間維持相1の開始から2年間)の治療を受ける。これは、標準の治療法と比較して男児に対して1年の治療期間短縮を示している。
高リスクおよび超高リスクのALL
- COG-AALL1521(NCT02723994)(ALL小児を対象としたルキソリチニブと化学療法の第II相研究):この非ランダム化研究は、次の遺伝子異常のいずれかを認めるNCI高リスクB-ALL患者(1~21歳)の治療を目的に修正増強BFMレジメン(AALL1131に類似)と併用してルキソリチニブ(JAK阻害薬)を追加した場合を検証している:1)CRLF2再構成;2)JAK1またはJAK2の変異;3)JAK経路に関与するその他の変化(例、JAK2融合、EPO-R融合、SH2B3欠失、IL7RA変異)。患者は寛解導入相を完了した後に本研究に移行する。ルキソリチニブは、すべての寛解導入後治療相と並行して投与される。主要目的は、この併用療法の安全性、忍容性、および有効性を評価することである。
-
COG-AALL1721(NCT03876769)(高リスクB-ALLで地固め療法終了時のMRDが陽性の患者におけるtisagenlecleucelの有効性および安全性に関する研究):このプロトコルは、年齢が1~25歳のNCI高リスクのB-ALLで、寛解導入療法終了時に形態学的CRであり、地固め療法終了時のMRDが0.01%以上の患者を対象としている。本試験の主要目的は、この患者集団における根治療法として、tisagenlecleucel(CD19を標的としたキメラ抗原受容体[CAR]T細胞)の有効性を評価すること、特にtisagenlecleucel治療による5年DFS率が55%を超えるかどうかを判定することである。
本試験に登録された患者は白血球アフェレーシスを受け、自己T細胞を収集してtisagenlecleucelの製造者に送られる。製造完了を待つ間に、患者は中間維持相1(大量メトトレキサート)に進む;この相は、製品が利用可能になり次第、中止される場合がある。利用可能になり次第、患者はリンパ球枯渇化学療法およびtisagenlecleucelの注入を受ける。tisagenlecleucelの後に抗白血病治療をさらに実施することはない。注入後定期的に骨髄サンプルが採取され、tisagenlecleucel投与後29日目から疾患状態の評価が開始される;B細胞形成不全の証拠をスクリーニングするために、末梢血検査も送付される。
患者を試験に登録するには、診断時にCD19陽性の証拠が得られていなければならない。寛解導入療法終了時にM3骨髄、地固め療法終了時にM2/M3骨髄、低二倍体(染色体数が44未満)、Ph+ ALL、またはチロシンキナーゼ阻害薬による治療歴がある患者は、登録から除外される。
-
COG-AALL1732(NCT03959085)(新たに診断された高リスクB-ALLに対するイノツズマブ オゾガマイシンの第III相ランダム化試験;高リスクB-ALL、混合表現型急性白血病[MPAL]、播種性B-リンパ芽球性リンパ腫に対するリスク調整寛解導入後療法):このプロトコルは、非ダウン症候群のNCI高リスクB-ALL患者、MPALのすべての患者、および播種性B-リンパ芽球性リンパ腫患者を対象としている。NCI標準リスクB-ALLで、以前にステロイドで治療を受けた患者、CNS3状態の患者、または診断時に精巣病変を認める患者もまたこの研究に適格とされる。
B-ALL患者に対するこのプロトコルでは、修正BFMの基本骨格への2ブロックのイノツズマブ オゾガマイシン追加によりDFSが改善するかどうか、および男児における治療期間短縮(中間維持相1の開始後3年から同相の開始後2年へ)がDFSに悪影響を及ぼさないかどうかが検証される。この研究はまた、MPALおよび播種性B-リンパ芽球性リンパ腫で、高リスクALLに対する標準の化学療法レジメンで治療された患者のEFSを明らかにすることを目的としている。
すべての患者が、4剤併用寛解導入療法(ダウノルビシンを含む)を受ける。寛解導入療法完了後の治療は、年齢、生物学、および治療への反応によって異なる。
すべての患者が、同じ期間(中間維持相1の開始から2年間)の治療を受ける。これは、標準の治療法と比較して男児に対して1年の治療期間短縮を示している。EOC MRDが0.01%以上のNCI高リスクB-ALL患者はプロトコルの治療から除外され、COG-AALL1721試験(上記を参照のこと)への登録が適格となる。EOC MRDが1%以上のNCI標準リスク患者はプロトコルの治療から除外され、COG-AALL1721試験への登録も適格とされない。
その他の研究
-
St. Jude Total 17研究(TOT17、NCT03117751)(ALLまたはリンパ腫患者の治療における併用化学療法):
この試験には以下の4つの主要目的がある:
- 難治性B-ALL患者に対するチロシンキナーゼ阻害薬またはCAR T細胞/ブリナツモマブおよび標的可能病変がない患者に対するプロテアソーム阻害薬ボルテゾミブを含む分子的および免疫療法的アプローチの追加により、遺伝的または免疫学的に標的可能な病変を有するか、15日目のMRDが5%以上または寛解導入療法終了時のMRDが1%以上の暫定的に標準リスクまたは高リスクの患者のEFSを改善すること。
- pegaspargaseとシクロホスファミドによる治療の最適化、標的可能なゲノム異常(例、活性化チロシンキナーゼまたはJAK/STAT変異)を有する患者に対する新規薬剤の追加、または治療に対する早期反応が不良であるが、標的可能病変がない患者に対するボルテゾミブの追加、および診断時の脳脊髄液に白血病細胞がみられるか、寛解導入療法終了時のMRDが0.01%以上のT-ALL患者に対するネララビン投与によって、T-ALL患者の全体的な治療転帰を改善すること。
- ランダム化研究デザインで、B-ALLの小児に対して早期寛解導入療法中にリツキシマブの2つの用量を投与することで、pegaspargaseに対するアレルギー反応が軽減されるかどうかについて検討すること。
- ランダム化研究デザインで、高リスクCEP72 TT遺伝子型の患者でビンクリスチンの用量を減量することによって、またはCEP72 CCもしくはCT遺伝子型の患者でビンクリスチンの投与期間を短縮することによって、急性ビンクリスチン誘発性末梢神経障害の発生率および/または重症度を低減できるかどうかについて検討すること。
-
DFCI ALL Consortium 16-001(NCT03020030)(ALLの小児および青年に対してより良好な治療法選択肢を特定するリスク分類スキーム):
この試験には以下の2つの主要目的がある:
- ALLの小児および青年に対する新規のリスク分類スキームを検証すること。
- 治療による血清アスパラギナーゼ活性レベルを維持する一方で、非アレルギー性アスパラギナーゼ関連毒性を潜在的に低減させることを目標として、寛解導入後療法相で用量を減量した(血清アスパラギナーゼ活性レベルに基づき用量を調節した)pegaspargase投与の実現可能性を検証すること。
患者は治療10日目までに初期リスク群に割り当てられる。次のいずれかが認められる場合に患者は最初に超高リスクとみなされる:IKZF1欠失、KMT2A遺伝子再構成、TCF3-HLF融合(t(17;19))、または低度の低二倍体(染色体数が40未満)。次の基準をすべて満たす場合に患者は初期低リスクとみなされる:B細胞ALL、1歳から15歳未満、WBC数が50x109/L未満、CNS1またはCNS2、iAMP21がみられない、および超高リスクの特徴がみられない。初期高リスクの患者には、T-ALLのすべての患者を含め、超高リスクの特徴がみられない他のすべての患者が含まれる。
寛解導入療法の強度は初期リスク群に依存する。初期低リスクの患者は、3剤併用寛解導入療法(アントラサイクリンを含まない)を受ける。他のすべての患者は、4剤併用寛解導入療法(アントラサイクリン系薬剤を含む)を受ける。
寛解導入後療法の強度を決定する最終リスクグループは、寛解導入療法終了時点(32日目;第1時点)および治療10週目(第2時点)でのMRD(次世代の塩基配列決定法により評価)に基づいて割り当てられる。
すべてのリスク群に対する治療には、寛解導入後治療期に30週間のpegaspargase(2週間ごとに15回投与)が含まれる。最終低リスク/高リスクのすべての患者は、寛解導入後のpegaspargase投与:標準用量(1回当たり2,500IU/m2)または薬物動態による減量調節(開始用量;2,000IU/m2)のランダム化比較への参加に適格である。すべての患者で、pegaspargaseの各投与前に血清中アスパラギナーゼ活性最低値(NSAA)がチェックされる;NSAAが検出できないことが明らかになった患者は、Erwinia菌由来アスパラギナーゼへ切り替える。薬物動態による減量調節群で、pegaspargaseを4回投与後にNSAAが極端に高い(1.0IU/mLを超える)ことが明らかになった場合、用量をさらに1,750IU/m2まで減量できる;すべての時点で低い(0.4IU/mL未満)NSAAが検出可能な場合、用量を標準用量(2,500IU/m2)まで増量する。この試験は、pegaspargaseに対するグレード2の過敏症反応を認める患者に再投与して、薬物動態モニタリングにより、このような患者ではErwiniaに切り替えるか、前投薬とともにpegaspargaseの投与を継続してもよいかどうかを決定する戦略のパイロット研究でもあった。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.[PUBMED Abstract]
- Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.[PUBMED Abstract]
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.[PUBMED Abstract]
- Oudot C, Auclerc MF, Levy V, et al.: Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol 26 (9): 1496-503, 2008.[PUBMED Abstract]
- Salzer WL, Devidas M, Carroll WL, et al.: Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia 24 (2): 355-70, 2010.[PUBMED Abstract]
- Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.[PUBMED Abstract]
- Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.[PUBMED Abstract]
- Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.[PUBMED Abstract]
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.[PUBMED Abstract]
- McNeer JL, Nachman JB: The optimal use of steroids in paediatric acute lymphoblastic leukaemia: no easy answers. Br J Haematol 149 (5): 638-52, 2010.[PUBMED Abstract]
- Silverman LB, Supko JG, Stevenson KE, et al.: Intravenous PEG-asparaginase during remission induction in children and adolescents with newly diagnosed acute lymphoblastic leukemia. Blood 115 (7): 1351-3, 2010.[PUBMED Abstract]
- Rizzari C, Citterio M, Zucchetti M, et al.: A pharmacological study on pegylated asparaginase used in front-line treatment of children with acute lymphoblastic leukemia. Haematologica 91 (1): 24-31, 2006.[PUBMED Abstract]
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.[PUBMED Abstract]
- Asselin BL, Whitin JC, Coppola DJ, et al.: Comparative pharmacokinetic studies of three asparaginase preparations. J Clin Oncol 11 (9): 1780-6, 1993.[PUBMED Abstract]
- Avramis VI, Sencer S, Periclou AP, et al.: A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 99 (6): 1986-94, 2002.[PUBMED Abstract]
- Tram Henriksen L, Gottschalk Højfeldt S, Schmiegelow K, et al.: Prolonged first-line PEG-asparaginase treatment in pediatric acute lymphoblastic leukemia in the NOPHO ALL2008 protocol-Pharmacokinetics and antibody formation. Pediatr Blood Cancer 64 (12): , 2017.[PUBMED Abstract]
- Jeha S, Pei D, Choi J, et al.: Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol 37 (35): 3377-3391, 2019.[PUBMED Abstract]
- van der Sluis IM, Vrooman LM, Pieters R, et al.: Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica 101 (3): 279-85, 2016.[PUBMED Abstract]
- Bleyer A, Asselin BL, Koontz SE, et al.: Clinical application of asparaginase activity levels following treatment with pegaspargase. Pediatr Blood Cancer 62 (6): 1102-5, 2015.[PUBMED Abstract]
- Tong WH, Pieters R, Kaspers GJ, et al.: A prospective study on drug monitoring of PEGasparaginase and Erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood 123 (13): 2026-33, 2014.[PUBMED Abstract]
- Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.[PUBMED Abstract]
- Li RJ, Jin R, Liu C, et al.: FDA Approval Summary: Calaspargase Pegol-mknl For Treatment of Acute Lymphoblastic Leukemia in Children and Young Adults. Clin Cancer Res 26 (2): 328-331, 2020.[PUBMED Abstract]
- Angiolillo AL, Schore RJ, Devidas M, et al.: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children's Oncology Group Study AALL07P4. J Clin Oncol 32 (34): 3874-82, 2014.[PUBMED Abstract]
- Vrooman LM, Blonquist TM, Supko JG, et al.: Efficacy and toxicity of pegaspargase and calaspargase pegol in childhood acute lymphoblastic leukemia/lymphoma: results of DFCI 11-001. [Abstract] J Clin Oncol 37 (Suppl 15): A-10006, 2019. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Salzer WL, Asselin B, Supko JG, et al.: Erwinia asparaginase achieves therapeutic activity after pegaspargase allergy: a report from the Children's Oncology Group. Blood 122 (4): 507-14, 2013.[PUBMED Abstract]
- Vrooman LM, Kirov II, Dreyer ZE, et al.: Activity and Toxicity of Intravenous Erwinia Asparaginase Following Allergy to E. coli-Derived Asparaginase in Children and Adolescents With Acute Lymphoblastic Leukemia. Pediatr Blood Cancer 63 (2): 228-33, 2016.[PUBMED Abstract]
- Escherich G, Zimmermann M, Janka-Schaub G, et al.: Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr Blood Cancer 60 (2): 254-7, 2013.[PUBMED Abstract]
- Pui CH, Sandlund JT, Pei D, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood 104 (9): 2690-6, 2004.[PUBMED Abstract]
- Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.[PUBMED Abstract]
- Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.[PUBMED Abstract]
- Prucker C, Attarbaschi A, Peters C, et al.: Induction death and treatment-related mortality in first remission of children with acute lymphoblastic leukemia: a population-based analysis of the Austrian Berlin-Frankfurt-Münster study group. Leukemia 23 (7): 1264-9, 2009.[PUBMED Abstract]
- Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.[PUBMED Abstract]
- Silverman LB, Gelber RD, Young ML, et al.: Induction failure in acute lymphoblastic leukemia of childhood. Cancer 85 (6): 1395-404, 1999.[PUBMED Abstract]
- Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.[PUBMED Abstract]
- Gaynon PS, Desai AA, Bostrom BC, et al.: Early response to therapy and outcome in childhood acute lymphoblastic leukemia: a review. Cancer 80 (9): 1717-26, 1997.[PUBMED Abstract]
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.[PUBMED Abstract]
- Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.[PUBMED Abstract]
- van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.[PUBMED Abstract]
- Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.[PUBMED Abstract]
- Conter V, Bartram CR, Valsecchi MG, et al.: Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 115 (16): 3206-14, 2010.[PUBMED Abstract]
- Wood B, Wu D, Crossley B, et al.: Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 131 (12): 1350-1359, 2018.[PUBMED Abstract]
- Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.[PUBMED Abstract]
- Coustan-Smith E, Sancho J, Behm FG, et al.: Prognostic importance of measuring early clearance of leukemic cells by flow cytometry in childhood acute lymphoblastic leukemia. Blood 100 (1): 52-8, 2002.[PUBMED Abstract]
- Basso G, Veltroni M, Valsecchi MG, et al.: Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol 27 (31): 5168-74, 2009.[PUBMED Abstract]
- Schrappe M, Valsecchi MG, Bartram CR, et al.: Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118 (8): 2077-84, 2011.[PUBMED Abstract]
- Karsa M, Dalla Pozza L, Venn NC, et al.: Improving the identification of high risk precursor B acute lymphoblastic leukemia patients with earlier quantification of minimal residual disease. PLoS One 8 (10): e76455, 2013.[PUBMED Abstract]
- Nachman JB, Sather HN, Sensel MG, et al.: Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med 338 (23): 1663-71, 1998.[PUBMED Abstract]
- Seibel NL, Steinherz PG, Sather HN, et al.: Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 111 (5): 2548-55, 2008.[PUBMED Abstract]
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.[PUBMED Abstract]
- Veerman AJ, Hählen K, Kamps WA, et al.: High cure rate with a moderately intensive treatment regimen in non-high-risk childhood acute lymphoblastic leukemia. Results of protocol ALL VI from the Dutch Childhood Leukemia Study Group. J Clin Oncol 14 (3): 911-8, 1996.[PUBMED Abstract]
- Chauvenet AR, Martin PL, Devidas M, et al.: Antimetabolite therapy for lesser-risk B-lineage acute lymphoblastic leukemia of childhood: a report from Children's Oncology Group Study P9201. Blood 110 (4): 1105-11, 2007.[PUBMED Abstract]
- Gustafsson G, Kreuger A, Clausen N, et al.: Intensified treatment of acute childhood lymphoblastic leukaemia has improved prognosis, especially in non-high-risk patients: the Nordic experience of 2648 patients diagnosed between 1981 and 1996. Nordic Society of Paediatric Haematology and Oncology (NOPHO) Acta Paediatr 87 (11): 1151-61, 1998.[PUBMED Abstract]
- Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.[PUBMED Abstract]
- Mahoney DH, Shuster JJ, Nitschke R, et al.: Intensification with intermediate-dose intravenous methotrexate is effective therapy for children with lower-risk B-precursor acute lymphoblastic leukemia: A Pediatric Oncology Group study. J Clin Oncol 18 (6): 1285-94, 2000.[PUBMED Abstract]
- Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.[PUBMED Abstract]
- Silverman LB, Gelber RD, Dalton VK, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97 (5): 1211-8, 2001.[PUBMED Abstract]
- Pession A, Valsecchi MG, Masera G, et al.: Long-term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukemia. J Clin Oncol 23 (28): 7161-7, 2005.[PUBMED Abstract]
- Coustan-Smith E, Sancho J, Hancock ML, et al.: Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood 100 (7): 2399-402, 2002.[PUBMED Abstract]
- Stow P, Key L, Chen X, et al.: Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood 115 (23): 4657-63, 2010.[PUBMED Abstract]
- Gaynon PS, Angiolillo AL, Carroll WL, et al.: Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children's Oncology Group Report. Leukemia 24 (2): 285-97, 2010.[PUBMED Abstract]
- Riehm H, Gadner H, Henze G, et al.: Results and significance of six randomized trials in four consecutive ALL-BFM studies. Hamatol Bluttransfus 33: 439-50, 1990.[PUBMED Abstract]
- Hutchinson RJ, Gaynon PS, Sather H, et al.: Intensification of therapy for children with lower-risk acute lymphoblastic leukemia: long-term follow-up of patients treated on Children's Cancer Group Trial 1881. J Clin Oncol 21 (9): 1790-7, 2003.[PUBMED Abstract]
- Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.[PUBMED Abstract]
- Maloney KW, Devidas M, Wang C, et al.: Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0331. J Clin Oncol 38 (6): 602-612, 2020.[PUBMED Abstract]
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.[PUBMED Abstract]
- Schrappe M, Bleckmann K, Zimmermann M, et al.: Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J Clin Oncol 36 (3): 244-253, 2018.[PUBMED Abstract]
- Pui CH, Mahmoud HH, Rivera GK, et al.: Early intensification of intrathecal chemotherapy virtually eliminates central nervous system relapse in children with acute lymphoblastic leukemia. Blood 92 (2): 411-5, 1998.[PUBMED Abstract]
