

ご利用について
医療専門家向けの本PDQがん情報要約では、小児髄芽腫およびその他の中枢神経系胚芽腫の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 髄芽腫および他の中枢神経系(CNS)胚芽腫に関する一般情報
-
CNS胚芽腫および松果体芽腫に対する世界保健機関(WHO)分類
胚芽腫は生物学的に異種性の病変の集まりで、いずれも脳脊髄液(CSF)経路を介して神経系の至るところに播種する傾向を有する。これらの腫瘍には大きな差があるが、少なくとも部分的に過染色性細胞(標準の染色で青色細胞腫瘍)と少量の細胞質(高密度に集積し、高度の細胞分裂能を示す)で構成されることから、これらの腫瘍は組織学的には一緒に分類される。他の組織学的および免疫組織化学的特徴(同定可能な細胞系列[上衣腫、グリア系腫瘍など]での明らかな細胞形質転換の程度など)は、これらの腫瘍をある程度分類するのに使用できる。しかしながら、WHOに受け入れられている慣例ではまた、これらの腫瘍を中枢神経系(CNS)内で推定される原発巣の位置に基づいて分類している。分子的研究により、脳の異なる領域で発生した腫瘍間での差が実証され、この分類アプローチの信頼性を部分的に高めている。[ 1 ]
2016年時点で、WHOはCNS腫瘍について表現型と遺伝子型を用いた分類システムを統合するように提案しており、この分類システムでは、WHO分類悪性度、組織学的分類、および分子分類を用いて診断が階層化される。[ 2 ]最新のWHO分類の診断用語から原始神経外胚葉性腫瘍(PNET)という用語が削除されているが、一部のまれな疾患実体(例、髄上皮腫)は残されている。分子的に異なる疾患実体、多層性ロゼットを有する胎児性腫瘍(ETMR)、C19MC変異型が追加され、神経網および真性ロゼットに富む胎児性腫瘍(ETANTR)、上衣芽腫、および髄上皮腫が包含されている。WHO分類は、分子的に異なる他の疾患実体が定義されるにつれて更新される。
胚芽腫の病理診断は主として、組織学的および免疫組織学的な顕微鏡的特徴に基づく。しかしながら、胚芽腫を下位分類するための分子遺伝学的検査の使用が増えている。こうした分子遺伝学的所見はまた、リスク層別化や治療計画の立案でも用いられている。[ 3 ][ 4 ][ 5 ][ 6 ]
胚芽腫に対する2016年のWHO分類は以下の通りである:[ 2 ]
以前は慣習的に胚芽腫とともに分類されていた松果体芽腫は、WHOにより松果体実質細胞腫瘍として分類されている。松果体芽腫に対する治療法が胚芽腫に用いられている治療法に非常に類似していることを考慮して、松果体芽腫についても本要約で考察する。いくぶん密接して並んだ腫瘍である中分化型松果体実質細胞腫瘍が同定されているが、胚芽腫とは考えられておらず、主として成人に発生する。[ 2 ]
解剖学
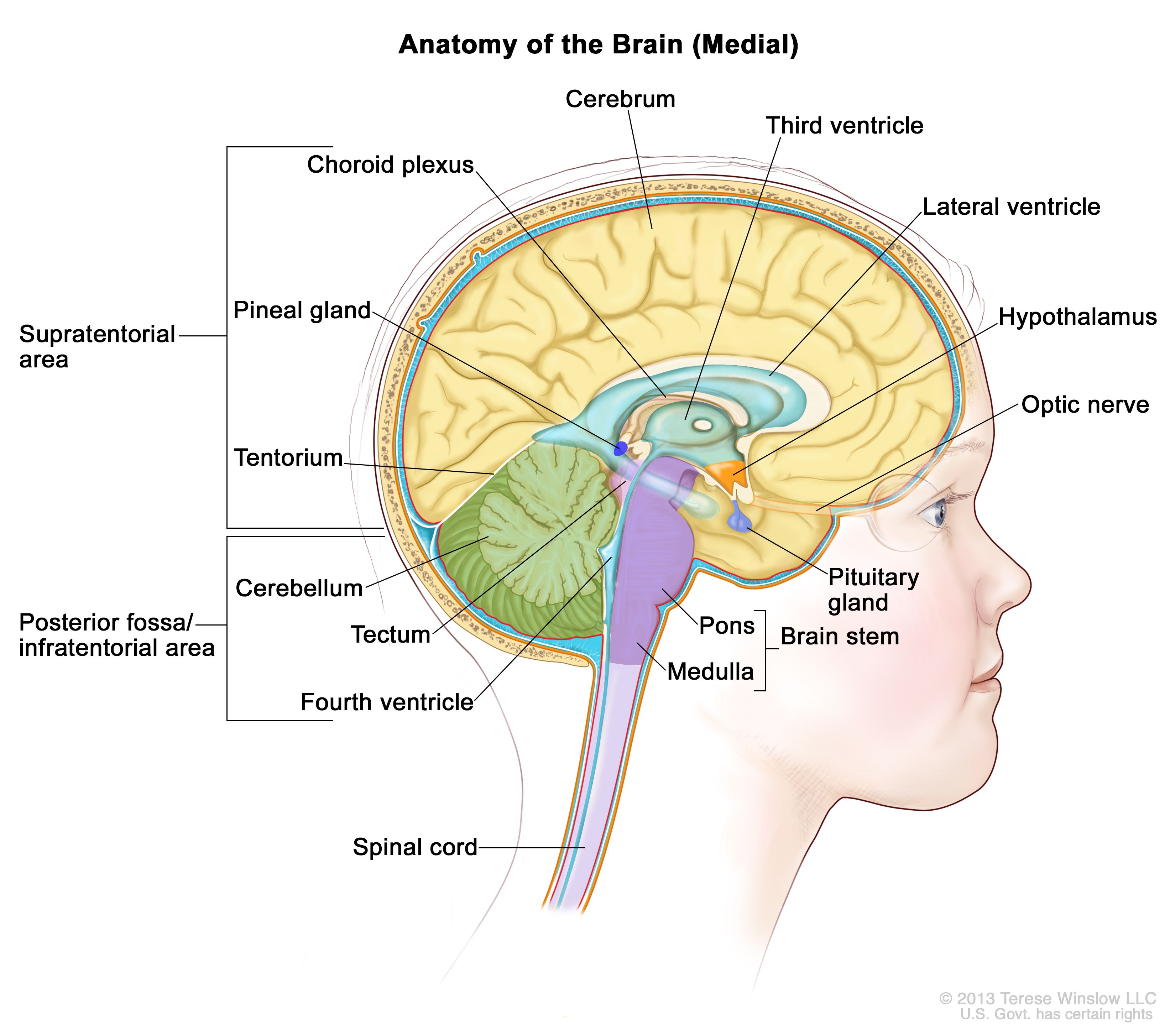
図1.脳内部の解剖図で、松果体、下垂体、視神経、脳室(脳脊髄液を水色で示している)など、脳の構成要素が示されている。後頭蓋窩はテント下の領域で、大脳皮質と小脳を分けており、基本的には脳幹、小脳、および第四脳室を含む領域を示す。 発生率