- Mattano LA, Sather HN, Trigg ME, et al.: Osteonecrosis as a complication of treating acute lymphoblastic leukemia in children: a report from the Children's Cancer Group. J Clin Oncol 18 (18): 3262-72, 2000.[PUBMED Abstract]
- Aricò M, Valsecchi MG, Conter V, et al.: Improved outcome in high-risk childhood acute lymphoblastic leukemia defined by prednisone-poor response treated with double Berlin-Frankfurt-Muenster protocol II. Blood 100 (2): 420-6, 2002.[PUBMED Abstract]
- Steinherz PG, Seibel NL, Sather H, et al.: Treatment of higher risk acute lymphoblastic leukemia in young people (CCG-1961), long-term follow-up: a report from the Children's Oncology Group. Leukemia 33 (9): 2144-2154, 2019.[PUBMED Abstract]
- Mattano LA, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012.[PUBMED Abstract]
- Lipshultz SE, Scully RE, Lipsitz SR, et al.: Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol 11 (10): 950-61, 2010.[PUBMED Abstract]
- Barry EV, Vrooman LM, Dahlberg SE, et al.: Absence of secondary malignant neoplasms in children with high-risk acute lymphoblastic leukemia treated with dexrazoxane. J Clin Oncol 26 (7): 1106-11, 2008.[PUBMED Abstract]
- Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016.[PUBMED Abstract]
- Schultz KR, Pullen DJ, Sather HN, et al.: Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109 (3): 926-35, 2007.[PUBMED Abstract]
- Schrauder A, Reiter A, Gadner H, et al.: Superiority of allogeneic hematopoietic stem-cell transplantation compared with chemotherapy alone in high-risk childhood T-cell acute lymphoblastic leukemia: results from ALL-BFM 90 and 95. J Clin Oncol 24 (36): 5742-9, 2006.[PUBMED Abstract]
- Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007.[PUBMED Abstract]
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.[PUBMED Abstract]
- Conter V, Valsecchi MG, Parasole R, et al.: Childhood high-risk acute lymphoblastic leukemia in first remission: results after chemotherapy or transplant from the AIEOP ALL 2000 study. Blood 123 (10): 1470-8, 2014.[PUBMED Abstract]
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.[PUBMED Abstract]
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.[PUBMED Abstract]
- Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.[PUBMED Abstract]
- Bhatia S, Landier W, Hageman L, et al.: 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 124 (15): 2345-53, 2014.[PUBMED Abstract]
- Schmiegelow K, Glomstein A, Kristinsson J, et al.: Impact of morning versus evening schedule for oral methotrexate and 6-mercaptopurine on relapse risk for children with acute lymphoblastic leukemia. Nordic Society for Pediatric Hematology and Oncology (NOPHO). J Pediatr Hematol Oncol 19 (2): 102-9, 1997 Mar-Apr.[PUBMED Abstract]
- Clemmensen KK, Christensen RH, Shabaneh DN, et al.: The circadian schedule for childhood acute lymphoblastic leukemia maintenance therapy does not influence event-free survival in the NOPHO ALL92 protocol. Pediatr Blood Cancer 61 (4): 653-8, 2014.[PUBMED Abstract]
- Landier W, Hageman L, Chen Y, et al.: Mercaptopurine Ingestion Habits, Red Cell Thioguanine Nucleotide Levels, and Relapse Risk in Children With Acute Lymphoblastic Leukemia: A Report From the Children's Oncology Group Study AALL03N1. J Clin Oncol 35 (15): 1730-1736, 2017.[PUBMED Abstract]
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.[PUBMED Abstract]
- Andersen JB, Szumlanski C, Weinshilboum RM, et al.: Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr 87 (1): 108-11, 1998.[PUBMED Abstract]
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.[PUBMED Abstract]
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.[PUBMED Abstract]
- Zhou H, Li L, Yang P, et al.: Optimal predictor for 6-mercaptopurine intolerance in Chinese children with acute lymphoblastic leukemia: NUDT15, TPMT, or ITPA genetic variants? BMC Cancer 18 (1): 516, 2018.[PUBMED Abstract]
- Escherich G, Richards S, Stork LC, et al.: Meta-analysis of randomised trials comparing thiopurines in childhood acute lymphoblastic leukaemia. Leukemia 25 (6): 953-9, 2011.[PUBMED Abstract]
- Broxson EH, Dole M, Wong R, et al.: Portal hypertension develops in a subset of children with standard risk acute lymphoblastic leukemia treated with oral 6-thioguanine during maintenance therapy. Pediatr Blood Cancer 44 (3): 226-31, 2005.[PUBMED Abstract]
- De Bruyne R, Portmann B, Samyn M, et al.: Chronic liver disease related to 6-thioguanine in children with acute lymphoblastic leukaemia. J Hepatol 44 (2): 407-10, 2006.[PUBMED Abstract]
- Vora A, Mitchell CD, Lennard L, et al.: Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet 368 (9544): 1339-48, 2006.[PUBMED Abstract]
- Jacobs SS, Stork LC, Bostrom BC, et al.: Substitution of oral and intravenous thioguanine for mercaptopurine in a treatment regimen for children with standard risk acute lymphoblastic leukemia: a collaborative Children's Oncology Group/National Cancer Institute pilot trial (CCG-1942). Pediatr Blood Cancer 49 (3): 250-5, 2007.[PUBMED Abstract]
- Stork LC, Matloub Y, Broxson E, et al.: Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: report of the Children's Oncology Group CCG-1952 clinical trial. Blood 115 (14): 2740-8, 2010.[PUBMED Abstract]
- Felice MS, Rossi JG, Gallego MS, et al.: No advantage of a rotational continuation phase in acute lymphoblastic leukemia in childhood treated with a BFM back-bone therapy. Pediatr Blood Cancer 57 (1): 47-55, 2011.[PUBMED Abstract]
- Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007.[PUBMED Abstract]
- Pui CH, Ribeiro RC, Hancock ML, et al.: Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 325 (24): 1682-7, 1991.[PUBMED Abstract]
- Bleyer WA, Sather HN, Nickerson HJ, et al.: Monthly pulses of vincristine and prednisone prevent bone marrow and testicular relapse in low-risk childhood acute lymphoblastic leukemia: a report of the CCG-161 study by the Childrens Cancer Study Group. J Clin Oncol 9 (6): 1012-21, 1991.[PUBMED Abstract]
- Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children. Childhood ALL Collaborative Group. Lancet 347 (9018): 1783-8, 1996.[PUBMED Abstract]
- Eden TO, Pieters R, Richards S, et al.: Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia - an individual patient data meta-analysis involving 5,659 children. Br J Haematol 149 (5): 722-33, 2010.[PUBMED Abstract]
- Conter V, Valsecchi MG, Silvestri D, et al.: Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-risk acute lymphoblastic leukaemia: a multicentre randomised trial. Lancet 369 (9556): 123-31, 2007.[PUBMED Abstract]
- De Moerloose B, Suciu S, Bertrand Y, et al.: Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood 116 (1): 36-44, 2010.[PUBMED Abstract]
- Strauss AJ, Su JT, Dalton VM, et al.: Bony morbidity in children treated for acute lymphoblastic leukemia. J Clin Oncol 19 (12): 3066-72, 2001.[PUBMED Abstract]
- Warris LT, van den Heuvel-Eibrink MM, Aarsen FK, et al.: Hydrocortisone as an Intervention for Dexamethasone-Induced Adverse Effects in Pediatric Patients With Acute Lymphoblastic Leukemia: Results of a Double-Blind, Randomized Controlled Trial. J Clin Oncol 34 (19): 2287-93, 2016.[PUBMED Abstract]
- Bhatia S, Landier W, Hageman L, et al.: Systemic Exposure to Thiopurines and Risk of Relapse in Children With Acute Lymphoblastic Leukemia: A Children's Oncology Group Study. JAMA Oncol 1 (3): 287-95, 2015.[PUBMED Abstract]
- Landier W, Chen Y, Hageman L, et al.: Comparison of self-report and electronic monitoring of 6MP intake in childhood ALL: a Children's Oncology Group study. Blood 129 (14): 1919-1926, 2017.[PUBMED Abstract]
- 小児ALLに対するCNSに向けた治療
-
CNSに向けた治療レジメンの概要
診断時に患者の約3%は、中枢神経系3(CNS3)疾患(脳脊髄液[CSF]検体にリンパ芽球を含む白血球[WBC]が5個/μL以上認められ、かつ/または脳神経麻痺が存在する)である。しかしながら、CNSに対して特異的な治療を実施しない限り、初回診断時の脊髄液にリンパ芽球が検出されるかどうかにかかわらず、最終的にほとんどの小児が顕性のCNS白血病を発症する。したがって、急性リンパ芽球性白血病(ALL)患児はすべて、全身性多剤併用化学療法とともに、何らかのCNS予防療法を受ける必要がある。
CNSは聖域部位(すなわち、典型的にALLの治療に用いられる全身性投与の化学療法薬の多くが浸透しにくい解剖学的空間)であるため、すべての患者で診断時に臨床的に明らかなCNS病変を除去し、CNS再燃を防ぐために、治療の早期に特定のCNSに向けた治療法を策定しなければならない。歴史的に、CNSに向けた治療法が治療レジメンに追加されてからは、ALL患児の生存率は劇的に改善した。
CNSに向けた治療の標準治療法の選択肢には以下のものがある:
これらの治療法のすべてがCNS白血病の治療および予防に役立っている。髄腔内化学療法とCNSに向けた全身化学療法を合わせた併用療法が標準である;頭蓋照射療法は、厳選した状況でのみ使用される。[ 1 ]
使用されるCNS療法の種類は、患者のCNS疾患再燃リスクに基づき、リスクが高い患者ほど強力な治療を受ける。以下の患者グループは、CNS再燃のリスクが高いことがデータから示唆されている:
新規診断小児ALLに対するCNSに向けた治療レジメンを表11に示している。
表11.新規診断小児ALLに対するCNSに向けた治療レジメン 病態 標準治療法の選択肢 ALL = 急性リンパ芽球性白血病;CNS = 中枢神経系;CNS3 = 脳脊髄液で白血球が5/μL以上、サイトスピンで芽球陽性、または脳神経麻痺。 a 薬物自体はCNSに浸透しないが、脳脊髄液のアスパラギン枯渇をもたらす。 標準リスクのALL 髄腔内化学療法 メトトレキサート単独 メトトレキサートにシタラビンとヒドロコルチゾンを併用 CNSに向けた全身化学療法 デキサメタゾン L-アスパラギナーゼa 高用量メトトレキサートにロイコボリン救援を併用 漸増用量の静脈内投与メトトレキサート(ロイコボリン救援を伴わない) 高リスクおよび超高リスクのALL 髄腔内化学療法 メトトレキサート単独 メトトレキサートにシタラビンとヒドロコルチゾンを併用 CNSに向けた全身化学療法 デキサメタゾン L-アスパラギナーゼa 高用量メトトレキサートにロイコボリン救援を併用 頭蓋照射療法 現在のALL臨床試験の主な目的は、神経毒性およびその他の晩期合併症(晩期障害)を最小限に抑えながら、有効なCNS療法を提供することにある。
髄腔内化学療法
小児ALLに対するすべての治療レジメンに髄腔内化学療法が含まれる。髄腔内化学療法は通常、寛解導入相の初めから開始し、地固め相で強化するが、多くのプロトコルで維持相を通して継続している。
髄腔内化学療法は、典型的に以下のいずれかにより構成される:[ 5 ]
- メトトレキサート単独。
- メトトレキサートにシタラビンとヒドロコルチゾンを併用(トリプル髄腔内化学療法)。
髄腔内シタラビン投与とは異なり、髄腔内メトトレキサート投与には有意な全身的効果があり、骨髄再燃の防止に寄与する可能性がある。[ 6 ]
CNSに向けた全身化学療法
脳および脊髄液に直接施行される治療に加えて、全身的な薬剤投与もCNS発症を効果的に予防する重要な手段である。以下の全身的に投与される薬剤により、ある程度のCNS予防効果が得られる:
証拠(CNSに向けた全身化学療法):
- 標準リスクの患者を対象としたランダム化Children's Cancer Group(CCG)研究では、すべての患者が同一の用量とスケジュールで髄腔内メトトレキサート投与を受け、頭蓋照射は受けなかったが、経口デキサメタゾン投与に伴い、経口プレドニゾン投与と比較してCNS再燃率に50%の低下が認められた。[ 7 ]
- 別の標準リスクALLの試験(COG-1991)では、ロイコボリン救援を伴わない漸増用量のIVメトトレキサートにより、2回の中間維持相のそれぞれで投与する標準低量経口メトトレキサートと比べ、CNS再燃率が有意に減少した。[ 8 ]
- 旧Pediatric Oncology Groupにより実施されたランダム化臨床試験では、T-ALL患者に高用量のメトトレキサートを投与することで、高用量のメトトレキサートを投与しなかった患者よりもCNS再燃率が有意に低いことが認められた。[ 9 ]
頭蓋照射療法
頭蓋照射療法を受ける患者の割合は次第に著しく低下してきている。現在では、新たにALLと診断された小児のほとんどが、頭蓋照射療法を使用しないで治療を受けている。診断時にCNS白血病(前述の定義の通り)(芽球を伴うWBCが5/μL以上;CNS3)と認定された患者および/または初診時のWBC数が多いT細胞表現型の患者など、治療後のCNS再燃リスクが最も高いとみなされる患者に対してのみ頭蓋照射療法を施行しているグループが多い。[ 10 ]実際に放射線療法を施行する患者では、頭蓋照射線量を大幅に低くしており、脊髄照射の実施は非標準である。
実施中の試験では、生存に悪影響を与えたり、毒性の発生率が増加したりすることなく、初期療法から救援療法まで、新たにALLと診断されたすべての小児の治療から放射線照射療法を省略することができるかどうかを判定しようとしている。[ 11 ][ 12 ]CNSに向けた治療のランダム化試験を対象としたメタアナリシスでは、新たにALLと診断された患者のほとんどで放射線療法が髄腔内化学療法に置き換えられることが確認されている。使用した薬剤および強度にもよるが、全身性治療の追加が必要になる場合がある。[ 13 ];[ 1 ][証拠レベル:1iDi]
標準リスクの患者に対するCNS療法
頭蓋照射療法を併用しない髄腔内化学療法に加え、適切な場合に全身化学療法を施行することで、標準リスクのALL小児ではCNS再燃率が5%未満となる。[ 11 ][ 12 ][ 14 ][ 15 ][ 16 ][ 17 ]
これらの患者に対する頭蓋照射療法の使用はCNS向けの治療法に必要な要素ではない。[ 18 ][ 19 ]トリプル髄腔内化学療法(メトトレキサート、シタラビン、およびヒドロコルチゾン)を使用しているレジメンもあるが、治療を通して髄腔内メトトレキサートのみを使用しているレジメンもある。
証拠(トリプル髄腔内化学療法 vs 髄腔内メトトレキサート):
- 米国国立がん研究所(NCI)標準リスクの患者を対象としたCCG-1952研究では、放射線照射を受けなかった患者における単独の髄腔内投与薬剤としてのメトトレキサートに対して、トリプル髄腔内化学療法(メトトレキサート、シタラビン、およびヒドロコルチゾン)の相対的な有効性および毒性が比較された。[
20
]
- CNS毒性または非CNS毒性のいずれについても有意差は認められなかった。
- トリプル髄腔内化学療法に伴って、孤立性CNS再燃率(髄腔内メトトレキサートの5.9%±1.2%と比較して3.4%±1.0%;P = 0.004)が低下したが、イベントフリー生存(EFS)に差は認められなかった。
- 2群における神経認知機能の追跡研究では、臨床的有意差は認められなかった。[ 23 ][証拠レベル:1iiC]
CNS病変のない高リスクおよび超高リスク患者に対するCNS療法
高リスクおよび超高リスク患者では頭蓋照射療法による治療を実施すべきであるという点に関しては異論があるが、これらの患者のほとんどで頭蓋照射療法は必要ないというコンセンサスが得られつつある。[ 13 ]一部の治療レジメンで頭蓋照射療法が適応となるのは以下の患者である:[ 10 ]
放射線療法を受ける患者の割合および照射される放射線量は、いずれも過去20年間にわたって減少している。
証拠(頭蓋照射療法):
- 1990年から1995年に実施された試験で、ベルリン-フランクフルト-ミュンスター(BFM)グループは、線量を減弱した放射線(18Gyの代わりに12Gy)を予防照射することで、高リスクの患者において効果的なCNS予防法が得られることを示した。[ 24 ]
- 1995年から2000年にBFMグループが実施した追跡試験(BFM-95)では、T細胞表現型、初期反応の遅延(ステロイドによる1週間の前治療後の末梢血芽球数により判定)、および/または有害な細胞遺伝学的異常を認める患者を含め、約20%の患者(以前の試験では70%の患者)に対して頭蓋照射療法が施行された。[ 17 ]
- St. Jude Children's Research Hospital(SJCRH)、Dutch Childhood Oncology Group(DCOG)、およびEuropean Organization for Research and Treatment of Cancer(EORTC)を含むいくつかのグループが、高リスクのサブセットを含むすべての患者に対して頭蓋照射療法を省略した試験の結果を公表している。[ 11 ][ 12 ][ 25 ]これらの試験のほとんどが寛解導入後の地固め療法期間に高用量メトトレキサートを4回以上投与し、髄腔内化学療法の頻度を高めている。SJCRHおよびDCOGの研究でも、高頻度のビンクリスチン/デキサメタゾンの律動的投与および強化用量のpegaspargaseが含まれていたが[ 11 ][ 12 ]、EORTC試験では、CNS3の患者(CSFにWBCが5個/μL以上みられ、遠沈で芽球陽性)に対して寛解導入後治療相で高用量のメトトレキサートの追加投与および高用量のシタラビンの反復投与が含まれていた。 [ 25 ]
- 10組の共同グループにより1996年から2007年に治療を受けた16,000人を超える患者から併合したデータのメタアナリシスにおいて、頭蓋照射療法の使用は5年OS率または何らかのイベントの累積発生率に影響を及ぼさないと考えられた。[ 13 ]
診断時にCNS3の病態を示す患者に対するCNS療法
診断時に臨床的に明らかなCNS病変(高倍率視野当たりWBCが5個以上、サイトスピンで芽球陽性;CNS3)を認めるALL患者に対する治療には、典型的に髄腔内化学療法と頭蓋照射療法(通常の線量は18Gy)が含まれる。[ 17 ][ 19 ]現在。脊髄照射は使用されていない。
証拠(頭蓋照射療法):
- SJCRH、DCOG、およびEORTCは、高リスクのサブセットを含むすべての患者に対して頭蓋照射療法を省略した試験の結果を公表している。[ 11 ][ 25 ]これらの試験には、寛解導入後の地固め療法期間に高用量メトトレキサートを4回以上投与すること、および髄腔内化学療法の頻度を高めることが含まれている。SJCRHの研究には、小児腫瘍学グループ(COG)試験より累積用量が高いアントラサイクリン系薬剤に加え、頻回のビンクリスチン/デキサメタゾンの律動的投与および強化用量のpegaspargaseも含まれていたが[ 11 ]、EORTC試験には、CNS3(CSFにWBCが5個/μL以上、遠沈で芽球陽性)の患者に対する寛解導入後治療相で高用量メトトレキサートの追加投与および高用量シタラビンの反復投与が含まれていた。 [ 25 ]
- 10組の共同グループにより1996年から2007年に治療を受けた16,000人を超える患者から併合したデータのメタアナリシスでは、高リスク患者のサブセットで頭蓋照射療法の使用により転帰に影響が現れるかどうかが評価された。[ 13 ]
CNS3の患者において頭蓋照射療法を省略することによる安全性を完全に解明するには、大規模なプロスペクティブ研究が必要である。
臨床評価段階にある発症前CNS療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
- SJCRH Total 17研究(TOT17;NCT03117751)(ALLまたはリンパ腫患者の治療における併用化学療法):患者に対して髄腔内化学療法および大量メトトレキサートを併用し、放射線療法は使用しない。高リスクの特徴を示す特定の患者では、強化髄腔内療法を実施する。
- DFCI ALL 16-001(NCT03020030)(ALLの小児および青年に対してより優れた治療法選択肢を特定するリスク分類スキーム):診断時にCNS3状態の患者(5%未満の患者)のみが頭蓋照射療法(18Gy)を受ける。その他のすべての患者では、髄腔内化学療法および大量メトトレキサートを併用し、放射線療法は使用しない。T-ALLの患者は、継続相で過剰用量の髄腔内化学療法を受ける。
CNSに向けた治療の毒性
小児ALLに対するCNSに向けた治療法の毒性作用は、急性および亜急性または晩期発現の可能性がある。(詳しい情報については、小児がん治療の晩期合併症(晩期障害)に関するPDQ要約の中枢神経系の晩期合併症(晩期障害)のセクションを参照のこと。)
急性および亜急性毒性
髄腔内化学療法の単独施行に伴って最も多くみられる急性の副作用は痙攣発作である。髄腔内化学療法薬を頻繁に投与する治療を受け、放射線療法を受けていないALL患児では、治療中に少なくとも1回の痙攣発作が最大5%にみられる。[ 11 ]髄腔内化学療法に加えて、中用量の静脈内(IV)メトトレキサート(1g/m2)を2週間ごとに投与する12コースを含む地固め療法レジメンで、痙攣発作の高い発生率が観察された。[ 26 ]髄腔内および高用量IVメトトレキサートに伴って、ほとんどの場合に可逆性とみられる脳卒中様症候群も認められている。[ 27 ]
治療中に痙攣発作を起こしたALL患児および抗痙攣薬療法を受けているALL患児に対して抗痙攣薬治療としてフェノパルビタールまたはフェニトインを投与すると、一部の化学療法薬のクリアランスを増大させ、転帰に有害な影響を及ぼす可能性があるため投与すべきではない。[ 28 ]ガバペンチンおよびバルプロ酸は代用可能な坑痙攣薬で、酵素誘導能が低い。[ 28 ]
遅発性毒性
CNSに向けた治療法に伴う晩期合併症(晩期障害)には、二次腫瘍、神経内分泌障害、白質脳症、および神経認知的障害がある。
二次腫瘍は、主に頭蓋照射療法を受けた生存者に観察される。髄膜腫が多くみられ、典型的に悪性化の可能性は低いが、高悪性度の病変も発生する。再燃したことのない1,290人を超えるALL患者を対象としたSJCRHのレトロスペクティブ研究で、CNSに発生した二次腫瘍の30年累積発生率は3%であった;髄膜腫を除くと、30年累積発生率は1.17%であった。[ 29 ]これらのCNS二次腫瘍のほぼすべてが過去に放射線照射を受けた患者に発生した。
神経認知的障害は、重症度および機能的帰結に幅があり、放射線療法の有無を問わず治療を受けた長期ALL生存者で明らかになっている。一般に、頭蓋照射療法なしで治療を受けた患者は、頭蓋照射療法を受けた患者よりも重度の神経認知的後遺症が少なく、実際に発生する障害は、いくつかの限定された領域において比較的軽度の神経心理学的機能低下をもたらす。[ 30 ][ 31 ][ 32 ][ 33 ]頭蓋照射療法を受けた患者では、毒性の頻度および重症度が用量依存性であると考えられる;頭蓋照射療法で線量が18Gyであった患者では、線量が24Gy以上の治療を受けた患者と比較して重度障害のリスクが低いと考えられた。診断時年齢が若い、および女性であることが神経認知的晩期合併症(晩期障害)の高いリスクに関係することが多くの研究で報告されている。[ 34 ]
数件の研究で神経認知的晩期障害の発生に対する他の治療要素の影響も評価されている。メトトレキサート vs トリプル髄腔内化学療法による治療を受けた患者を対象に神経認知機能の転帰を比較した場合、臨床的意義のある差は示されなかった。[ 23 ][証拠レベル:3iiiC]デキサメタゾンの投与を受けている患者では神経認知的障害のリスクが高いかどうかという問題に関しては見解の一致をみていない。[ 35 ]放射線照射を受けていない長期生存者を対象にしたSJCRH研究で、デキサメタゾンによる治療は、注意および実行機能の障害の高いリスクと関連していた。[ 36 ]対照的に、標準リスクのALLの既往があり、治療中にデキサメタゾンまたはプレドニゾンのいずれかの投与を受けた92人の小児を対象とした長期の神経認知機能の検証では、コルチコステロイドによるランダム化に基づいた認知機能において何らかの意義のある差は示されなかった。[ 37 ]
証拠(頭蓋照射の神経認知的晩期合併症[晩期障害]):
- 小児ALLの成人までの長期生存者567人(診断からの平均期間、26年)を対象としたSJCRH研究では、神経認知機能検査を実施した。[ 36 ]
- 18Gyの頭蓋照射療法を受けた患者(n = 127) vs 24Gyの頭蓋照射療法を受けた患者(n = 138)で、記憶障害を比較した研究がある。[ 38 ]
- 標準リスクALL患者の放射線(線量18Gy)照射群と非照射群を比較したランダム化試験で、以下が観察された;[ 30 ][証拠レベル:1iiC]
- あるランダム化試験において、多分割照射療法(線量は18Gy)では、従来の分割照射療法と比較して神経学的晩期障害が低下しなかった;両群とも認知機能に重大な障害はみられなかった。[ 39 ]
証拠(放射線照射を受けていない患者における神経認知的晩期合併症[晩期障害]):
参考文献- Richards S, Pui CH, Gayon P, et al.: Systematic review and meta-analysis of randomized trials of central nervous system directed therapy for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 60 (2): 185-95, 2013.[PUBMED Abstract]
- Mahmoud HH, Rivera GK, Hancock ML, et al.: Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 329 (5): 314-9, 1993.[PUBMED Abstract]
- Bürger B, Zimmermann M, Mann G, et al.: Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 21 (2): 184-8, 2003.[PUBMED Abstract]
- Gajjar A, Harrison PL, Sandlund JT, et al.: Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 96 (10): 3381-4, 2000.[PUBMED Abstract]
- Pullen J, Boyett J, Shuster J, et al.: Extended triple intrathecal chemotherapy trial for prevention of CNS relapse in good-risk and poor-risk patients with B-progenitor acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 11 (5): 839-49, 1993.[PUBMED Abstract]
- Thyss A, Suciu S, Bertrand Y, et al.: Systemic effect of intrathecal methotrexate during the initial phase of treatment of childhood acute lymphoblastic leukemia. The European Organization for Research and Treatment of Cancer Children's Leukemia Cooperative Group. J Clin Oncol 15 (5): 1824-30, 1997.[PUBMED Abstract]
- Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.[PUBMED Abstract]
- Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.[PUBMED Abstract]
- Asselin BL, Devidas M, Wang C, et al.: Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118 (4): 874-83, 2011.[PUBMED Abstract]
- Pui CH, Howard SC: Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol 9 (3): 257-68, 2008.[PUBMED Abstract]
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.[PUBMED Abstract]
- Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.[PUBMED Abstract]
- Vora A, Andreano A, Pui CH, et al.: Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 34 (9): 919-26, 2016.[PUBMED Abstract]
- Pui CH, Sandlund JT, Pei D, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood 104 (9): 2690-6, 2004.[PUBMED Abstract]
- Tubergen DG, Gilchrist GS, O'Brien RT, et al.: Prevention of CNS disease in intermediate-risk acute lymphoblastic leukemia: comparison of cranial radiation and intrathecal methotrexate and the importance of systemic therapy: a Childrens Cancer Group report. J Clin Oncol 11 (3): 520-6, 1993.[PUBMED Abstract]
- Conter V, Aricò M, Valsecchi MG, et al.: Extended intrathecal methotrexate may replace cranial irradiation for prevention of CNS relapse in children with intermediate-risk acute lymphoblastic leukemia treated with Berlin-Frankfurt-Münster-based intensive chemotherapy. The Associazione Italiana di Ematologia ed Oncologia Pediatrica. J Clin Oncol 13 (10): 2497-502, 1995.[PUBMED Abstract]
- Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.[PUBMED Abstract]
- Clarke M, Gaynon P, Hann I, et al.: CNS-directed therapy for childhood acute lymphoblastic leukemia: Childhood ALL Collaborative Group overview of 43 randomized trials. J Clin Oncol 21 (9): 1798-809, 2003.[PUBMED Abstract]
- Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.[PUBMED Abstract]
- Matloub Y, Lindemulder S, Gaynon PS, et al.: Intrathecal triple therapy decreases central nervous system relapse but fails to improve event-free survival when compared with intrathecal methotrexate: results of the Children's Cancer Group (CCG) 1952 study for standard-risk acute lymphoblastic leukemia, reported by the Children's Oncology Group. Blood 108 (4): 1165-73, 2006.[PUBMED Abstract]
- Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.[PUBMED Abstract]
- Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.[PUBMED Abstract]
- Kadan-Lottick NS, Brouwers P, Breiger D, et al.: Comparison of neurocognitive functioning in children previously randomly assigned to intrathecal methotrexate compared with triple intrathecal therapy for the treatment of childhood acute lymphoblastic leukemia. J Clin Oncol 27 (35): 5986-92, 2009.[PUBMED Abstract]
- Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.[PUBMED Abstract]
- Sirvent N, Suciu S, Rialland X, et al.: Prognostic significance of the initial cerebro-spinal fluid (CSF) involvement of children with acute lymphoblastic leukaemia (ALL) treated without cranial irradiation: results of European Organization for Research and Treatment of Cancer (EORTC) Children Leukemia Group study 58881. Eur J Cancer 47 (2): 239-47, 2011.[PUBMED Abstract]
- Mahoney DH, Shuster JJ, Nitschke R, et al.: Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: an association with intermediate-dose intravenous methotrexate and intrathecal triple therapy--a Pediatric Oncology Group study. J Clin Oncol 16 (5): 1712-22, 1998.[PUBMED Abstract]
- Bhojwani D, Sabin ND, Pei D, et al.: Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J Clin Oncol 32 (9): 949-59, 2014.[PUBMED Abstract]
- Relling MV, Pui CH, Sandlund JT, et al.: Adverse effect of anticonvulsants on efficacy of chemotherapy for acute lymphoblastic leukaemia. Lancet 356 (9226): 285-90, 2000.[PUBMED Abstract]
- Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007.[PUBMED Abstract]
- Waber DP, Turek J, Catania L, et al.: Neuropsychological outcomes from a randomized trial of triple intrathecal chemotherapy compared with 18 Gy cranial radiation as CNS treatment in acute lymphoblastic leukemia: findings from Dana-Farber Cancer Institute ALL Consortium Protocol 95-01. J Clin Oncol 25 (31): 4914-21, 2007.[PUBMED Abstract]
- Jansen NC, Kingma A, Schuitema A, et al.: Neuropsychological outcome in chemotherapy-only-treated children with acute lymphoblastic leukemia. J Clin Oncol 26 (18): 3025-30, 2008.[PUBMED Abstract]
- Espy KA, Moore IM, Kaufmann PM, et al.: Chemotherapeutic CNS prophylaxis and neuropsychologic change in children with acute lymphoblastic leukemia: a prospective study. J Pediatr Psychol 26 (1): 1-9, 2001 Jan-Feb.[PUBMED Abstract]
- Copeland DR, Moore BD, Francis DJ, et al.: Neuropsychologic effects of chemotherapy on children with cancer: a longitudinal study. J Clin Oncol 14 (10): 2826-35, 1996.[PUBMED Abstract]
- von der Weid N, Mosimann I, Hirt A, et al.: Intellectual outcome in children and adolescents with acute lymphoblastic leukaemia treated with chemotherapy alone: age- and sex-related differences. Eur J Cancer 39 (3): 359-65, 2003.[PUBMED Abstract]
- Waber DP, Carpentieri SC, Klar N, et al.: Cognitive sequelae in children treated for acute lymphoblastic leukemia with dexamethasone or prednisone. J Pediatr Hematol Oncol 22 (3): 206-13, 2000 May-Jun.[PUBMED Abstract]
- Krull KR, Brinkman TM, Li C, et al.: Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J Clin Oncol 31 (35): 4407-15, 2013.[PUBMED Abstract]
- Kadan-Lottick NS, Brouwers P, Breiger D, et al.: A comparison of neurocognitive functioning in children previously randomized to dexamethasone or prednisone in the treatment of childhood acute lymphoblastic leukemia. Blood 114 (9): 1746-52, 2009.[PUBMED Abstract]
- Armstrong GT, Reddick WE, Petersen RC, et al.: Evaluation of memory impairment in aging adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiotherapy. J Natl Cancer Inst 105 (12): 899-907, 2013.[PUBMED Abstract]
- Waber DP, Silverman LB, Catania L, et al.: Outcomes of a randomized trial of hyperfractionated cranial radiation therapy for treatment of high-risk acute lymphoblastic leukemia: therapeutic efficacy and neurotoxicity. J Clin Oncol 22 (13): 2701-7, 2004.[PUBMED Abstract]
- Jacola LM, Krull KR, Pui CH, et al.: Longitudinal Assessment of Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated on a Contemporary Chemotherapy Protocol. J Clin Oncol 34 (11): 1239-47, 2016.[PUBMED Abstract]
- 特定のALLサブグループに対する寛解導入後療法
-
T-ALL
歴史的に見て、T-急性リンパ芽球性白血病(ALL)の患者の予後は、B-ALLの小児より不良である。小児腫瘍学グループ(COG)試験で治療を受けた多数の患者を15年にわたって追跡したレビューによると、多変量解析でT細胞免疫表現型は依然として不良な予後因子であることが明らかにされた。[ 1 ]しかしながら、現行の治療レジメンにより、T-ALLの小児の転帰は、B-ALLの小児で達成できる転帰に現在接近してきている。例えば、Dana Farber Cancer Institute(DFCI)のDFCI-95001(NCT00004034)試験で治療を受けたT-ALL患児では、10年全生存(OS)率が90.1%であったのに対して、B-ALL患児では88.7%であった。[ 2 ]その他の例として、T-ALLに対するCOG試験(AALL0434 [NCT00408005])で、5年イベントフリー生存(EFS)率が83.8%、OS率が89.5%であった。[ 3 ]
T-ALLに対する治療法の選択肢
T-ALLに対する治療法の選択肢には以下のものがある:
- 化学療法および予防的頭蓋照射療法。
証拠(化学療法および予防的放射線療法):
- 旧Pediatric Oncology Group(POG)のプロトコルでは、B-ALL患児と異なる方法でT-ALL患児の治療を実施した。T-ALL患児を対象としたPOG-9404プロトコルは、大量メトトレキサート療法の役割を評価することを目的としてデザインされたものである。このプロトコルの多剤併用化学療法レジメンは、DFCI-87001レジメンに基づいていた。[ 4 ]
- POG-9404研究では、デクスラゾキサン併用または非併用にてドキソルビシンを投与する群に患者がランダムに割り付けられ、晩期の心臓死を予防するデクスラゾキサンの有効性が判定された。[ 6 ][証拠レベル:1iiDi]
- 旧Children's Cancer Group(CCG)のプロトコルで、B-ALL患児と同じ治療レジメンでT-ALL患児が治療を受けた;プロトコルおよび治療割り付けは、患者の臨床的特徴(例えば、年齢および白血球[WBC]数)および初期療法への疾患の反応に基づいていた。T-ALL患児のほとんどが米国国立がん研究所(NCI)の高リスク基準を満たしていた。
- COGでは、T-ALL患児に対してB-ALL患児と同じプロトコルによる治療を実施していない。
- COGによるパイロット研究では、新たにT-ALLと診断された患者に対してBFMレジメンが使用されている状況下で、ネララビン(再燃および難治性のT細胞リンパ芽球性疾患の患者に効果が実証されているヌクレオシドアナログ)を組み込む実施可能性が実証されている。[ 12 ][ 13 ][ 14 ]
- COGのAALL0434(NCT00408005)試験では、T-ALL患者に対して強化BFMレジメンによる治療を実施し、ロイコボリン救援を伴う大量メトトレキサートまたはロイコボリン救援を伴わない漸増用量のメトトレキサート(Capizzi方式)のいずれかに患者をランダムに割り付けた。[ 3 ]ほぼすべての患者に対して、予防的(12Gy)または治療的(18Gy)頭蓋照射が施行された;低リスクと考えられる患者のわずか10%が照射を受けなかった。Capizzi方式のメトトレキサート群に割り付けられた患者は、高用量メトトレキサート群に割り付けられた患者より早期(8週目 vs 26週目)に頭蓋照射療法を受けた。Capizzi方式のメトトレキサート群の患者は、pegaspargaseの2回の追加投与も受けた。結果は以下の通りであった:[ 3 ]
T-ALL患者の治療における予防的な頭蓋照射療法の使用は減少している。St. Jude Children's Research Hospital(SJCRH)およびDutch Childhood Oncology Group(DCOG)など、一部のグループでは、ALLの第一選択治療に頭蓋照射療法を使用しておらず、他のDFCI、COG、およびBFMなどのグループでは、現時点で超高リスクの特徴またはCNS3疾患を認める患者に放射線療法を制限している。
T-ALLに対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
ALL乳児
乳児のALLはまれであり、小児ALL症例の約2~4%を占める。[ 16 ]ALLの乳児は特有な生物学的特徴を示し、白血病の再発リスクが高いことから、この患者集団に対して特化してデザインされたプロトコルによる治療を実施する。ALL乳児の治療に用いられる強化化学療法レジメンの一般的な治療のテーマは、大量のシタラビンおよびメトトレキサートを用いた寛解導入後強化コースを含めることである。[ 17 ][ 18 ][ 19 ]
生後数ヵ月以内に診断された乳児の転帰は特に不良である。1件の研究では、生後1ヵ月以内に診断された患者の2年OS率は20%であった。[ 20 ][証拠レベル:2A]別の研究で、生後90日未満で診断された乳児の5年EFS率は16%であった。[ 19 ][証拠レベル:2A]
KMT2A(MLL)遺伝子再構成が認められる乳児では、4~5年でのEFS率が35%の範囲に留まっている。[ 17 ][ 18 ][ 19 ][ 21 ][証拠レベル:2A]KMT2A再構成を有する乳児で不良な転帰を予測する因子には以下のものがある:[ 18 ][ 19 ];[ 22 ][証拠レベル:3iDii];[ 23 ][証拠レベル:2A]
乳児は、より年齢の高いALL小児よりも再燃率が有意に高く、治療関連毒性、特に感染症を発症するリスクが高い。この集団に対して現在の治療アプローチを用いた場合、治療関連死は乳児の約10%に発生すると報告されており、この割合は、より年齢の高いALL小児よりもはるかに高い。[ 18 ][ 19 ]COG AALL0631(NCT00557193)試験では、強化された寛解導入レジメンによる寛解導入中死亡率が15.4%(患者26人中4人)であった;この試験はその後、強度を弱めた寛解導入と高度支持療法のガイドラインを含めるように修正され、寛解導入中死亡率が有意に低下し(1.6%、患者123人中2人)、有意に高い完全寛解(CR)率(94% vs 以前のより強度の高い寛解導入レジメンでは68%)が得られた。[ 24 ]
KMT2A再構成を有する乳児に対する治療法選択肢
KMT2A遺伝子再構成を有する乳児では、一般に年長のALL小児に対する初期治療で典型的に採用されない薬剤を用いた強化化学療法レジメンで治療を実施する。しかしながら、このような強化アプローチにもかかわらず、これらの患者ではEFS率が依然として劣っている。
証拠(KMT2A再構成を認める乳児に対する強化化学療法レジメン):
- 国際共同Interfant-99試験では、シタラビン強化化学療法レジメンを用い、治療の最初の数ヵ月間で低量および大量にかかわらずシタラビンに対する曝露を高めた。[ 18 ]
- COGは、大量メトトレキサート、シクロホスファミド、およびエトポシドの反復投与を含むレジメンによる治療の強化を検討した。[ 17 ]
- COG P9407(NCT00002756)試験では、乳児が短期(46週間)の強化化学療法レジメンで治療された。[ 19 ][証拠レベル:2A]
- 国際共同Interfant-06研究では、急性骨髄性白血病(AML)スタイルの地固め化学療法の方がALLスタイルの化学療法より優れているかどうかが検証された。[ 23 ][証拠レベル:2A]
KMT2A遺伝子再構成を認める乳児における第一寛解期の同種造血幹細胞移植(HSCT)の役割については、依然として意見の相違がある。
証拠(KMT2A再構成を認める乳児に対する第一寛解期における同種HSCT):
- 1998年から2002年に実施された日本の臨床試験で、KMT2A再構成を認めるすべての乳児は、診断から3~5ヵ月後に利用可能性が最も高いドナー(血縁者、非血縁者、または臍帯血)からの同種HSCTを受けるようになっていた。[ 25 ]
- 1996年から2000年にCCGまたはPOGによる乳児向けALLプロトコルで治療を受けた乳児189人を含むCOGの報告では、第一CR期にHSCTを受けた患者と化学療法のみを受けた患者の間で、EFSに差は認められなかった。[ 26 ]
- Interfantの臨床試験グループは、Interfant-99試験で第一CR期の同種HSCTまたは化学療法単独のいずれかによる治療を受けたKMT2A再構成を認める高リスク(プレドニゾン反応により判定)の乳児において、移植待機期間について調整した後でもDFSに差を認めなかった。[ 18 ]
- Interfant-06研究で、高リスク(以下のすべてを満たす:KMT2A再構成、年齢が生後6ヵ月未満、およびWBCが300,000/μL以上)とみなされた乳児は、第一CR期での同種HSCTに適格と判断された。[ 23 ][証拠レベル:2A]
第一CR期に移植を受けるALLの乳児では、全身照射(TBI)を行わないレジメンとTBIを基本としたレジメンで転帰はほぼ同じと考えられる。[ 26 ][ 28 ]
KMT2A再構成を認めない乳児に対する治療法選択肢
KMT2A再構成を認めない乳児に対する至適治療法も、年長の小児で使用される標準のALLレジメンの使用に関するデータが不足していることが1つの要因となり、依然として明らかになっていない。
- Interfant-99試験で、KMT2A再構成を認めない患者では、シタラビン強化療法レジメンを使用することで、比較的良好な転帰が達成された(4年EFS率が74%)。[ 18 ]
- 強化化学療法に関するCOG P9407(NCT00002756)試験では、KMT2A再構成を認めない乳児の5年EFS率が70%であったと報告された。[ 19 ][証拠レベル:2A]
- この患者サブセットの良好な転帰は、より年長のALL小児の治療に使用されるものと同様な治療法を用いた日本の研究で得られた[ 21 ];しかしながら、この研究は、被験者が少数(n = 22)で、性別分布が異常に偏っていた(男性が91%)ことから限界があった。
- Interfant-06研究では、KMT2A再構成を認めない乳児の6年EFS率が73.9%で、OS率が87.2%であった。[ 23 ][証拠レベル:2A]
ALL乳児に対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
- AALL15P1(NCT02828358)(KMT2A遺伝子再構成が認められるALLの乳児の治療におけるアザシチジンおよび併用化学療法):このCOGプロトコルは、Interfantによる化学療法の基本骨格へのアザシチジン(DNA脱メチル化薬)追加の実施可能性を検証している非ランダム化パイロット研究である。生後12ヵ月未満でB細胞ALLまたは細胞系列があいまいな急性白血病を新たに診断された患者が登録に適格とされている。患者は4週間の多剤併用導入相から治療が開始される。寛解導入後、KMT2A再構成が認められない乳児は、寛解導入相終了時に治療を中止したが、KMT2A再構成が認められる乳児は、本研究で治療を継続し、Interfant化学療法の基本骨格における寛解導入後化学療法の各主要ブロックの直前に、エピジェネティック・プライミングとしてアザシチジンによる5日間の治療コースを4回受けた。この試験の主要目的は、Interfantによる化学療法の基本骨格にアザシチジンを安全に組み込むことができるかどうかを明らかにすることである。
ALLの青年および若年成人
ALLの青年および若年成人は、数十年にわたって高リスクとみなされてきた。ほぼすべての治療研究において、この年齢層の転帰は10歳未満の小児と比べて不良である。[ 29 ][ 30 ][ 31 ]この違いにおける原因として、診断時に以下を含む有害な予後因子を示す頻度が高いことが挙げられる:
有害な予後因子の頻度が高いことに加え、この年齢層の患者では、治療関連死亡[ 30 ][ 31 ][ 32 ][ 33 ]および治療に対する不遵守の割合が高くなっている。[ 32 ][ 34 ]
青年および若年成人のALL患者に対する治療法の選択肢
米国およびフランスの研究は、治療レジメンに基づいて転帰の差を特定した最初のものである。[ 35 ]他の研究により、年長の青年および若年成人の患者は、成人向けのレジメンよりむしろ小児向けのレジメンで経過が良好なことが確認されている。[ 35 ][ 36 ][ 37 ][ 38 ][ 39 ][ 40 ][ 41 ][ 42 ];[ 43 ][証拠レベル:2A]これらの研究結果を表12に要約している。
これらの患者では、高リスク小児ALLに使用される化学療法レジメンで比較的良好な転帰が達成できることを考えると、ALLの青年および若年成人に対して第一寛解期における同種HSCTのルーチン使用が果たす役割はない。[ 31 ]
証拠(ALLの青年および若年成人に対する小児治療レジメンの使用):