胚芽腫は小児に発生する原発性CNS腫瘍(悪性脳腫瘍および毛様細胞性星細胞腫)の20~25%を占める。これらの腫瘍はいずれの年齢の小児でも発生するが、年少期に集中して発生する傾向がある。1~9歳の小児における胚芽腫の発生率は、成人における胚芽腫の発生率よりも5~10倍高い(表1を参照のこと)。[ 7 ][ 8 ]
表1.小児中枢神経系胚芽腫の年齢に応じた年間発生率a 年齢グループ(歳) 年間発生率(100万人当たりの症例数) a出典:Childhood cancer by the International Classification of Childhood Cancer [ 7 ] and Smoll et al.[ 8 ] <5 11 5–9 7 10–19 3–4 髄芽腫は小児胚芽腫の大多数を占めており、定義上、後頭蓋窩(図1を参照のこと)に発生し、すべての後頭蓋窩腫瘍の約40%を占める。他の型の胚芽腫はそれぞれ、小児脳腫瘍全体の2%以下を構成する。
診断的評価および病期評価
画像検査およびCSF分析が診断的評価および病期評価に含められる。
画像検査
診断は通常、磁気共鳴画像法(MRI)またはコンピュータ断層撮影(CT)のいずれかにより容易に下される。腫瘍と周囲の脳および腫瘍播種の解剖学的関係の視覚化に優れているため、MRIが好ましい。[ 9 ]
診断後の胚芽腫の評価は、実質的に腫瘍の組織学的サブタイプと位置に関係なく、きわめて類似している。これらの腫瘍が疾患経過の早期にCNS全体に播種する傾向があることを考慮して、脳および脊椎全体のMRIを用いた脳脊髄幹の画像評価が適応とされる。術後のアーチファクト(特に血液)を回避するため、こうした画像法は術前の実施が望ましい。こうした画像検査は解釈が困難なことがあり、造影増強(ガドリニウム)を併用したおよび併用しない少なくとも2平面画像上で行うべきである。[ 10 ]髄芽腫の小児における脊髄MRIでの曖昧な所見の意義に関する1件の研究で、患者100人中48人(48%)に曖昧な所見が確認された。この研究では以下の結果が報告された:
しかしながら、診断時に転移病変が認められず、23.4Gyの頭蓋脊髄照射で治療された患者における経験について記述した1編の論文において、曖昧な所見は比較的不良なイベントフリー生存(EFS)およびOSに関連していた。[ 10 ]中枢神経軸全体の術前の評価や残存腫瘍量に関する術後評価を含めて、時期および神経画像検査技術に関して複数のコンセンサスガイドラインが提案されている。[ 12 ]
手術後、残存病変の範囲を判断するために原発腫瘍部位の画像検査が適応となる。
CSF分析
手術後、安全であると考えられる場合は、腰椎部のCSF分析が実施される。10%もの患者が、MRIスキャンで軟髄膜病変の明確な証拠がないのにCSF内に腫瘍細胞が浮遊している証拠を示すことから、神経画像検査およびCSF評価は相補的なものと考えられる。[ 13 ]
CSF分析は慣習的に手術の14~21日後に施行される。手術後14日以内にCSFを採取し、脊髄液内に腫瘍細胞が検出された場合は、外科的手技に関係している可能性がある。ほとんどの病期分類システムでは、術後数日間のうちに脳脊髄液を採取して腫瘍細胞が陽性であった場合、診断的に重要とみなすためにはその後に脊椎穿刺により陽性を確定する必要がある。腰椎部の脊椎穿刺による脊髄液の採取が安全ではないと考えられる場合は、脳室液を採取してもよい;しかしながら、この方法は腰椎部脊髄液の評価ほど感度が高くない可能性がある。[ 13 ]