- CALGB-10403(NCT00558519)試験では、ALLを新たに診断された青年および若年成人患者に対して(内科腫瘍医が実施する)小児治療レジメン使用の実施可能性と効力がプロスペクティブに研究された。登録された318人の患者のうち、295人が適格で、反応について評価可能であった。年齢中央値は24歳(範囲、17~39歳)であった。[ 44 ]
- 研究者らは、CCG研究(小児ALLレジメン)で治療を受けた16~21歳の患者197人について、Cancer and Leukemia Group B(CALGB)研究(成人ALLレジメン)で治療を受けた青年および若年成人124人と比較して報告した。[ 35 ]
- カナダの集団ベース・コホート研究で、ALLの青年および若年成人に適応させた小児プロトコルの有効性が20年の期間で判定された。[ 45 ]
青年および若年成人では、小児向けレジメンにより達成される転帰が優れている理由は不明であるが、考えられる説明として以下のものが挙げられる:[ 36 ]
表12.ALLの青年および若年成人に対する治療プロトコルよる転帰 施設および研究グループ 青年および若年成人の患者数 平均年齢(歳) 生存率(%) ALL = 成人急性リンパ芽球性白血病;EFS = イベントフリー生存;OS = 全生存。 AIEOP = Associazione Italiana di Ematologia e Oncologia Pediatrica;CALGB = Cancer and Leukemia Group B;CCG = Children's Cancer Group;DCOG = Dutch Childhood Oncology Group;FRALLE = French Acute Lymphoblastic Leukaemia Study Group;GIMEMA = Gruppo Italiano Malattie EMatologiche dell'Adulto;HOVON = Dutch-Belgian Hemato-Oncology Cooperative Group;LALA = France-Belgium Group for Lymphoblastic Acute Leukemia in Adults;MRC = Medical Research Council(英国);NOPHO = Nordic Society for Pediatric Hematology and Oncology;UKALL = United Kingdom Acute Lymphoblastic Leukaemia。 米国 [ 35 ] CCG(小児) 197 16 7年OS率で、67 CALGB(成人) 124 19 46 フランス [ 40 ] FRALLE 93(小児) 77 16 EFS率で、67 LALA 94 100 18 41 イタリア [ 46 ] AIEOP(小児) 150 15 2年OS率で、80 GIMEMA(成人) 95 16 71 オランダ [ 47 ] DCOG(小児) 47 12 EFS率で、71 HOVON 44 20 38 スウェーデン [ 48 ] NOPHO 92(小児) 36 16 5年OS率で、74 成人ALL 99 18 39 英国 [ 38 ] MRC ALL(小児) 61 15–17 5年OS率で、71 UKALL XII(成人) 67 15–17 56 UKALL 2003 [ 49 ] 229 16–24 EFS率で、72 青年および若年成人のALL患者に対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
- A041501(NCT03150693)(新たにB細胞ALLと診断された若年成人の治療におけるイノツズマブ・オゾガマイシンおよびフロントライン化学療法):これは、ALLの若年成人を対象に小児向け化学療法(pediatric-inspired chemotherapy)骨格の使用経験をさらに拡大するNational Clinical Trials Network試験である。適格性には、年齢が18~39歳で、新たにCD22陽性のALLと診断された患者が含まれる。寛解導入後に寛解に達した患者を地固め療法の開始前に、小児向け治療骨格に加え、イノツズマブ・オゾガマイシン(毒素結合抗CD22モノクローナル抗体)の2コースを併用する群または併用しない群にランダムに割り付ける。
- COG-AALL1521(NCT02723994)(ALLの小児を対象とした化学療法と併用するルキソリチニブの第II相研究):これは、B-ALLの治療に対する標準の多剤併用化学療法レジメンと併用するルキソリチニブの非ランダム化研究である。本研究のパート1では、化学療法レジメンと併用する治験薬(ルキソリチニブ)の最適用量を決定する。パート2では、パート1で決定された推奨用量で化学療法とルキソリチニブの併用療法の有効性を評価する。
- COG-AALL1721(NCT03876769)(高リスクB-ALLで地固め療法終了後のMRDが陽性の患者を対象としたtisagenlecleucelの有効性および安全性に関する研究):本研究の目的は、地固め療法終了時のMRDが陽性の患者を対象にCD19キメラ抗原受容体(CAR)T細胞療法(tisagenlecleucel)の有効性について5年EFS率の測定により評価することである。その他の目的としては、1年で同種移植なしに無病状態である被験者の割合、OS、およびtisagenlecleucel後の3ヵ月でMRD陰性のCRまたはCRIに達する被験者の割合がある。
- COG-AALL1731(NCT03914625)(標準リスクB-ALL向け治療を施行した場合の限局性B細胞リンパ芽球性リンパ腫患者の転帰を判定する研究):この研究では、標準化学療法にブリナツモマブを追加することで、DFSが改善するかどうかが検証される。すべてのダウン症候群患者(青年および31歳未満の若年成人患者を含む)が登録に適格である。ダウン症候群で高リスクの特徴を示す患者は、強化治療要素を省略した化学療法の基本骨格に加えてブリナツモマブを投与するように非ランダム的に割り付けられる。ダウン症候群で高リスクの特徴を示さない患者は、ブリナツモマブ併用または非併用の化学療法へのランダム化に適格となる。Murphy病期がI期およびII期のB細胞リンパ芽球性リンパ腫の患者は、ブリナツモマブなしの標準のB-ALL療法を受ける。
- COG-AALL1732(NCT03959085)(新たに診断された高リスクB-ALLに対するイノツズマブ オゾガマイシンの第III相ランダム化試験;高リスクB-ALL、混合表現型急性白血病[MPAL]、播種性B-リンパ芽球性リンパ腫に対するリスク調整寛解導入後療法):このプロトコルは、次の診断のいずれかを満たす診断時年齢が25歳未満の患者を対象としている:非ダウン症候群のNCI高リスクB-ALL、MPAL、および播種性B-リンパ芽球性リンパ腫。B-ALL患者について、このプロトコルでは修正BFMの基本骨格への2ブロックのイノツズマブ オゾガマイシンの追加により、DFSが改善するかどうかが検証されている。MPALおよび播種性B-リンパ芽球性リンパ腫患者について、研究では、標準の高リスクB-ALL修正BFM基本骨格を用いる治療でのEFSを明らかにすることを目的としている。
フィラデルフィア染色体陽性(BCR-ABL1陽性)ALL
フィラデルフィア染色体陽性(Ph+)ALLは、小児ALL症例の約3%にみられ、青年ではこれより多く、成人では15~25%にみられる。過去に、このALL亜型はきわめて治療が困難で転帰不良であるとみなされてきた。2000年に国際小児白血病グループは、7年EFS率が25%で、OS率が36%であることを報告した。[ 53 ]2010年に同グループは、チロシンキナーゼ阻害薬を使用しない治療を受けた場合のPh+ ALL患者で7年EFS率が31%、OS率が44%であることを報告した。[ 54 ]このサブグループの治療は、積極的な化学療法に重点を置いたものから骨髄移植に進展しており、現在では化学療法にチロシンキナーゼ阻害薬を加えた併用療法に進歩している。
Ph+ ALL患者に対する治療法の選択肢
Ph+ ALL患者に対する標準の治療法には、第一CR期での同種HSCTの有無を問わず、細胞毒性化学療法と併用するチロシンキナーゼ阻害薬(例、イマチニブまたはダサチニブ)の使用がある。
メシル酸イマチニブは、BCR-ABLプロテインキナーゼの選択的阻害薬である。再燃または難治性のPh+ ALLの小児および成人を対象としたイマチニブ単剤の第I相および第II相研究により、奏効率は比較的高いが、これらの反応は持続期間が短い傾向にあることが示されている。[ 55 ][ 56 ]
Ph+ ALLの成人および小児における複数の臨床試験により、多剤併用化学療法と併用するメシル酸イマチニブ投与の可能性が示唆されている。[ 57 ][ 58 ][ 59 ]Ph+ ALL患者では、イマチニブを移植の前後に投与した場合、HSCT後の転帰が良好なことが明らかになった。[ 60 ][ 61 ][ 62 ][ 63 ][ 64 ]Ph+ ALLの小児患者の多くは、移植を受けるよりも、化学療法とチロシンキナーゼ阻害薬の併用により同程度のEFSが得られることが臨床試験でも実証されている。[ 64 ][ 65 ]
Ph+ ALLの治療において第二世代のチロシンキナーゼ阻害薬であるダサチニブも研究されている。ダサチニブは、マウスモデルでも、CNS陽性の白血病患者を対象としたシリーズでも、CNSにおいて顕著な効果を示している。[ 66 ]小児患者におけるダサチニブの第I相試験の結果によると、1日1回の投与でグレード3または4の非血液学的有害事象はほとんどなく、毒性プロファイルは受け入れられるものであることが示された。[ 67 ]
証拠(チロシンキナーゼ阻害薬):
- Ph+ ALLの小児患者30人を対象にした1件のレトロスペクティブ研究(1991年から2004年までは19人の患者がチロシンキナーゼ阻害薬なしの治療を受け、2004年から2012年までは11人の患者がイマチニブまたはダサチニブのいずれかによる治療を受けた)によると、チロシンキナーゼ阻害薬を寛解導入療法の途中で開始した場合に、寛解導入療法終了時のMRDレベルが低くなることが示された。[ 68 ]
- COG-AALL0031研究では、Ph+ ALLの小児に対する強化化学療法レジメンにメシル酸イマチニブを組み込めるかどうかが評価された。患者は、寛解導入後療法中に化学療法とともにメシル酸イマチニブの投与を受けた。メシル酸イマチニブを併用した2サイクルの地固め化学療法後に同種HSCTに進む患者もいたが、全治療期間にわたって化学療法と併用してメシル酸イマチニブの投与を受ける患者もいた。[ 59 ][ 64 ]
- COG-AALL0622(NCT00720109)研究では、COG-AALL0031で使用されたものと類似の化学療法骨格とのダサチニブの併用が検証された。[ 69 ][証拠レベル:2A]この試験では、寛解導入療法の15日目にダサチニブの投与が開始され、寛解導入期間が完了するまでイマチニブが開始されなかったAALL0031と比較すると、CR率が高く、寛解導入終了時のMRDが少ない患者の割合が高いという結果が得られた。
- EsPhALL2004試験では、強化化学療法と合わせてイマチニブを投与(不連続的に投与)することで、小児Ph+ ALL患者の転帰が改善するかどうかが検証されたが、患者のほとんど(80%)が第一CR期に同種HSCTを受けた。寛解導入療法終了時点で早期反応判定および寛解状態に基づいて、患者は良好リスクまたは不良リスクのいずれかに分類された。良好リスクの患者(n = 90)は、イマチニブ投与群またはイマチニブ非投与群にランダムに割り付けられた;不良リスクの患者(n = 70)は、イマチニブによる治療に直接割り付けられた。この研究では、良好リスク患者におけるランダム化割り付けで非コンプライアンス率が高く、イマチニブが化学療法と併用して連続的に投与されているCOG AALL0031試験の結果が公表されたことで、目標の登録数に達するまでに早期に閉鎖されたことから、解釈が限定される。[ 65 ]
- その後のEsPhALL2010(NCT00287105)試験は2004試験への修正の結果であり、寛解導入療法15日目にイマチニブ療法をより早期に開始し、治療終了または移植後1年までイマチニブの投与が継続された。この試験のその後の修正ではまた、第一CR期のHSCTの適応が不良リスクの患者のみに変更された。この結果、寛解導入療法終了時のCR率は(以前の試験の78%から)97%に増加し、HSCTに割り付けられる患者は少なくなった(修正された試験で38% vs 最初の試験で81%)。[ 71 ]
Ph+ ALLに対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
-
AALL1631(NCT03007147)(Ph+ ALLを新たに診断された患者の治療におけるメシル酸イマチニブと併用化学療法):AALL1631は、COGおよびEuropean EsPhALLグループにより実施された国際共同プロトコルである。Ph+ ALL患者は、寛解導入IAの15日目に試験に加わり、その時点でイマチニブ1日1回投与を開始する。寛解導入IB相(10~12週目)後に、免疫グロブリンH/T細胞受容体(IgH-TCR)PCRを用いてMRDが評価され、患者は標準リスク(MRD < 0.05%)または高リスク(MRD > 0.05%)に分類される。標準リスクの患者は、次の2種類の細胞毒性化学療法基本骨格のいずれかを受けるようにランダムに割り付けられる:
両群の標準リスクの患者は、計画されたすべての化学療法(2年間の治療)が完了するまでイマチニブを継続して投与される。標準リスクのランダム化の目的は、強度の弱い化学療法基本骨格は標準療法(EsPhALL化学療法基本骨格)と比較して同等のDFSが得られながら、治療関連毒性の割合を低く抑えられるかどうかを明らかにすることである。
高リスク患者(患者の約15~20%)は3つの地固め化学療法ブロック完了後にHSCTに進む。イマチニブはこの薬物のHSCT後の投与の実施可能性を検証するために、HSCT後に再開され、56日以降から365日以降まで投与され、この方法で治療された患者の転帰が報告される予定である。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Hunger SP, Lu X, Devidas M, et al.: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 30 (14): 1663-9, 2012.[PUBMED Abstract]
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.[PUBMED Abstract]
- Winter SS, Dunsmore KP, Devidas M, et al.: Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children's Oncology Group AALL0434 Methotrexate Randomization. J Clin Oncol 36 (29): 2926-2934, 2018.[PUBMED Abstract]
- LeClerc JM, Billett AL, Gelber RD, et al.: Treatment of childhood acute lymphoblastic leukemia: results of Dana-Farber ALL Consortium Protocol 87-01. J Clin Oncol 20 (1): 237-46, 2002.[PUBMED Abstract]
- Asselin BL, Devidas M, Wang C, et al.: Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118 (4): 874-83, 2011.[PUBMED Abstract]
- Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016.[PUBMED Abstract]
- Chow EJ, Asselin BL, Schwartz CL, et al.: Late Mortality After Dexrazoxane Treatment: A Report From the Children's Oncology Group. J Clin Oncol 33 (24): 2639-45, 2015.[PUBMED Abstract]
- Seibel NL, Asselin BL, Nachman JB, et al.: Treatment of high risk T-cell acute lymphoblastic leukemia (T-ALL): comparison of recent experience of the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG). [Abstract] Blood 104 (11): A-681, 2004.[PUBMED Abstract]
- Seibel NL, Steinherz PG, Sather HN, et al.: Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 111 (5): 2548-55, 2008.[PUBMED Abstract]
- Hastings C, Gaynon PS, Nachman JB, et al.: Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (≥200 × 10(9) /l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol 168 (4): 533-46, 2015.[PUBMED Abstract]
- Matloub Y, Stork L, Asselin B, et al.: Outcome of Children with Standard-Risk T-Lineage Acute Lymphoblastic Leukemia--Comparison among Different Treatment Strategies. Pediatr Blood Cancer 63 (2): 255-61, 2016.[PUBMED Abstract]
- Berg SL, Blaney SM, Devidas M, et al.: Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23 (15): 3376-82, 2005.[PUBMED Abstract]
- Kurtzberg J, Ernst TJ, Keating MJ, et al.: Phase I study of 506U78 administered on a consecutive 5-day schedule in children and adults with refractory hematologic malignancies. J Clin Oncol 23 (15): 3396-403, 2005.[PUBMED Abstract]
- Winter SS, Dunsmore KP, Devidas M, et al.: Safe integration of nelarabine into intensive chemotherapy in newly diagnosed T-cell acute lymphoblastic leukemia: Children's Oncology Group Study AALL0434. Pediatr Blood Cancer 62 (7): 1176-83, 2015.[PUBMED Abstract]
- Dunsmore KP, Devidas M, Linda SB, et al.: Pilot study of nelarabine in combination with intensive chemotherapy in high-risk T-cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. J Clin Oncol 30 (22): 2753-9, 2012.[PUBMED Abstract]
- Silverman LB: Acute lymphoblastic leukemia in infancy. Pediatr Blood Cancer 49 (7 Suppl): 1070-3, 2007.[PUBMED Abstract]
- Hilden JM, Dinndorf PA, Meerbaum SO, et al.: Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108 (2): 441-51, 2006.[PUBMED Abstract]
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.[PUBMED Abstract]
- Dreyer ZE, Hilden JM, Jones TL, et al.: Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer 62 (3): 419-26, 2015.[PUBMED Abstract]
- van der Linden MH, Valsecchi MG, De Lorenzo P, et al.: Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood 114 (18): 3764-8, 2009.[PUBMED Abstract]
- Tomizawa D, Koh K, Sato T, et al.: Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 21 (11): 2258-63, 2007.[PUBMED Abstract]
- Van der Velden VH, Corral L, Valsecchi MG, et al.: Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia 23 (6): 1073-9, 2009.[PUBMED Abstract]
- Pieters R, De Lorenzo P, Ancliffe P, et al.: Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated With the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J Clin Oncol 37 (25): 2246-2256, 2019.[PUBMED Abstract]
- Salzer WL, Jones TL, Devidas M, et al.: Decreased induction morbidity and mortality following modification to induction therapy in infants with acute lymphoblastic leukemia enrolled on AALL0631: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (3): 414-8, 2015.[PUBMED Abstract]
- Kosaka Y, Koh K, Kinukawa N, et al.: Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 104 (12): 3527-34, 2004.[PUBMED Abstract]
- Dreyer ZE, Dinndorf PA, Camitta B, et al.: Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the Children's Oncology Group. J Clin Oncol 29 (2): 214-22, 2011.[PUBMED Abstract]
- Mann G, Attarbaschi A, Schrappe M, et al.: Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood 116 (15): 2644-50, 2010.[PUBMED Abstract]
- Kato M, Hasegawa D, Koh K, et al.: Allogeneic haematopoietic stem cell transplantation for infant acute lymphoblastic leukaemia with KMT2A (MLL) rearrangements: a retrospective study from the paediatric acute lymphoblastic leukaemia working group of the Japan Society for Haematopoietic Cell Transplantation. Br J Haematol 168 (4): 564-70, 2015.[PUBMED Abstract]
- Nachman J: Clinical characteristics, biologic features and outcome for young adult patients with acute lymphoblastic leukaemia. Br J Haematol 130 (2): 166-73, 2005.[PUBMED Abstract]
- Pui CH, Pei D, Campana D, et al.: Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29 (4): 386-91, 2011.[PUBMED Abstract]
- Nachman JB, La MK, Hunger SP, et al.: Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children's oncology group. J Clin Oncol 27 (31): 5189-94, 2009.[PUBMED Abstract]
- Pichler H, Reismüller B, Steiner M, et al.: The inferior prognosis of adolescents with acute lymphoblastic leukaemia (ALL) is caused by a higher rate of treatment-related mortality and not an increased relapse rate--a population-based analysis of 25 years of the Austrian ALL-BFM (Berlin-Frankfurt-Münster) Study Group. Br J Haematol 161 (4): 556-65, 2013.[PUBMED Abstract]
- Burke MJ, Gossai N, Wagner JE, et al.: Survival differences between adolescents/young adults and children with B precursor acute lymphoblastic leukemia after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 19 (1): 138-42, 2013.[PUBMED Abstract]
- Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.[PUBMED Abstract]
- Stock W, La M, Sanford B, et al.: What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112 (5): 1646-54, 2008.[PUBMED Abstract]
- Ramanujachar R, Richards S, Hann I, et al.: Adolescents with acute lymphoblastic leukaemia: emerging from the shadow of paediatric and adult treatment protocols. Pediatr Blood Cancer 47 (6): 748-56, 2006.[PUBMED Abstract]
- Barry E, DeAngelo DJ, Neuberg D, et al.: Favorable outcome for adolescents with acute lymphoblastic leukemia treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol 25 (7): 813-9, 2007.[PUBMED Abstract]
- Ramanujachar R, Richards S, Hann I, et al.: Adolescents with acute lymphoblastic leukaemia: outcome on UK national paediatric (ALL97) and adult (UKALLXII/E2993) trials. Pediatr Blood Cancer 48 (3): 254-61, 2007.[PUBMED Abstract]
- Ram R, Wolach O, Vidal L, et al.: Adolescents and young adults with acute lymphoblastic leukemia have a better outcome when treated with pediatric-inspired regimens: systematic review and meta-analysis. Am J Hematol 87 (5): 472-8, 2012.[PUBMED Abstract]
- Boissel N, Auclerc MF, Lhéritier V, et al.: Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 21 (5): 774-80, 2003.[PUBMED Abstract]
- Huguet F, Leguay T, Raffoux E, et al.: Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol 27 (6): 911-8, 2009.[PUBMED Abstract]
- DeAngelo DJ, Stevenson KE, Dahlberg SE, et al.: Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia 29 (3): 526-34, 2015.[PUBMED Abstract]
- Ribera JM, Oriol A, Sanz MA, et al.: Comparison of the results of the treatment of adolescents and young adults with standard-risk acute lymphoblastic leukemia with the Programa Español de Tratamiento en Hematología pediatric-based protocol ALL-96. J Clin Oncol 26 (11): 1843-9, 2008.[PUBMED Abstract]
- Stock W, Luger SM, Advani AS, et al.: A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood 133 (14): 1548-1559, 2019.[PUBMED Abstract]
- Gupta S, Pole JD, Baxter NN, et al.: The effect of adopting pediatric protocols in adolescents and young adults with acute lymphoblastic leukemia in pediatric vs adult centers: An IMPACT Cohort study. Cancer Med 8 (5): 2095-2103, 2019.[PUBMED Abstract]
- Testi AM, Valsecchi MG, Conter V, et al.: Difference in outcome of adolescents with acute lymphoblastic leukemia (ALL) enrolled in pediatric (AIEOP) and adult (GIMEMA) protocols. [Abstract] Blood 104: A-1954, 2004.[PUBMED Abstract]
- de Bont JM, van der Holt B, Dekker AW, et al.: [Adolescents with acute lymphatic leukaemia achieve significantly better results when treated following Dutch paediatric oncology protocols than with adult protocols]. Ned Tijdschr Geneeskd 149 (8): 400-6, 2005.[PUBMED Abstract]
- Hallböök H, Gustafsson G, Smedmyr B, et al.: Treatment outcome in young adults and children >10 years of age with acute lymphoblastic leukemia in Sweden: a comparison between a pediatric protocol and an adult protocol. Cancer 107 (7): 1551-61, 2006.[PUBMED Abstract]
- Hough R, Rowntree C, Goulden N, et al.: Efficacy and toxicity of a paediatric protocol in teenagers and young adults with Philadelphia chromosome negative acute lymphoblastic leukaemia: results from UKALL 2003. Br J Haematol 172 (3): 439-51, 2016.[PUBMED Abstract]
- Mattano LA, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012.[PUBMED Abstract]
- Mogensen SS, Harila-Saari A, Mäkitie O, et al.: Comparing osteonecrosis clinical phenotype, timing, and risk factors in children and young adults treated for acute lymphoblastic leukemia. Pediatr Blood Cancer 65 (10): e27300, 2018.[PUBMED Abstract]
- Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.[PUBMED Abstract]
- Aricò M, Valsecchi MG, Camitta B, et al.: Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med 342 (14): 998-1006, 2000.[PUBMED Abstract]
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.[PUBMED Abstract]
- Champagne MA, Capdeville R, Krailo M, et al.: Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104 (9): 2655-60, 2004.[PUBMED Abstract]
- Ottmann OG, Druker BJ, Sawyers CL, et al.: A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood 100 (6): 1965-71, 2002.[PUBMED Abstract]
- Thomas DA, Faderl S, Cortes J, et al.: Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 103 (12): 4396-407, 2004.[PUBMED Abstract]
- Yanada M, Takeuchi J, Sugiura I, et al.: High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol 24 (3): 460-6, 2006.[PUBMED Abstract]
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.[PUBMED Abstract]
- Burke MJ, Trotz B, Luo X, et al.: Allo-hematopoietic cell transplantation for Ph chromosome-positive ALL: impact of imatinib on relapse and survival. Bone Marrow Transplant 43 (2): 107-13, 2009.[PUBMED Abstract]
- Lee S, Kim YJ, Min CK, et al.: The effect of first-line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 105 (9): 3449-57, 2005.[PUBMED Abstract]
- de Labarthe A, Rousselot P, Huguet-Rigal F, et al.: Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood 109 (4): 1408-13, 2007.[PUBMED Abstract]
- Rives S, Estella J, Gómez P, et al.: Intermediate dose of imatinib in combination with chemotherapy followed by allogeneic stem cell transplantation improves early outcome in paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (ALL): results of the Spanish Cooperative Group SHOP studies ALL-94, ALL-99 and ALL-2005. Br J Haematol 154 (5): 600-11, 2011.[PUBMED Abstract]
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.[PUBMED Abstract]
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.[PUBMED Abstract]
- Porkka K, Koskenvesa P, Lundán T, et al.: Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 112 (4): 1005-12, 2008.[PUBMED Abstract]
- Zwaan CM, Rizzari C, Mechinaud F, et al.: Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31 (19): 2460-8, 2013.[PUBMED Abstract]
- Jeha S, Coustan-Smith E, Pei D, et al.: Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 120 (10): 1514-9, 2014.[PUBMED Abstract]
- Slayton WB, Schultz KR, Kairalla JA, et al.: Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0622. J Clin Oncol 36 (22): 2306-2314, 2018.[PUBMED Abstract]
- Biondi A, Cario G, De Lorenzo P, et al.: Long-term follow up of pediatric Philadelphia positive acute lymphoblastic leukemia treated with the EsPhALL2004 study: high white blood cell count at diagnosis is the strongest prognostic factor. Haematologica 104 (1): e13-e16, 2019.[PUBMED Abstract]
- Biondi A, Gandemer V, De Lorenzo P, et al.: Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): a prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol 5 (12): e641-e652, 2018.[PUBMED Abstract]
- 再燃小児ALLの治療
-
小児ALLの初回再燃後の予後因子
急性リンパ芽球性白血病(ALL)が再発した小児の予後は、多くの因子に左右される。[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ][ 6 ][ 7 ][ 8 ][ 9 ][ 10 ][ 11 ][ 12 ][ 13 ][ 14 ];[ 15 ][証拠レベル:3iiDi]
小児ALLの初回再燃後に重要な以下の2つの危険因子は、予後および治療アプローチを決定する鍵となる:
その他の予後因子には以下のものがある:
再燃部位
孤立性髄外再燃の患者は、骨髄に浸潤した再燃の患者より経過が良好である。一部の研究によると、骨髄再燃/髄外再燃を併発した患者は、骨髄再燃のみの患者より予後が良好であった;しかしながら、他の研究ではこの知見を確認できていない。[ 5 ][ 13 ][ 16 ]
診断から再燃までの時間
B-ALLの再燃患者において、早期の再燃は遅発再燃よりも経過が不良であり、骨髄再燃は孤立性の髄外再燃よりも経過が不良である。例えば、生存率は、診断後18ヵ月以内に骨髄再燃を起こした患者の20%未満から、診断後36ヵ月を過ぎてから再燃を起こした患者の60%を超えるまでに及んでいる。[ 5 ][ 13 ][ 17 ]
孤立性中枢神経系(CNS)再燃を認める患者では、全生存(OS)率が、早期(診断から18ヵ月未満)再燃で40~50%、晩期(診断から18ヵ月以上)再燃で75~80%である。[ 13 ][ 18 ]治療を行っていない患者では、監視(全血球数または骨髄検査)の頻度を高め、再燃を早期に発見することにより転帰が改善するという証拠はない。[ 19 ]
患者特性
診断時および再燃時の年齢が10歳以上は、不良な転帰の独立した予測因子として報告されている。[ 13 ][ 16 ]さらに小児腫瘍学グループ(COG)の研究によると、初回診断時に10~15歳の患者は1~9歳の患者より予後が悪いが(再燃後3年生存率が35% vs 48%)、15歳を超える患者ではさらに悪い(3年OS率、15%;P = 0.001)ことが示された。[ 20 ]
18歳以下で診断され、晩期再燃がみられたB-ALL患者で、四分位別に解析した場合、年齢はその後の転帰の有意な予測因子ではなかった。しかしながら、再燃時点で18歳以上の患者の転帰は、18歳未満の年齢で再燃した患者の転帰に対して有意に劣っていた(39.5% vs 68.7%;P = 0.0001)。[ 21 ]
ベルリン-フランクフルト-ミュンスター(BFM)グループも、遅発性骨髄再燃の患者において再燃時の末梢血芽球数が多い(10,000/μLを超える)ことが不良な転帰と関連していたことを報告している。[ 10 ]
ALLが再燃したダウン症候群の小児では、一般に寛解導入中の死亡、治療関連死亡、および再燃の増加により転帰が劣っていた。
初回診断時のリスクグループ分類
COGは、初回診断時のリスクグループ分類は再燃後の予後に重要であることを報告した;初回診断時に米国国立がん研究所(NCI)の標準リスク基準を満たす患者は、NCIの高リスク患者よりも再燃後の予後が良好であった。[ 13 ]
再寛解導入療法に対する反応
再寛解導入療法開始から1ヵ月終了時点で形態学的に病変が残存している骨髄再燃の患者は、その後に第二完全寛解(CR)に達したとしても、予後がきわめて不良である。[ 24 ][証拠レベル:2Di];[ 25 ][証拠レベル:3iiiA]第二CR達成後の微小残存病変(MRD)レベルは再燃ALLにおいて予後的に重要なことがいくつかの研究で示されている。[ 21 ][ 24 ][ 26 ][ 27 ][ 28 ];[ 29 ][証拠レベル:3iiiDi]再寛解導入療法終了時およびその後の時点でMRDレベルが高いことは、その後のきわめて高い再燃リスクと関連している。[ 21 ]
細胞遺伝学的/ゲノム変化
診断から再燃までの期間で、変異プロファイルの変化が遺伝子塩基配列決定法で確認されている。[ 30 ][ 31 ]発がん遺伝子の融合(例、TCF3-PBX1、ETV6-RUNX1)は、初回診断時から再燃までにみられる一方で、一塩基多様体およびコピー数多様体は、診断時にみられることがあるが、再燃時にはみられず、その逆の場合もある。[ 30 ]例えば、RAS家族性変異は診断時と再燃時の両方で一般的にみられる一方、特定のRAS家族性変異は、特異的な白血病サブクローンが治療期間中に増加および減少するために、診断から再燃までの間に変化することがある。[ 30 ]対照的にNT5C2(ヌクレオチド代謝に関与する遺伝子)における再燃特異的な変異は、早期再燃がみられるALL症例の実に45%に示されている。[ 30 ][ 32 ][ 33 ]
TP53の変化(変異および/またはコピー数の変化)は、ALL患者の初回再燃時点で約10%に認められ、最初の再寛解導入療法後も白血病が持続する可能性が高いこと、およびイベントフリー生存(EFS)率が低いこととの関連が示されている。[ 21 ][ 34 ]ある研究で、TP53変化の約半数は初回診断時に認められていたもので、半数が再燃時に新たに確認されたものであった。[ 34 ]
IKZF1欠失でも、骨髄再燃を初めて来したB-ALL患者における予後不良との関連が報告されている。[ 35 ]しかしながら、晩期に最初の骨髄再燃が認められたB-ALL患者のBFM研究では、IKZF1欠失に予後的意義が認められなかった。[ 21 ]
RAS経路の変異(KRAS、NRAS、FLT3、およびPTPN11)は、B-ALL患者における再燃時によくみられ、206人の小児を対象にした1件の研究では、初回再燃時の患者の約40%に認められた。[ 30 ][ 36 ]診断時に観察されているように、再燃時のRAS経路の変異の頻度は細胞遺伝学的サブタイプによって異なる(例、高二倍体症例では頻度が高く、ETV6-RUNX1症例では頻度が低い)。再燃時のRAS経路の変異の存在は早期再燃と関連していた。しかしながら、再燃時のRAS経路の変異の存在は転帰の独立した予測因子ではなかった。
ETV6-RUNX1陽性ALL患者は初回再燃時の予後が比較的良好と考えられ、こうした患者では診断後36ヵ月以上経過してから再燃する割合が高いことと一致している。[ 35 ][ 37 ]
小児ALLの初回骨髄再燃に対する標準治療法の選択肢
初回骨髄再燃に対する標準治療法の選択肢には以下のものがある:
再寛解導入化学療法
再燃に対する最初の治療は、第二CRを達成するための再寛解導入療法である。4剤併用の再寛解導入レジメン(新規診断高リスク患者に投与されるレジメンと同様)または大量メトトレキサートおよび大量シタラビンを含む代替レジメンのいずれかを用いることで、骨髄再燃患者の約85%が治療開始1ヵ月時点で第二CRに達する。[ 5 ];[ 38 ][証拠レベル:2A];[ 24 ][証拠レベル:2Di]骨髄再燃が早い患者では、形態学的第二CRに達する割合(約70%)が、骨髄再燃が遅い患者(約95%)より低い。[ 24 ][ 38 ]
証拠(再寛解導入化学療法):
- COG研究では、HSCTまたは化学療法継続のいずれかを行う前に、最初にドキソルビシンを含む4剤併用療法を行った後に強化地固めブロックを2回行う3ブロックの強化再寛解導入療法を使用した。[ 24 ]
- 英国をベースとしたランダム化試験では、初回再燃ALL患者を対象に、4剤併用療法による再寛解導入療法として用いたイダルビシン vs ミトキサントロンを比較した。[
39
][証拠レベル:1iiA]
再燃ALLレジメンにおいてミトキサントロンから得られる可能性がある有益性については、さらに研究する必要がある。
- ALL-REZ BFMグループの研究者は、大量メトトレキサートを含む6剤併用アプローチを用いた。[ 40 ]
- クロファラビン、シクロホスファミド、およびエトポシドの併用により、難治性病態または多数回再燃した患者の42~56%に寛解が得られることが報告された。[ 41 ][ 42 ];[ 43 ][証拠レベル:2A]
- ボルテゾミブを追加したビンクリスチン、デキサメタゾン、pegaspargase、およびドキソルビシンの併用療法により、多数回再燃したB-ALL患者の70~80%に完全奏効(血小板回復の有無を問わない)が得られることが報告されている。[ 44 ][証拠レベル:3iiiA];[ 45 ][証拠レベル:3iiiDiv]
T-ALLが再燃した患者では、標準の再寛解導入レジメンにより第二CRに達する割合が、B細胞表現型を示す患者よりはるかに低い。[ 24 ]骨髄に初めてT-ALLの再燃が認められる小児に対するT細胞選択的薬剤ネララビンを用いた単剤療法による治療で、約50%の奏効率が得られている。[ 46 ]再燃/難治性のT-ALL患者では、ネララビン、シクロホスファミド、およびエトポシドの併用療法も使用されている。[ 47 ]
再寛解導入失敗は予後不良因子であるが、その後の寛解を得ようとする試みが成功し、HSCT後の生存につながる可能性があり、特にMRDが低値または検出不能になった場合に顕著である(MRDリスク層別化に関する詳しい情報については、本要約の遅発性再燃のB-ALLのセクションを参照のこと)。伝統的に、治療アプローチでは最初の試みとは異なる薬物の併用が用いられている;これらのレジメンには臨床試験で研究段階にある新たな薬物がしばしば含まれる。それぞれの試みの後で生存の可能性は次第に低くなるが、追加の試みはしばしば2~4回続行され、各試みの後に測定される成功の水準が減少していく。[ 48 ]キメラ抗原受容体(CAR)T細胞、ブリナツモマブ、およびイノツズマブの研究では、複数回再発した化学療法難治性のB-ALL患者で高い寛解率につながることが示されているため、これらの薬剤を初回再燃後に検証する試験が進行中である(詳しい情報については、本要約の難治性ALLに対する免疫療法アプローチのセクションを参照のこと)。
第二完全寛解に達した患者に対する再寛解導入療法
早発性再燃のB-ALL
早期に骨髄再燃を来したB-ALL患者では、HLA適合同胞ドナーまたは適合非血縁ドナーによる同種移植を第二寛解期に実施することで、化学療法アプローチより無白血病生存率が高くなることがほとんどの研究で報告されている。[ 7 ][ 29 ][ 49 ][ 50 ][ 51 ][ 52 ][ 53 ][ 54 ][ 55 ][ 56 ][ 57 ]しかし、移植をしたとしても、早期に骨髄再燃を来す患者の生存率は50%未満である。(詳しい情報については、本要約の初回およびその後の骨髄再燃に対する造血幹細胞移植のセクションを参照のこと。)
遅発性再燃のB-ALL
骨髄再燃が遅いB-ALL患者の過去の研究では、第二CR達成後の一次化学療法アプローチにより、約50%の生存率が得られることが報告されたが、同種移植により優れた治癒率につながるかどうかは明らかとなっていない。[ 5 ][ 9 ][ 39 ][ 58 ][ 59 ][ 60 ];[ 61 ][証拠レベル:3iiA]その後のデータでは、再寛解導入療法終了時のMRDの存在により、第二CR期で(HSCTではなく)化学療法単独による治療を受けた場合に再燃が確実となる高リスク患者が識別されることが示されている。再寛解導入療法終了時のMRDが多いものの骨髄再燃が遅い患者は、MRD状態が低値または検出不能に達した後の第二CR期に同種HSCTを受けた場合により良好な転帰が得られることが多くの研究で示されている。[ 17 ][ 62 ]
証拠(遅発性再燃のB-ALLに対するMRDベースのリスク層別化):
- St. Jude Children's Research Hospitalの研究には、第二CR期に化学療法を受けた晩期再燃の患者23人が含まれていた。[ 26 ]
- BFM研究で、患者が遅発性の孤立性骨髄再燃または早発性もしくは遅発性の骨髄/髄外併発型再燃であれば、中リスクとみなされる。このグループのALL-REZ BFM P95/96研究では、中リスクB-ALLが再燃した小児に対して化学療法単独(HSCTではない)による治療を実施して第二CRに達した場合、再寛解導入療法終了時のMRD(ポリメラーゼ連鎖反応をベースとした検査で評価)により転帰が有意に予測された。[ 28 ]
- その後のBFM研究(ALL-REZ BFM 2002 [NCT00114348])において、中リスクの再燃患者が治療開始から1ヵ月後におけるMRDレベルが高い場合に、同種HSCTに割り付けられた。再寛解導入療法終了時のMRDレベルが低い患者は化学療法単独(HSCTなし)で治療された。[ 62 ]
- 英国のALLR3試験では、第一選択治療の完了後6ヵ月を過ぎて再燃した患者を再寛解導入療法終了時のMRDが0.01%以上の場合にHSCTへ、またはMRDが0.01%未満の場合に化学療法へ割り付けた。[ 17 ]
2回目およびその後の骨髄再燃に対する治療法の選択肢
第三CR期またはその後のCR期の患者を対象に化学療法とHSCTを直接比較した研究はないが、化学療法単独による治癒はまれであるため、一般に寛解に達している患者に対して移植が妥当なアプローチとみなされてきている。2回目の再燃後のALL患者では、長期生存率が特に不良であり、10%未満から20%の範囲となっている。[ 55 ]この主な理由の1つは、第三寛解が得られないことである。新規併用療法アプローチが数多く検討されているにもかかわらず、2回目の再燃を来した小児が寛解を達成するのは約40%に過ぎない。[ 64 ]しかしながら、多数回再燃した難治性患者における標準の再寛解導入療法の薬物にボルテゾミブを追加した2件の研究で、70~80%のCR率が得られている。[ 44 ][証拠レベル:3iiiA];[ 45 ][証拠レベル:3iiiDiv]このような患者がCRに達したとしても、HSCTによる治癒率は20~35%で、再燃率および移植関連死亡率が高いために治療に失敗することが示されている。[ 65 ][ 66 ][ 67 ][ 68 ][ 69 ][証拠レベル:3iiA]
複数回再燃したB-ALL患者で、化学療法とその後のHSCTによる治療を受けた場合の転帰が不良なことを考慮して、この集団でCAR T細胞療法が検証されており、高い寛解率および短期生存の改善が得られている(長期追跡の結果はまだ得られていない;詳しい情報については本要約のCAR T細胞療法のセクションを参照のこと)。ブリナツモマブおよびイノツズマブなどの免疫療法でも寛解達成が大きく促進されているが、一般に後でHSCTが施行されている。[ 70 ][ 71 ]免疫療法と細胞療法アプローチの比較研究は、この集団でまだ実施されていないため、初回治療に対する至適アプローチまたは治療の順序に関する情報を得るにはデータが不足している。
初回およびその後の骨髄再燃に対する造血幹細胞移植
移植プロセスの要素
HSCTの適応に関する専門家委員会のレビューが2012年に発表された。[ 72 ]ALL患児に対するHSCTによる転帰の改善または予測に重要なことが示されている移植プロセスの要素には以下のものがある:
- 全身放射線照射(TBI)を含む移植前処置レジメン。
- 移植直前のMRD検出。
- 移植後のMRD検出。
- ドナー種別およびHLA適合性。
- ALLにおけるGVHD/移植片対白血病(GVL)の役割と再燃を予防する移植後の免疫調節。
TBIを含む移植前処置レジメン
同種HSCTを受ける患者では、TBIが前処置レジメンの重要な要素であると考えられている。2件のレジストリー研究および1件の小規模なランダム化試験では、TBIを含む移植前処置レジメンの方が化学療法のみによる移植前処置レジメンよりも高い治癒率をもたらすことが示された。[ 49 ][ 73 ][ 74 ]プロスペクティブ試験からのデータセットおよび単一施設データを組み合わせた国際共同研究(米国、欧州、およびオーストラリア)で、TBIを含まないレジメンの使用は、転帰不良の独立した危険因子であることが示された。最も若い小児(3歳未満から4歳)を除く全例に対するTBIは、北米および欧州のほとんどのセンターで依然として標準の医療である。[ 63 ][ 68 ][ 75 ]
分割TBI(総線量12~14Gy)は、シクロホスファミド、エトポシド、チオテパ、またはこれらの併用療法と組み合わせることが多い。これらの併用療法による研究結果では、一般に同程度の生存率が得られているが[ 76 ][ 77 ][ 78 ]、1件の研究では、シクロホスファミドを他の化学療法薬と併用しない場合は、TBI線量を高くする必要があることが示唆された。[ 79 ]多くの標準レジメンで、線量が13.2~14GyのTBIと併用して、シクロホスファミドが使用されている。一方で、シクロホスファミドおよびエトポシドをTBIと併用した場合は、線量が12Gyを超えると過剰な毒性のために生存率が悪化した。[ 77 ]
移植直前のMRD検出
移植時の疾患の寛解状態は、転帰の重要な予測因子であることが古くから知られており、HSCT時にCRに達していない患者の生存率はきわめて低い。[ 80 ]数件の研究でも、CR期に同種HSCTを受けるALL患児において移植時のMRDレベルが重要な危険因子であることが実証されている。[ 27 ][ 81 ][ 82 ][ 83 ][ 84 ][ 85 ][ 86 ][ 87 ][ 88 ][証拠レベル:3iiA];[ 89 ][証拠レベル:3iiB];[ 21 ][ 75 ]移植前にMRD陽性の患者における生存率は、20~47%であるのと比較して、MRD陰性の患者では60~88%であることが報告されている。
MRD陰性の寛解を得ることを目的として患者が化学療法を2~3サイクル受けている場合、MRD陰性を達成するためにさらに強化療法を受ける有益性については、重大な毒性の可能性と比較検討しなければならない。さらに、複数サイクルの治療を受けている患者におけるMRD陽性は、修正できない転帰不良を示す生物学的病勢マーカーであること、またはこのような患者をMRD陰性の寛解に導くために実施するさらなる介入により、この危険因子が克服され、生存が改善するかどうかを示す明らかな証拠はない。
移植後のMRD検出
HSCT後の検出可能なMRDの存在はその後の再燃リスクの増加に関連している。[ 88 ][ 90 ][ 91 ][ 92 ][ 93 ]HSCT前にMRDが検出可能な患者について、HSCT後にMRDがいずれのレベルであっても検出された場合には、治療失敗のリスクが非常に高く(90%超)なる。[ 75 ]HSCTからの経過時間が長くなるにつれて再燃を予測するMRDの正確度が高まり、いずれかの所定の時点で検出されたMRDのレベルが高い患者ほど、再燃リスクが高い。1件の研究により、HSCT後早期ではフローサイトメトリーよりも次世代の塩基配列決定法を用いた場合に再燃を予測する感度が高いことが示された。[ 92 ]
ドナー種別およびHLA適合性
適合非血縁ドナーおよび臍帯血移植後の生存率は、過去10年間で著しく改善してきており、適合同胞ドナー移植で得られるものとほぼ同程度の転帰が得られる。[ 53 ][ 94 ][ 95 ][ 96 ][ 97 ];[ 98 ][ 99 ][証拠レベル:2A];[ 100 ][証拠レベル:3iiiA];[ 101 ][証拠レベル:3iiiDii]非血縁者ドナー移植後の臨床的に広範囲のGVHDの発生率および治療関連死亡率は、適合同胞ドナー移植と比べて依然として高い。[ 54 ][ 65 ][ 94 ]しかしながら、適合非血縁ドナー移植では再燃率が低い可能性を示す証拠がいくつか得られており、National Marrow Donor ProgramおよびCIBMTRによる解析では、GVHDの発生率、治療関連死亡率、およびOSが時間の経過とともに改善していることが実証されている。[ 102 ][ 103 ][ 104 ];[ 105 ][ 106 ][証拠レベル:3iiA]
別のCIBMTR研究により、HLA抗原が1座または2座不一致の臍帯血移植後の転帰は、適合家族ドナーまたは適合非血縁ドナーによる転帰と同程度であることが示唆された。[ 107 ]適合ドナーがみつからないか、即時移植がきわめて重要であると考えられる一部の症例においては、大量の幹細胞を用いるハプロタイプ一致移植が検討されることもある。[ 108 ]α-β鎖T細胞受容体(TCR)/CD19枯渇法または移植後のシクロホスファミド投与を用いてハプロタイプ一致HSCTを改善したアプローチでは、他の幹細胞ソースを用いた研究と同程度の生存率が示されている。[ 109 ]イタリアからの大規模な多施設共同試験では、α-β鎖TCR/CD19枯渇法によるハプロタイプ一致ドナーを用いた場合、適合した非血縁ドナーと比較して転帰が同程度で、GVHDの発生率が低いことが示された。[ 110 ]
ALLにおけるGVHD/GVLの役割と再燃を予防する移植後の免疫調節
小児および若年成人患者を対象にしてこの問題に取り組んでいる研究のほとんどで、再燃を減少させる上で急性および慢性の両GVHDの効果が示唆されている。[ 94 ][ 111 ][ 112 ][ 113 ]
このGVL効果を利用して、移植後の再燃を防ぐために多くのアプローチが研究されており、その中には、免疫抑制剤の中止またはドナーリンパ球輸注のほか、モノクローナル抗体およびナチュラルキラー細胞療法のような標的免疫療法などがある。[ 114 ][ 115 ]欧州および米国の試験によると、レシピエントのキメリズム増加(すなわち、レシピエントDNAマーカーの割合が増加)に基づいて再燃リスクが高いと判定された患者では、過剰な毒性を伴わないで免疫抑制の中止を問題なく実施可能なことが示されている。[ 116 ][ 117 ]
ALL再燃に対する同種HSCT後の再燃