胚芽腫が診断時に骨、骨髄をはじめとする他の身体部位に転移していることはきわめてまれであるため、骨髄穿刺、胸部X線、骨スキャンなどの検査は、こうした臓器への転移を示唆する症状または徴候がない限り適応とならない。
予後因子
さまざまな臨床的および生物学的パラメータが治療後の胚芽腫における疾患制御の可能性に関連することが示されている。[ 4 ]これらの因子の多くが髄芽腫の予測に重要であることが示されているが、他の胚芽腫についてのリスクの指定にある程度用いられるものもある。転帰の予測に最も頻繁に用いられるパラメータには以下のものがある:[ 14 ][ 15 ]
転帰はまた腫瘍の分子的特徴に関係しているということが特に髄芽腫で次第に明らかになっているが、このことは他の胚芽腫では明確には示されていない。[ 1 ][ 5 ][ 6 ][ 16 ][ 17 ][ 18 ]OS率は、髄芽腫の分子的サブタイプおよびおそらく診断時の播種の範囲や切除範囲など、他の因子によって40~90%に及ぶ。5年生存が得られた髄芽腫の小児は、腫瘍が治癒したとみなされる。他の胚芽腫の生存率は一般的にこれより不良で、5%未満から50%であり、個別の生存率は本要約の各サブグループ内で考察する。[ 19 ][ 20 ][ 21 ][ 22 ]
過去の研究では、髄芽腫の小児に脳幹病変が存在する場合は予後因子であることが認められていた;放射線と化学療法を併用したその後の研究では、脳幹病変の存在に予測価値があるとは示されていない。[ 10 ][ 14 ]
正確な診断は胚芽腫患者にとって非常に重要な問題である。例えば、高リスク髄芽腫、テント上CNS-PNET腫瘍、および松果体芽腫の患者80人が登録したACNS0332(NCT00392327)試験において、60人の患者では評価のために十分な組織を得られていた。31個の腫瘍は位置が松果体以外で、このうち22個(71%)は、試験に適格とされない腫瘍であった(18個が高悪性度グリオーマ、2個が非定型奇形腫様/ラブドイド腫瘍、および2個が上衣腫)。腫瘍タイプ間の転帰は著しく異なっていた。テント上胚芽腫/松果体芽腫患者は62.8%(95%信頼区間[CI]、43.4%-82.2%)の5年EFS率および78.5%(95%CI、62.2%-94.8%)のOS率を示した一方、高悪性度グリオーマと分子的に分類された患者のEFS率は5.6%(95%CI、0%-13%)およびOS率は12.0%(95%CI、0%-24.7%)であった。高悪性度グリオーマ患者の生存率は、頭蓋脊髄照射および強化化学療法が回避されていた過去の研究に登録された患者の生存率とほぼ同じであった。したがって、CNS-PNET/松果体芽腫患者の予後は、分子的に確認された高悪性度グリオーマを除外すると、以前に想定されていたよりもかなり良好である。[ 23 ]
髄上皮腫およびETMRを有する患者の予後は不良であり、5年生存率は0~30%である。[ 24 ][ 25 ][ 26 ]38人の患者を対象にした1件のレトロスペクティブ多変量解析では、完全切除またはほぼ完全切除、放射線療法の使用、および大量化学療法の使用が予後の改善に関連した。[ 27 ][証拠レベル:3iA]
診断時のCNS病変の範囲