同種HSCT後に再燃し、免疫抑制剤から問題なく離脱でき、GVHDがみられないB-ALLの患者では、tisagenlecleucelおよび他の4-1BB CAR T細胞アプローチにより、12ヵ月で50%超えるEFS率が得られている。[ 124 ]再燃したT-ALL患者またはCAR T細胞療法を受けられないB-ALL患者に対しては、2回目の骨髄破壊的同種HSCTが実施可能な場合がある。しかしながら、寛解達成失敗、毒性による早期死亡、または救援化学療法に伴う重度の臓器毒性のために、2回目のHSCTを受けることができない患者が多い。[ 125 ]2回目の骨髄破壊的同種HSCTを受けることができる厳選された患者グループでは、約10~30%が長期のEFSを達成する。[ 125 ][ 126 ][ 127 ][ 128 ][ 129 ][ 130 ];[ 69 ][ 131 ][証拠レベル:3iiA]初回HSCT後の寛解持続期間がより長い患者および2回目のHSCT時にCR期の患者の予後はより良好である。[ 127 ][ 128 ][ 132 ]さらに、2回目のHSCT後に急性GVHDが発生した場合、特に初回の移植では発生しなかった場合には生存が改善することが、ある研究で示された。[ 133 ]
2回目の同種移植アプローチとして強度縮小アプローチを用いた場合、一定の割合の患者に治癒をもたらす可能性もあるが、フローサイトメトリーによってCR達成が確認された患者に限定される。[ 134 ][証拠レベル:2A]同種HSCT後に再燃するALL患者に対するドナーリンパ球輸注の有益性は限られている。[ 135 ];[ 136 ][証拠レベル:3iiiA]
HSCT後の孤立したCNS再燃および精巣再燃の治療で、2回目の同種移植が必要かどうかは不明である。小規模なシリーズでは、化学療法単独または化学療法後の2回目の移植を用いることを選択した患者で生存が示されている。[ 137 ][証拠レベル:3iA]
難治性ALLに対する免疫療法アプローチ
難治性ALLの治療を目的とした免疫療法アプローチには、モノクローナル抗体療法およびCAR T細胞療法がある。
モノクローナル抗体療法
難治性B-ALL患者の治療を目的に、以下の2つの免疫療法薬が研究されている:
CAR T細胞療法
キメラ抗原受容体(CAR)T細胞療法は、難治性疾患または2回以上再燃した小児B-ALL患者に対する治療戦略である。この治療には、T細胞の特異性および機能の方向を変えるCARを用いたT細胞のエンジニアリングが含まれる。[ 142 ]CAR修飾T細胞の広く用いられている標的の1つは、ほぼすべての正常なB細胞とほとんどのB細胞性悪性腫瘍に発現しているCD19抗原である。
CAR T細胞療法に伴う毒性
CAR T細胞による治療は、致死的となりうるサイトカイン放出症候群と関連している。[ 143 ][ 144 ]サイトカイン放出症候群は、発熱、頭痛、筋肉痛、高血圧、毛細血管漏出、低酸素症、および腎機能障害として現れる。重度のサイトカイン放出症候群は、抗インターロイキン-6受容体(IL-6R)抗体のトシリズマブにより有効に治療されている。[ 143 ]CAR T細胞の長期間の持続は、免疫グロブリンの補給を要するB細胞低形成につながる場合がある。[ 143 ]
CAR T細胞療法では、失語症、精神状態の変化、痙攣発作などの神経毒性も観察されており、これらの症状は通常自然に回復する。[ 145 ]CNS症状に対して、IL-6R標的薬または他のアプローチでは効果が得られていない。
CAR T細胞療法の他の副作用には、凝血異常、血球貪食性リンパ組織球症に似た臨床検査値変化、心機能障害などがある。患者の20~40%が集中治療室での治療(特に昇圧サポート)を必要とし、10~20%が挿管および/または透析を必要とする。[ 142 ][ 143 ][ 146 ]
CD19標的CAR T細胞療法
再燃/難治性ALLにおいてCD19を標的にしたCAR T細胞の数件の臨床試験が実施されており、結果は勇気付けられるものである。4-1BBおよびCD28という2種類の共刺激性分子の使用を検討している試験が公表されている。CD28ベースのアプローチは、高い寛解率につながっているが、これらの試験でCAR T細胞が1~2ヵ月より長く持続することはまれであり、長期生存にはHSCTを必要とする。[ 147 ]4-1BB共刺激を用いた試験の多くでは、長期間にわたるCAR T細胞の持続および長期の奏効が得られている。[ 124 ][ 146 ]
証拠(CD19標的CAR T細胞療法):
- Children's Hospital of Philadelphia(CHOP)およびHospital of the University of Pennsylvaniaで実施されたパイロット臨床試験では、多数回再燃した、または難治性のCD19陽性ALLの小児および成人30人(このうち25人が22歳以下であった)に対して、CD19標的4-1BB CARレンチウイルスベクターで形質導入されたT細胞が投与された。[ 143 ][証拠レベル:3iiiDi]
- 再燃/難治性のCD19陽性B-ALLの小児および若年成人45人を対象に、4-1BBをベースにしたレンチウイルスベクター拡張CAR T細胞を用いた第I相試験の3つ目の報告により、以下が示された:[ 146 ]
- CHOPおよびUniversity of Pennsylvaniaで開発された抗CD19 4-1BBベクターのグローバル第II相試験は、複数回再発または難治性B-ALLの小児に対するtisagenlecleucelの米国食品医薬品局による承認につながった。[ 124 ]
- NCIのPediatric Oncology Branchからの報告では、遺伝子形質導入にレトロウイルスベクターを用いたCD28共刺激領域を有する別のCD19標的CAR T細胞製剤の使用が記述された。[ 147 ]
- 別の報告により、抗CD19、抗CD28z CAR T細胞で治療された小児および若年成人25人を対象にした1件の多施設試験について記述された。研究者らは試験中のリンパ球除去シクロホスファミドの用量を増加させ、低用量および高用量の前処置期のほか、MRDの存在 vs 治療前の疾患の形態学的証拠に基づいて転帰を解析した。[ 148 ][証拠レベル:1iiA]
CD22標的CAR T細胞療法
CD19を標的としたCAR T細胞療法後の再発のうち50%以上は、抗原エスケープによるもので、これはCAR T細胞作成で使用される結合部位を除去するCD19蛋白の変異に関連することが示されている。[ 149 ]抗原エスケープ後の救援は、第二のリンパ系抗原であるCD22を標的とした細胞療法および免疫療法アプローチで報告されている。特にCD19陰性再燃のイノツズマブ救援を検討した研究は公表されていないが、一般にCD22 CAR T細胞療法に続けてHSCTによる治療を行った場合に、その後の寛解達成率および生存率が高いことを2つのグループが報告している。[ 150 ][ 151 ]CD22抗原は下方制御されることがあるため、長期CAR T細胞反応のためにCD22のみを標的とすることに懸念がある;そのため、このアプローチは、しばしばHSCTと併用される。
証拠(CD22標的CAR T細胞療法):
孤立性髄外再燃の治療
ALL患児の治療成功率が向上するにつれて、孤立性髄外再燃の発生率が低下している。孤立性CNS再燃の発生率は5%未満で、精巣再燃は1%未満から2%である。[ 152 ][ 153 ][ 154 ]骨髄再燃および併発型再燃と同様に、孤立性髄外再燃では、初回診断から再燃までの時間が重要な予後因子である。[ 155 ]さらに、1件の研究において、孤立性髄外再燃患者に対する予後不良因子として、再燃時に6歳を超えていることが指摘されたが、別の研究ではより良好なカットオフとして10歳が提唱された。[ 16 ][ 156 ]注目すべき点として、孤立性髄外再燃を認める小児のほとんどで、感度の高い分子解析技術を用いると超顕微鏡的な骨髄病変が明らかになることがあり[ 157 ]、治療戦略を成功させるには局所性病変とともに全身性病変を効果的にコントロールしなければならない。孤立性CNS再燃を来し、形態学的に骨髄が正常でもMRDが0.01%を超える患者の予後(5年EFS率、30%)は、MRDが認められないか、MRDが0.01%未満の患者(5年EFS率、60%)より不良である。[ 157 ]
孤立性CNS再燃
CNSに再発した小児ALLに対する標準治療法の選択肢には以下のものがある:
- 全身性および髄腔内化学療法。
- 頭蓋照射または頭蓋脊髄照射。
- HSCT。
これまで孤立性のCNS再燃がみられる小児の予後はきわめて不良であったが、積極的な全身療法および髄腔内化学療法後の頭蓋照射または頭蓋脊髄照射により、特に第一寛解期に頭蓋照射を受けなかった患児では、見通しが明るくなっている。[ 18 ][ 155 ][ 158 ][ 159 ]
証拠(化学療法および放射線療法):
- この治療戦略を用いたPediatric Oncology Group(POG)の研究によると、過去に放射線療法を受けておらず、第一寛解期が18ヵ月以上続いた小児では、4年EFS率が約80%であったのに対して、診断から18ヵ月以内にCNS再燃が認められた小児では、4年EFS率が約45%であった。[ 155 ]
- POGの追跡研究では、過去に放射線療法を受けておらず、第一寛解期が18ヵ月以上続いた小児が、強化全身性および髄腔内化学療法を1年間受けた後に、18Gyの頭蓋照射単独による治療を受けた。[ 18 ]
孤立性CNS再燃に対する治療に関しては、HSCTについて報告した多くケースシリーズが発表されている。[ 160 ][ 161 ]一部の報告では、非常に早期の再燃およびT細胞病変を伴う孤立性CNS疾患を有する患者に対するHSCTの潜在的役割が示唆されているが、早期の再燃におけるHSCTの必要性に関する証拠は少なく、晩期再燃における証拠は得られていない。診断から18ヵ月未満で認められた孤立性CNS再燃、特にT細胞CNS再燃を治療するための移植の使用については、さらなる研究が必要である。
証拠(HSCT):
- レトロスペクティブ登録ベース研究で、HLA適合同胞移植または上記のPOG研究と同様の化学放射線療法のいずれかの治療を受けた患者の転帰が比較された。[ 162 ][証拠レベル:3iiiDii]この研究には、早期再燃(診断から18ヵ月未満)および遅発再燃の両方に対する移植が含まれていた。
- MRC ALLR3試験では、再燃ALL患者においてミトキサントロン vs イダルビシンによる強力な寛解導入療法が検証され、ミトキサントロンを用いた場合に優れた転帰が明らかにされた。孤立性CNS再燃によりこの試験に登録された患者80人のサブアナリシスには、非常に早期(初回診断から18ヵ月未満と定義)に再燃した患者13人、早期(初回診断から18ヵ月を超えるが、治療中止から6ヵ月以内と定義)に再燃した患者55人、および晩期再燃の患者12人が含まれていた。[ 16 ][証拠レベル:2A]
孤立性精巣再燃
孤立性精巣再燃例に対する治療結果は再燃時期に左右される。治療中に顕性の精巣再燃が認められた男児の3年EFS率は約40%である;晩期に精巣の再燃が認められた男児では約85%である。[ 163 ]
北米における精巣に再発した小児ALLに対する標準治療法の選択肢には以下のものがある:
- 化学療法。
- 放射線療法。
北米における孤立性精巣再燃を治療するための標準アプローチには、強化化学療法を併用した局所放射線療法がある。欧州の一部の臨床試験グループでは、放射線療法の代わりに罹患精巣の精巣摘除術を実施している。再燃した時点で他方の精巣の生検を実施し、局所コントロール(外科的切除または放射線療法)を実施すべきかどうか判断する。第一選択治療終了時に精巣生検を検討した研究では、潜伏病変の早期発見による患者の生存利益を実証できなかった。[ 164 ]
放射線療法または精巣摘除術を使用しない場合の転帰に関する臨床データは限られている。このアプローチを検証している治療プロトコルでは、化学療法薬(例、大量メトトレキサート)の強化投与法を組み入れており、精巣で抗白血病効果をある程度達成できる可能性がある。
証拠(精巣再燃の治療):
- COG AALL02P2(NCT00096135)試験では、遅発性の孤立性精巣再燃(診断から18ヵ月を超えてから発生)の患者に対して放射線療法が省略できるかどうか検証された。[ 165 ]この試験では、大量メトトレキサートを含む再寛解導入化学療法の最初の1ヵ月後に精巣の大きさが再評価された。精巣が依然として大きい場合は、生検を実施し、可能であれば、患者は局所放射線療法による治療を受けた。精巣の大きさが正常化した患者または精巣の生検で陰性であった患者は、放射線療法なしの治療を受けた。すべての患者(放射線照射の有無を問わない)に対する寛解導入後化学療法には、高用量のメトトレキサートの複数回コースが含まれた。[ 166 ]
- オランダの研究者らは、晩期精巣再燃を認めた男児5人を対象に、大量メトトレキサートを寛解導入期間中(12g/m2)、およびその他の治療期間では一定間隔で(6g/m2)使用して治療し、精巣放射線療法は併用しなかった。[ 165 ]
- 過去のALLの全身性再燃に対してHSCTを実施後に孤立性精巣再燃が認められた男児を対象とした小規模シリーズでは、男児7人中5人が2回目のHSCTを受けずにEFSの延長を示した。[ 137 ][証拠レベル:3iA]
再燃小児ALLに対して臨床評価段階にある治療法の選択肢
ALLの初回再燃に対する試験
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
- COG-AALL1331;NCI-2014-00631(NCT02101853)(小児B-ALLの初回再燃に対するブリナツモマブのリスク層別化ランダム化第III相試験):この試験では、B-ALLの初回再燃患者に対してブリナツモマブを組み込むことでDFSを改善できるかどうかが評価されている。ブリナツモマブはほぼすべてのB-ALL細胞に発現したCD19抗原とT細胞に発現したCD3抗原に結合する二重特異性抗体である;このため、ブリナツモマブはBリンパ芽球と患者自身のT細胞を近接させて、白血病細胞の溶解を促進する。再燃部位(骨髄再燃 vs 孤立性髄外再燃)、再燃までの期間、および治療開始から1ヵ月後におけるMRD状態に基づいて患者のリスク層別化が行われる。この試験の化学療法の基本骨格は英国のALLR3レジメンに基づいている。[ 39 ]治療開始から1ヵ月後に高リスクおよび中リスク患者は、地固め化学療法ブロックを2回行うか、2サイクルのブリナツモマブのいずれかを受ける群にランダムに割り付けられた。これらの患者は続いて、HSCTに進む。低リスク患者は移植なしで治療される;これらの患者はALLR3プロトコルに基づく対照群または同じ化学療法の基本骨格に3サイクルのブリナツモマブも含める研究群のいずれかにランダムに割り付けられる。
- TACL 2012-002(NCT02879643)(ALLが再燃した小児、青年、および若年成人に対してUKALL R3寛解導入化学療法と併用する硫酸ビンクリスチンリポソーム注入):この試験では、初回、2回目、または3回目の再燃を来したALL患者(B-ALLまたはT-ALL)に対するUKALL R3寛解導入レジメンにおける標準のビンクリスチンの代替として硫酸ビンクリスチンリポソーム注入の安全性と実施可能性を評価している。M2(5~24%の芽球)またはM3(25%超の芽球)骨髄病変を有する患者が適格である。
ALLの2回目以降の再燃または難治性ALLに対する試験
2回目以降の再燃または難治性のALL患児に対しては、新規薬剤および新たな多剤併用療法を研究している多くの臨床試験が利用可能であり、参加を検討すべきである。これらの試験ではALLに特異的な標的療法を検証しており、その中にはモノクローナル抗体に基づく治療、および白血病細胞の増殖と生存に必要なシグナル伝達経路を阻害する薬剤がある。新薬、新しい併用法、および免疫療法アプローチを研究する多くの臨床試験が利用できる。(詳しい情報については、ClinicalTrials.govウェブサイトを参照のこと。)
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Reismüller B, Attarbaschi A, Peters C, et al.: Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria--a population-based report of the Austrian Berlin-Frankfurt-Münster (BFM) Study Group. Br J Haematol 144 (4): 559-70, 2009.[PUBMED Abstract]
- Uderzo C, Conter V, Dini G, et al.: Treatment of childhood acute lymphoblastic leukemia after the first relapse: curative strategies. Haematologica 86 (1): 1-7, 2001.[PUBMED Abstract]
- Chessells JM, Veys P, Kempski H, et al.: Long-term follow-up of relapsed childhood acute lymphoblastic leukaemia. Br J Haematol 123 (3): 396-405, 2003.[PUBMED Abstract]
- Rivera GK, Zhou Y, Hancock ML, et al.: Bone marrow recurrence after initial intensive treatment for childhood acute lymphoblastic leukemia. Cancer 103 (2): 368-76, 2005.[PUBMED Abstract]
- Einsiedel HG, von Stackelberg A, Hartmann R, et al.: Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Münster Group 87. J Clin Oncol 23 (31): 7942-50, 2005.[PUBMED Abstract]
- Schroeder H, Garwicz S, Kristinsson J, et al.: Outcome after first relapse in children with acute lymphoblastic leukemia: a population-based study of 315 patients from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Med Pediatr Oncol 25 (5): 372-8, 1995.[PUBMED Abstract]
- Wheeler K, Richards S, Bailey C, et al.: Comparison of bone marrow transplant and chemotherapy for relapsed childhood acute lymphoblastic leukaemia: the MRC UKALL X experience. Medical Research Council Working Party on Childhood Leukaemia. Br J Haematol 101 (1): 94-103, 1998.[PUBMED Abstract]
- Buchanan GR, Rivera GK, Pollock BH, et al.: Alternating drug pairs with or without periodic reinduction in children with acute lymphoblastic leukemia in second bone marrow remission: a Pediatric Oncology Group Study. Cancer 88 (5): 1166-74, 2000.[PUBMED Abstract]
- Rivera GK, Hudson MM, Liu Q, et al.: Effectiveness of intensified rotational combination chemotherapy for late hematologic relapse of childhood acute lymphoblastic leukemia. Blood 88 (3): 831-7, 1996.[PUBMED Abstract]
- Bührer C, Hartmann R, Fengler R, et al.: Peripheral blast counts at diagnosis of late isolated bone marrow relapse of childhood acute lymphoblastic leukemia predict response to salvage chemotherapy and outcome. Berlin-Frankfurt-Münster Relapse Study Group. J Clin Oncol 14 (10): 2812-7, 1996.[PUBMED Abstract]
- Roy A, Cargill A, Love S, et al.: Outcome after first relapse in childhood acute lymphoblastic leukaemia - lessons from the United Kingdom R2 trial. Br J Haematol 130 (1): 67-75, 2005.[PUBMED Abstract]
- Rizzari C, Valsecchi MG, Aricò M, et al.: Outcome of very late relapse in children with acute lymphoblastic leukemia. Haematologica 89 (4): 427-34, 2004.[PUBMED Abstract]
- Nguyen K, Devidas M, Cheng SC, et al.: Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's Oncology Group study. Leukemia 22 (12): 2142-50, 2008.[PUBMED Abstract]
- Locatelli F, Schrappe M, Bernardo ME, et al.: How I treat relapsed childhood acute lymphoblastic leukemia. Blood 120 (14): 2807-16, 2012.[PUBMED Abstract]
- Malempati S, Gaynon PS, Sather H, et al.: Outcome after relapse among children with standard-risk acute lymphoblastic leukemia: Children's Oncology Group study CCG-1952. J Clin Oncol 25 (36): 5800-7, 2007.[PUBMED Abstract]
- Masurekar AN, Parker CA, Shanyinde M, et al.: Outcome of central nervous system relapses in childhood acute lymphoblastic leukaemia--prospective open cohort analyses of the ALLR3 trial. PLoS One 9 (10): e108107, 2014.[PUBMED Abstract]
- Parker C, Krishnan S, Hamadeh L, et al.: Outcomes of patients with childhood B-cell precursor acute lymphoblastic leukaemia with late bone marrow relapses: long-term follow-up of the ALLR3 open-label randomised trial. Lancet Haematol 6 (4): e204-e216, 2019.[PUBMED Abstract]
- Barredo JC, Devidas M, Lauer SJ, et al.: Isolated CNS relapse of acute lymphoblastic leukemia treated with intensive systemic chemotherapy and delayed CNS radiation: a pediatric oncology group study. J Clin Oncol 24 (19): 3142-9, 2006.[PUBMED Abstract]
- Rubnitz JE, Hijiya N, Zhou Y, et al.: Lack of benefit of early detection of relapse after completion of therapy for acute lymphoblastic leukemia. Pediatr Blood Cancer 44 (2): 138-41, 2005.[PUBMED Abstract]
- Freyer DR, Devidas M, La M, et al.: Postrelapse survival in childhood acute lymphoblastic leukemia is independent of initial treatment intensity: a report from the Children's Oncology Group. Blood 117 (11): 3010-5, 2011.[PUBMED Abstract]
- Eckert C, Groeneveld-Krentz S, Kirschner-Schwabe R, et al.: Improving Stratification for Children With Late Bone Marrow B-Cell Acute Lymphoblastic Leukemia Relapses With Refined Response Classification and Integration of Genetics. J Clin Oncol 37 (36): 3493-3506, 2019.[PUBMED Abstract]
- Meyr F, Escherich G, Mann G, et al.: Outcomes of treatment for relapsed acute lymphoblastic leukaemia in children with Down syndrome. Br J Haematol 162 (1): 98-106, 2013.[PUBMED Abstract]
- Hitzler JK, He W, Doyle J, et al.: Outcome of transplantation for acute lymphoblastic leukemia in children with Down syndrome. Pediatr Blood Cancer 61 (6): 1126-8, 2014.[PUBMED Abstract]
- Raetz EA, Borowitz MJ, Devidas M, et al.: Reinduction platform for children with first marrow relapse in acute lymphoblastic lymphoma. J Clin Oncol 26 (24): 3971-8, 2008.[PUBMED Abstract]
- von Stackelberg A, Völzke E, Kühl JS, et al.: Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer 47 (1): 90-7, 2011.[PUBMED Abstract]
- Coustan-Smith E, Gajjar A, Hijiya N, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia after first relapse. Leukemia 18 (3): 499-504, 2004.[PUBMED Abstract]
- Sramkova L, Muzikova K, Fronkova E, et al.: Detectable minimal residual disease before allogeneic hematopoietic stem cell transplantation predicts extremely poor prognosis in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 48 (1): 93-100, 2007.[PUBMED Abstract]
- Eckert C, von Stackelberg A, Seeger K, et al.: Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia - long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 49 (6): 1346-55, 2013.[PUBMED Abstract]
- Paganin M, Zecca M, Fabbri G, et al.: Minimal residual disease is an important predictive factor of outcome in children with relapsed 'high-risk' acute lymphoblastic leukemia. Leukemia 22 (12): 2193-200, 2008.[PUBMED Abstract]
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.[PUBMED Abstract]
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.[PUBMED Abstract]
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.[PUBMED Abstract]
- Tzoneva G, Perez-Garcia A, Carpenter Z, et al.: Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 19 (3): 368-71, 2013.[PUBMED Abstract]
- Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.[PUBMED Abstract]
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.[PUBMED Abstract]
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.[PUBMED Abstract]
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.[PUBMED Abstract]
- Tallen G, Ratei R, Mann G, et al.: Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol 28 (14): 2339-47, 2010.[PUBMED Abstract]
- Parker C, Waters R, Leighton C, et al.: Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet 376 (9757): 2009-17, 2010.[PUBMED Abstract]
- von Stackelberg A, Hartmann R, Bührer C, et al.: High-dose compared with intermediate-dose methotrexate in children with a first relapse of acute lymphoblastic leukemia. Blood 111 (5): 2573-80, 2008.[PUBMED Abstract]
- Locatelli F, Testi AM, Bernardo ME, et al.: Clofarabine, cyclophosphamide and etoposide as single-course re-induction therapy for children with refractory/multiple relapsed acute lymphoblastic leukaemia. Br J Haematol 147 (3): 371-8, 2009.[PUBMED Abstract]
- Miano M, Pistorio A, Putti MC, et al.: Clofarabine, cyclophosphamide and etoposide for the treatment of relapsed or resistant acute leukemia in pediatric patients. Leuk Lymphoma 53 (9): 1693-8, 2012.[PUBMED Abstract]
- Hijiya N, Thomson B, Isakoff MS, et al.: Phase 2 trial of clofarabine in combination with etoposide and cyclophosphamide in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. Blood 118 (23): 6043-9, 2011.[PUBMED Abstract]
- Bertaina A, Vinti L, Strocchio L, et al.: The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br J Haematol 176 (4): 629-636, 2017.[PUBMED Abstract]
- Messinger YH, Gaynon PS, Sposto R, et al.: Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 120 (2): 285-90, 2012.[PUBMED Abstract]
- Berg SL, Blaney SM, Devidas M, et al.: Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23 (15): 3376-82, 2005.[PUBMED Abstract]
- Commander LA, Seif AE, Insogna IG, et al.: Salvage therapy with nelarabine, etoposide, and cyclophosphamide in relapsed/refractory paediatric T-cell lymphoblastic leukaemia and lymphoma. Br J Haematol 150 (3): 345-51, 2010.[PUBMED Abstract]
- Sun W, Malvar J, Sposto R, et al.: Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia 32 (11): 2316-2325, 2018.[PUBMED Abstract]
- Eapen M, Raetz E, Zhang MJ, et al.: Outcomes after HLA-matched sibling transplantation or chemotherapy in children with B-precursor acute lymphoblastic leukemia in a second remission: a collaborative study of the Children's Oncology Group and the Center for International Blood and Marrow Transplant Research. Blood 107 (12): 4961-7, 2006.[PUBMED Abstract]
- Barrett AJ, Horowitz MM, Pollock BH, et al.: Bone marrow transplants from HLA-identical siblings as compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission. N Engl J Med 331 (19): 1253-8, 1994.[PUBMED Abstract]
- Uderzo C, Valsecchi MG, Bacigalupo A, et al.: Treatment of childhood acute lymphoblastic leukemia in second remission with allogeneic bone marrow transplantation and chemotherapy: ten-year experience of the Italian Bone Marrow Transplantation Group and the Italian Pediatric Hematology Oncology Association. J Clin Oncol 13 (2): 352-8, 1995.[PUBMED Abstract]
- Harrison G, Richards S, Lawson S, et al.: Comparison of allogeneic transplant versus chemotherapy for relapsed childhood acute lymphoblastic leukaemia in the MRC UKALL R1 trial. MRC Childhood Leukaemia Working Party. Ann Oncol 11 (8): 999-1006, 2000.[PUBMED Abstract]
- Bunin N, Carston M, Wall D, et al.: Unrelated marrow transplantation for children with acute lymphoblastic leukemia in second remission. Blood 99 (9): 3151-7, 2002.[PUBMED Abstract]
- Borgmann A, von Stackelberg A, Hartmann R, et al.: Unrelated donor stem cell transplantation compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission: a matched-pair analysis. Blood 101 (10): 3835-9, 2003.[PUBMED Abstract]
- Saarinen-Pihkala UM, Heilmann C, Winiarski J, et al.: Pathways through relapses and deaths of children with acute lymphoblastic leukemia: role of allogeneic stem-cell transplantation in Nordic data. J Clin Oncol 24 (36): 5750-62, 2006.[PUBMED Abstract]
- Thomson B, Park JR, Felgenhauer J, et al.: Toxicity and efficacy of intensive chemotherapy for children with acute lymphoblastic leukemia (ALL) after first bone marrow or extramedullary relapse. Pediatr Blood Cancer 43 (5): 571-9, 2004.[PUBMED Abstract]
- Hahn T, Wall D, Camitta B, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 11 (11): 823-61, 2005.[PUBMED Abstract]
- Borgmann A, Baumgarten E, Schmid H, et al.: Allogeneic bone marrow transplantation for a subset of children with acute lymphoblastic leukemia in third remission: a conceivable alternative? Bone Marrow Transplant 20 (11): 939-44, 1997.[PUBMED Abstract]
- Schroeder H, Gustafsson G, Saarinen-Pihkala UM, et al.: Allogeneic bone marrow transplantation in second remission of childhood acute lymphoblastic leukemia: a population-based case control study from the Nordic countries. Bone Marrow Transplant 23 (6): 555-60, 1999.[PUBMED Abstract]
- van den Berg H, de Groot-Kruseman HA, Damen-Korbijn CM, et al.: Outcome after first relapse in children with acute lymphoblastic leukemia: a report based on the Dutch Childhood Oncology Group (DCOG) relapse all 98 protocol. Pediatr Blood Cancer 57 (2): 210-6, 2011.[PUBMED Abstract]
- Beck JC, Cao Q, Trotz B, et al.: Allogeneic hematopoietic cell transplantation outcomes for children with B-precursor acute lymphoblastic leukemia and early or late BM relapse. Bone Marrow Transplant 46 (7): 950-5, 2011.[PUBMED Abstract]
- Eckert C, Henze G, Seeger K, et al.: Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol 31 (21): 2736-42, 2013.[PUBMED Abstract]
- Burke MJ, Verneris MR, Le Rademacher J, et al.: Transplant Outcomes for Children with T Cell Acute Lymphoblastic Leukemia in Second Remission: A Report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant 21 (12): 2154-9, 2015.[PUBMED Abstract]
- Gaynon PS: Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol 131 (5): 579-87, 2005.[PUBMED Abstract]
- Woolfrey AE, Anasetti C, Storer B, et al.: Factors associated with outcome after unrelated marrow transplantation for treatment of acute lymphoblastic leukemia in children. Blood 99 (6): 2002-8, 2002.[PUBMED Abstract]
- Afify Z, Hunt L, Green A, et al.: Factors affecting the outcome of stem cell transplantation from unrelated donors for childhood acute lymphoblastic leukemia in third remission. Bone Marrow Transplant 35 (11): 1041-7, 2005.[PUBMED Abstract]
- Gassas A, Ishaqi MK, Afzal S, et al.: Outcome of haematopoietic stem cell transplantation for paediatric acute lymphoblastic leukaemia in third complete remission: a vital role for graft-versus-host-disease/ graft-versus-leukaemia effect in survival. Br J Haematol 140 (1): 86-9, 2008.[PUBMED Abstract]
- Nemecek ER, Ellis K, He W, et al.: Outcome of myeloablative conditioning and unrelated donor hematopoietic cell transplantation for childhood acute lymphoblastic leukemia in third remission. Biol Blood Marrow Transplant 17 (12): 1833-40, 2011.[PUBMED Abstract]
- Kato M, Horikoshi Y, Okamoto Y, et al.: Second allogeneic hematopoietic SCT for relapsed ALL in children. Bone Marrow Transplant 47 (10): 1307-11, 2012.[PUBMED Abstract]
- Bhojwani D, Sposto R, Shah NN, et al.: Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 33 (4): 884-892, 2019.[PUBMED Abstract]
- Pulsipher MA: Are CAR T cells better than antibody or HCT therapy in B-ALL? Hematology Am Soc Hematol Educ Program 2018 (1): 16-24, 2018.[PUBMED Abstract]
- Oliansky DM, Camitta B, Gaynon P, et al.: Role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of pediatric acute lymphoblastic leukemia: update of the 2005 evidence-based review. Biol Blood Marrow Transplant 18 (4): 505-22, 2012.[PUBMED Abstract]
- Davies SM, Ramsay NK, Klein JP, et al.: Comparison of preparative regimens in transplants for children with acute lymphoblastic leukemia. J Clin Oncol 18 (2): 340-7, 2000.[PUBMED Abstract]
- Bunin N, Aplenc R, Kamani N, et al.: Randomized trial of busulfan vs total body irradiation containing conditioning regimens for children with acute lymphoblastic leukemia: a Pediatric Blood and Marrow Transplant Consortium study. Bone Marrow Transplant 32 (6): 543-8, 2003.[PUBMED Abstract]
- Bader P, Salzmann-Manrique E, Balduzzi A, et al.: More precisely defining risk peri-HCT in pediatric ALL: pre- vs post-MRD measures, serial positivity, and risk modeling. Blood Adv 3 (21): 3393-3405, 2019.[PUBMED Abstract]
- Gassas A, Sung L, Saunders EF, et al.: Comparative outcome of hematopoietic stem cell transplantation for pediatric acute lymphoblastic leukemia following cyclophosphamide and total body irradiation or VP16 and total body irradiation conditioning regimens. Bone Marrow Transplant 38 (11): 739-43, 2006.[PUBMED Abstract]
- Tracey J, Zhang MJ, Thiel E, et al.: Transplantation conditioning regimens and outcomes after allogeneic hematopoietic cell transplantation in children and adolescents with acute lymphoblastic leukemia. Biol Blood Marrow Transplant 19 (2): 255-9, 2013.[PUBMED Abstract]
- Bakr M, Rasheed W, Mohamed SY, et al.: Allogeneic hematopoietic stem cell transplantation in adolescent and adult patients with high-risk T cell acute lymphoblastic leukemia. Biol Blood Marrow Transplant 18 (12): 1897-904, 2012.[PUBMED Abstract]
- Marks DI, Forman SJ, Blume KG, et al.: A comparison of cyclophosphamide and total body irradiation with etoposide and total body irradiation as conditioning regimens for patients undergoing sibling allografting for acute lymphoblastic leukemia in first or second complete remission. Biol Blood Marrow Transplant 12 (4): 438-53, 2006.[PUBMED Abstract]
- Duval M, Klein JP, He W, et al.: Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol 28 (23): 3730-8, 2010.[PUBMED Abstract]
- Shen Z, Gu X, Mao W, et al.: Influence of pre-transplant minimal residual disease on prognosis after Allo-SCT for patients with acute lymphoblastic leukemia: systematic review and meta-analysis. BMC Cancer 18 (1): 755, 2018.[PUBMED Abstract]
- Balduzzi A, Di Maio L, Silvestri D, et al.: Minimal residual disease before and after transplantation for childhood acute lymphoblastic leukaemia: is there any room for intervention? Br J Haematol 164 (3): 396-408, 2014.[PUBMED Abstract]
- Goulden N, Bader P, Van Der Velden V, et al.: Minimal residual disease prior to stem cell transplant for childhood acute lymphoblastic leukaemia. Br J Haematol 122 (1): 24-9, 2003.[PUBMED Abstract]
- Bader P, Kreyenberg H, Henze GH, et al.: Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol 27 (3): 377-84, 2009.[PUBMED Abstract]
- Leung W, Pui CH, Coustan-Smith E, et al.: Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood 120 (2): 468-72, 2012.[PUBMED Abstract]
- Ruggeri A, Michel G, Dalle JH, et al.: Impact of pretransplant minimal residual disease after cord blood transplantation for childhood acute lymphoblastic leukemia in remission: an Eurocord, PDWP-EBMT analysis. Leukemia 26 (12): 2455-61, 2012.[PUBMED Abstract]
- Bachanova V, Burke MJ, Yohe S, et al.: Unrelated cord blood transplantation in adult and pediatric acute lymphoblastic leukemia: effect of minimal residual disease on relapse and survival. Biol Blood Marrow Transplant 18 (6): 963-8, 2012.[PUBMED Abstract]
- Sutton R, Shaw PJ, Venn NC, et al.: Persistent MRD before and after allogeneic BMT predicts relapse in children with acute lymphoblastic leukaemia. Br J Haematol 168 (3): 395-404, 2015.[PUBMED Abstract]
- Sanchez-Garcia J, Serrano J, Serrano-Lopez J, et al.: Quantification of minimal residual disease levels by flow cytometry at time of transplant predicts outcome after myeloablative allogeneic transplantation in ALL. Bone Marrow Transplant 48 (3): 396-402, 2013.[PUBMED Abstract]
- Bader P, Kreyenberg H, von Stackelberg A, et al.: Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: results of the ALL-BFM-SCT 2003 trial. J Clin Oncol 33 (11): 1275-84, 2015.[PUBMED Abstract]
- Pulsipher MA, Langholz B, Wall DA, et al.: Risk factors and timing of relapse after allogeneic transplantation in pediatric ALL: for whom and when should interventions be tested? Bone Marrow Transplant 50 (9): 1173-9, 2015.[PUBMED Abstract]
- Pulsipher MA, Carlson C, Langholz B, et al.: IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 125 (22): 3501-8, 2015.[PUBMED Abstract]
- Liu J, Wang Y, Xu LP, et al.: Monitoring mixed lineage leukemia expression may help identify patients with mixed lineage leukemia--rearranged acute leukemia who are at high risk of relapse after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 20 (7): 929-36, 2014.[PUBMED Abstract]
- Locatelli F, Zecca M, Messina C, et al.: Improvement over time in outcome for children with acute lymphoblastic leukemia in second remission given hematopoietic stem cell transplantation from unrelated donors. Leukemia 16 (11): 2228-37, 2002.[PUBMED Abstract]
- Saarinen-Pihkala UM, Gustafsson G, Ringdén O, et al.: No disadvantage in outcome of using matched unrelated donors as compared with matched sibling donors for bone marrow transplantation in children with acute lymphoblastic leukemia in second remission. J Clin Oncol 19 (14): 3406-14, 2001.[PUBMED Abstract]
- Muñoz A, Diaz-Heredia C, Diaz MA, et al.: Allogeneic hemopoietic stem cell transplantation for childhood acute lymphoblastic leukemia in second complete remission-similar outcomes after matched related and unrelated donor transplant: a study of the Spanish Working Party for Blood and Marrow Transplantation in Children (Getmon). Pediatr Hematol Oncol 25 (4): 245-59, 2008.[PUBMED Abstract]
- Jacobsohn DA, Hewlett B, Ranalli M, et al.: Outcomes of unrelated cord blood transplants and allogeneic-related hematopoietic stem cell transplants in children with high-risk acute lymphocytic leukemia. Bone Marrow Transplant 34 (10): 901-7, 2004.[PUBMED Abstract]
- Kurtzberg J, Prasad VK, Carter SL, et al.: Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood 112 (10): 4318-27, 2008.[PUBMED Abstract]
- Peters C, Schrappe M, von Stackelberg A, et al.: Stem-cell transplantation in children with acute lymphoblastic leukemia: A prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003 trial. J Clin Oncol 33 (11): 1265-74, 2015.[PUBMED Abstract]
- Smith AR, Baker KS, Defor TE, et al.: Hematopoietic cell transplantation for children with acute lymphoblastic leukemia in second complete remission: similar outcomes in recipients of unrelated marrow and umbilical cord blood versus marrow from HLA matched sibling donors. Biol Blood Marrow Transplant 15 (9): 1086-93, 2009.[PUBMED Abstract]
- Zhang MJ, Davies SM, Camitta BM, et al.: Comparison of outcomes after HLA-matched sibling and unrelated donor transplantation for children with high-risk acute lymphoblastic leukemia. Biol Blood Marrow Transplant 18 (8): 1204-10, 2012.[PUBMED Abstract]
- Gassas A, Sung L, Saunders EF, et al.: Graft-versus-leukemia effect in hematopoietic stem cell transplantation for pediatric acute lymphoblastic leukemia: significantly lower relapse rate in unrelated transplantations. Bone Marrow Transplant 40 (10): 951-5, 2007.[PUBMED Abstract]
- Harvey J, Green A, Cornish J, et al.: Improved survival in matched unrelated donor transplant for childhood ALL since the introduction of high-resolution matching at HLA class I and II. Bone Marrow Transplant 47 (10): 1294-300, 2012.[PUBMED Abstract]
- Majhail NS, Chitphakdithai P, Logan B, et al.: Significant improvement in survival after unrelated donor hematopoietic cell transplantation in the recent era. Biol Blood Marrow Transplant 21 (1): 142-50, 2015.[PUBMED Abstract]
- MacMillan ML, Davies SM, Nelson GO, et al.: Twenty years of unrelated donor bone marrow transplantation for pediatric acute leukemia facilitated by the National Marrow Donor Program. Biol Blood Marrow Transplant 14 (9 Suppl): 16-22, 2008.[PUBMED Abstract]
- Davies SM, Wang D, Wang T, et al.: Recent decrease in acute graft-versus-host disease in children with leukemia receiving unrelated donor bone marrow transplants. Biol Blood Marrow Transplant 15 (3): 360-6, 2009.[PUBMED Abstract]
- Eapen M, Rubinstein P, Zhang MJ, et al.: Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet 369 (9577): 1947-54, 2007.[PUBMED Abstract]
- Klingebiel T, Handgretinger R, Lang P, et al.: Haploidentical transplantation for acute lymphoblastic leukemia in childhood. Blood Rev 18 (3): 181-92, 2004.[PUBMED Abstract]
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.[PUBMED Abstract]
- Bertaina A, Zecca M, Buldini B, et al.: Unrelated donor vs HLA-haploidentical α/β T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood 132 (24): 2594-2607, 2018.[PUBMED Abstract]
- Gustafsson Jernberg A, Remberger M, Ringdén O, et al.: Graft-versus-leukaemia effect in children: chronic GVHD has a significant impact on relapse and survival. Bone Marrow Transplant 31 (3): 175-81, 2003.[PUBMED Abstract]
- Dini G, Zecca M, Balduzzi A, et al.: No difference in outcome between children and adolescents transplanted for acute lymphoblastic leukemia in second remission. Blood 118 (25): 6683-90, 2011.[PUBMED Abstract]
- Pulsipher MA, Langholz B, Wall DA, et al.: The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children's Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood 123 (13): 2017-25, 2014.[PUBMED Abstract]
- Pulsipher MA, Bader P, Klingebiel T, et al.: Allogeneic transplantation for pediatric acute lymphoblastic leukemia: the emerging role of peritransplantation minimal residual disease/chimerism monitoring and novel chemotherapeutic, molecular, and immune approaches aimed at preventing relapse. Biol Blood Marrow Transplant 15 (1 Suppl): 62-71, 2008.[PUBMED Abstract]
- Lankester AC, Bierings MB, van Wering ER, et al.: Preemptive alloimmune intervention in high-risk pediatric acute lymphoblastic leukemia patients guided by minimal residual disease level before stem cell transplantation. Leukemia 24 (8): 1462-9, 2010.[PUBMED Abstract]
- Horn B, Soni S, Khan S, et al.: Feasibility study of preemptive withdrawal of immunosuppression based on chimerism testing in children undergoing myeloablative allogeneic transplantation for hematologic malignancies. Bone Marrow Transplant 43 (6): 469-76, 2009.[PUBMED Abstract]
- Pochon C, Oger E, Michel G, et al.: Follow-up of post-transplant minimal residual disease and chimerism in childhood lymphoblastic leukaemia: 90 d to react. Br J Haematol 169 (2): 249-61, 2015.[PUBMED Abstract]
- Bader P, Kreyenberg H, Hoelle W, et al.: Increasing mixed chimerism is an important prognostic factor for unfavorable outcome in children with acute lymphoblastic leukemia after allogeneic stem-cell transplantation: possible role for pre-emptive immunotherapy? J Clin Oncol 22 (9): 1696-705, 2004.[PUBMED Abstract]
- Gandemer V, Pochon C, Oger E, et al.: Clinical value of pre-transplant minimal residual disease in childhood lymphoblastic leukaemia: the results of the French minimal residual disease-guided protocol. Br J Haematol 165 (3): 392-401, 2014.[PUBMED Abstract]
- Rubin J, Vettenranta K, Vettenranta J, et al.: Use of intrathecal chemoprophylaxis in children after SCT and the risk of central nervous system relapse. Bone Marrow Transplant 46 (3): 372-8, 2011.[PUBMED Abstract]
- Thompson CB, Sanders JE, Flournoy N, et al.: The risks of central nervous system relapse and leukoencephalopathy in patients receiving marrow transplants for acute leukemia. Blood 67 (1): 195-9, 1986.[PUBMED Abstract]
- Oshima K, Kanda Y, Yamashita T, et al.: Central nervous system relapse of leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 14 (10): 1100-7, 2008.[PUBMED Abstract]
- Ruutu T, Corradini P, Gratwohl A, et al.: Use of intrathecal prophylaxis in allogeneic haematopoietic stem cell transplantation for malignant blood diseases: a survey of the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant 35 (2): 121-4, 2005.[PUBMED Abstract]
- Maude SL, Laetsch TW, Buechner J, et al.: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378 (5): 439-448, 2018.[PUBMED Abstract]
- Mehta J, Powles R, Treleaven J, et al.: Outcome of acute leukemia relapsing after bone marrow transplantation: utility of second transplants and adoptive immunotherapy. Bone Marrow Transplant 19 (7): 709-19, 1997.[PUBMED Abstract]
- Kuhlen M, Willasch AM, Dalle JH, et al.: Outcome of relapse after allogeneic HSCT in children with ALL enrolled in the ALL-SCT 2003/2007 trial. Br J Haematol 180 (1): 82-89, 2018.[PUBMED Abstract]
- Eapen M, Giralt SA, Horowitz MM, et al.: Second transplant for acute and chronic leukemia relapsing after first HLA-identical sibling transplant. Bone Marrow Transplant 34 (8): 721-7, 2004.[PUBMED Abstract]
- Bosi A, Laszlo D, Labopin M, et al.: Second allogeneic bone marrow transplantation in acute leukemia: results of a survey by the European Cooperative Group for Blood and Marrow Transplantation. J Clin Oncol 19 (16): 3675-84, 2001.[PUBMED Abstract]
- Willasch AM, Salzmann-Manrique E, Krenn T, et al.: Treatment of relapse after allogeneic stem cell transplantation in children and adolescents with ALL: the Frankfurt experience. Bone Marrow Transplant 52 (2): 201-208, 2017.[PUBMED Abstract]
- Yaniv I, Krauss AC, Beohou E, et al.: Second Hematopoietic Stem Cell Transplantation for Post-Transplantation Relapsed Acute Leukemia in Children: A Retrospective EBMT-PDWP Study. Biol Blood Marrow Transplant 24 (8): 1629-1642, 2018.[PUBMED Abstract]
- Nishikawa T, Inagaki J, Nagatoshi Y, et al.: The second therapeutic trial for children with hematological malignancies who relapsed after their first allogeneic SCT: long-term outcomes. Pediatr Transplant 16 (7): 722-8, 2012.[PUBMED Abstract]
- Bajwa R, Schechter T, Soni S, et al.: Outcome of children who experience disease relapse following allogeneic hematopoietic SCT for hematologic malignancies. Bone Marrow Transplant 48 (5): 661-5, 2013.[PUBMED Abstract]
- Schechter T, Avila L, Frangoul H, et al.: Effect of acute graft-versus-host disease on the outcome of second allogeneic hematopoietic stem cell transplant in children. Leuk Lymphoma 54 (1): 105-9, 2013.[PUBMED Abstract]
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.[PUBMED Abstract]
- Collins RH, Goldstein S, Giralt S, et al.: Donor leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplant 26 (5): 511-6, 2000.[PUBMED Abstract]
- Levine JE, Barrett AJ, Zhang MJ, et al.: Donor leukocyte infusions to treat hematologic malignancy relapse following allo-SCT in a pediatric population. Bone Marrow Transplant 42 (3): 201-5, 2008.[PUBMED Abstract]
- Bhadri VA, McGregor MR, Venn NC, et al.: Isolated testicular relapse after allo-SCT in boys with ALL: outcome without second transplant. Bone Marrow Transplant 45 (2): 397-9, 2010.[PUBMED Abstract]
- von Stackelberg A, Locatelli F, Zugmaier G, et al.: Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J Clin Oncol 34 (36): 4381-4389, 2016.[PUBMED Abstract]
- Kantarjian H, Thomas D, Jorgensen J, et al.: Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer 119 (15): 2728-36, 2013.[PUBMED Abstract]
- Kantarjian HM, DeAngelo DJ, Stelljes M, et al.: Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med 375 (8): 740-53, 2016.[PUBMED Abstract]
- Kebriaei P, Cutler C, de Lima M, et al.: Management of important adverse events associated with inotuzumab ozogamicin: expert panel review. Bone Marrow Transplant 53 (4): 449-456, 2018.[PUBMED Abstract]
- Grupp SA, Kalos M, Barrett D, et al.: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368 (16): 1509-18, 2013.[PUBMED Abstract]
- Maude SL, Frey N, Shaw PA, et al.: Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371 (16): 1507-17, 2014.[PUBMED Abstract]
- Fitzgerald JC, Weiss SL, Maude SL, et al.: Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit Care Med 45 (2): e124-e131, 2017.[PUBMED Abstract]
- Shalabi H, Wolters PL, Martin S, et al.: Systematic Evaluation of Neurotoxicity in Children and Young Adults Undergoing CD22 Chimeric Antigen Receptor T-Cell Therapy. J Immunother 41 (7): 350-358, 2018.[PUBMED Abstract]
- Gardner RA, Finney O, Annesley C, et al.: Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129 (25): 3322-3331, 2017.[PUBMED Abstract]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al.: T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385 (9967): 517-28, 2015.[PUBMED Abstract]
- Curran KJ, Margossian SP, Kernan NA, et al.: Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 134 (26): 2361-2368, 2019.[PUBMED Abstract]
- Sotillo E, Barrett DM, Black KL, et al.: Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5 (12): 1282-95, 2015.[PUBMED Abstract]
- Fry TJ, Shah NN, Orentas RJ, et al.: CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 24 (1): 20-28, 2018.[PUBMED Abstract]
- Pan J, Niu Q, Deng B, et al.: CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 33 (12): 2854-2866, 2019.[PUBMED Abstract]
- Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.[PUBMED Abstract]
- Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.[PUBMED Abstract]
- Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.[PUBMED Abstract]
- Ritchey AK, Pollock BH, Lauer SJ, et al.: Improved survival of children with isolated CNS relapse of acute lymphoblastic leukemia: a pediatric oncology group study . J Clin Oncol 17 (12): 3745-52, 1999.[PUBMED Abstract]
- Domenech C, Mercier M, Plouvier E, et al.: First isolated extramedullary relapse in children with B-cell precursor acute lymphoblastic leukaemia: results of the Cooprall-97 study. Eur J Cancer 44 (16): 2461-9, 2008.[PUBMED Abstract]
- Hagedorn N, Acquaviva C, Fronkova E, et al.: Submicroscopic bone marrow involvement in isolated extramedullary relapses in childhood acute lymphoblastic leukemia: a more precise definition of "isolated" and its possible clinical implications, a collaborative study of the Resistant Disease Committee of the International BFM study group. Blood 110 (12): 4022-9, 2007.[PUBMED Abstract]
- Ribeiro RC, Rivera GK, Hudson M, et al.: An intensive re-treatment protocol for children with an isolated CNS relapse of acute lymphoblastic leukemia. J Clin Oncol 13 (2): 333-8, 1995.[PUBMED Abstract]
- Kumar P, Kun LE, Hustu HO, et al.: Survival outcome following isolated central nervous system relapse treated with additional chemotherapy and craniospinal irradiation in childhood acute lymphoblastic leukemia. Int J Radiat Oncol Biol Phys 31 (3): 477-83, 1995.[PUBMED Abstract]
- Yoshihara T, Morimoto A, Kuroda H, et al.: Allogeneic stem cell transplantation in children with acute lymphoblastic leukemia after isolated central nervous system relapse: our experiences and review of the literature. Bone Marrow Transplant 37 (1): 25-31, 2006.[PUBMED Abstract]
- Harker-Murray PD, Thomas AJ, Wagner JE, et al.: Allogeneic hematopoietic cell transplantation in children with relapsed acute lymphoblastic leukemia isolated to the central nervous system. Biol Blood Marrow Transplant 14 (6): 685-92, 2008.[PUBMED Abstract]
- Eapen M, Zhang MJ, Devidas M, et al.: Outcomes after HLA-matched sibling transplantation or chemotherapy in children with acute lymphoblastic leukemia in a second remission after an isolated central nervous system relapse: a collaborative study of the Children's Oncology Group and the Center for International Blood and Marrow Transplant Research. Leukemia 22 (2): 281-6, 2008.[PUBMED Abstract]
- Wofford MM, Smith SD, Shuster JJ, et al.: Treatment of occult or late overt testicular relapse in children with acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 10 (4): 624-30, 1992.[PUBMED Abstract]
- Trigg ME, Steinherz PG, Chappell R, et al.: Early testicular biopsy in males with acute lymphoblastic leukemia: lack of impact on subsequent event-free survival. J Pediatr Hematol Oncol 22 (1): 27-33, 2000 Jan-Feb.[PUBMED Abstract]
- van den Berg H, Langeveld NE, Veenhof CH, et al.: Treatment of isolated testicular recurrence of acute lymphoblastic leukemia without radiotherapy. Report from the Dutch Late Effects Study Group. Cancer 79 (11): 2257-62, 1997.[PUBMED Abstract]
- Barredo JC, Hastings C, Lu X, et al.: Isolated late testicular relapse of B-cell acute lymphoblastic leukemia treated with intensive systemic chemotherapy and response-based testicular radiation: A Children's Oncology Group study. Pediatr Blood Cancer 65 (5): e26928, 2018.[PUBMED Abstract]
- 本要約の変更点(05/04/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
本要約は再編集された。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児急性リンパ芽球性白血病の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Childhood Acute Lymphoblastic Leukemia Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/leukemia/hp/child-all-treatment-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389206]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。