診断時に播種性CNS病変が認められる患者は、腫瘍再燃のリスクが最も高い。[ 13 ][ 14 ][ 15 ]髄芽腫患者の10~40%は診断時に播種性CNS病変が認められ、幼児で発生率が最も高く、青年および成人では最も低い。
髄芽腫以外の胚芽腫および松果体芽腫もまた診断時に播種が見られることがあるが、播種の発生率は髄芽腫よりもいくぶん低く、診断時の播種は患者の約10~20%で報告されている。[ 19 ][ 20 ]診断時に播種性病変が認められる髄芽腫以外の胚芽腫および松果体芽腫患者のOS率は不良であり、5年生存率は10~30%と報告されている。[ 19 ][ 20 ][ 21 ][ 22 ]
診断時年齢
診断時年齢が3歳未満の場合(線維形成性髄芽腫/MBENを除く)は、髄芽腫患者およびおそらく、他の胚芽腫患者でも不良な転帰が予測される。[ 28 ][ 29 ][ 30 ][ 31 ][ 32 ]
根治的手術後の残存腫瘍量
治療成績の予測因子として、根治的手術後の残存腫瘍量の術後のMRI測定は、手術後の切除範囲に取って代わられている。[ 10 ]
過去の研究で、髄芽腫に対する切除範囲は生存に関係することが明らかにされた。[ 14 ][ 15 ][ 33 ][ 34 ]340人の小児を対象にしたHIrnTumor and International Society of Paediatric Oncology(HIT-SIOP)研究による報告では、残存腫瘍(1.5cm2を超える)は、より不良な5年EFS率を暗示した。[ 35 ]手術後の切除範囲はなおも患者を各リスク群に分類するために使用されており、残存腫瘍が1.5cm2を超える患者は高リスク群に層別化され、頭蓋脊髄照射が36Gyに強化される。
1件のレトロスペクティブ国際共同研究には、さまざまな方法で治療された全年齢層の髄芽腫患者787人が含められ、解析には分子的亜型分類および臨床的因子が組み込まれた。多変量解析により、亜全切除(腫瘍残存が1.5cm2を超える)(ただし、ほぼ完全な切除[腫瘍残存が1.5cm2未満]ではない)は、肉眼的完全切除よりも無増悪生存が不良であったことが明らかにされた。この研究により、腫瘍を完全に切除する試みは、特に神経学的罹病の可能性が高い場合は、ほぼ完全な切除と比較して肉眼的完全切除の有益性がほとんどまたは全く得られないようであるため、妥当ではないことが示唆された。この研究から、1.5cm2超の病変を有する患者が高リスク患者と考えられる場合、現在のアプローチはある程度信頼できる。[ 36 ]分子的に定義されたサブグループにおいて切除範囲が治療成績に及ぼす影響をより良く定義するには、プロスペクティブ研究が必要である。
他の型の胚芽腫患者では、切除範囲が生存に影響することは明確には示されていない。[ 21 ]しかし、テント上胚芽腫の小児66人を対象にした小児腫瘍学グループの1件の研究では、切除範囲は診断時に限局性疾患であった患者について予後因子となることが明らかにされた。[ 37 ]
腫瘍の病理組織学
大細胞変異体、退形成、線維形成といった髄芽腫の病理組織学的特徴は、レトロスペクティブ解析において転帰と相関することが明らかにされている。[ 29 ][ 38 ][ 39 ]プロスペクティブ研究によると、診断時に3歳より年長の小児における免疫組織化学所見および病理組織所見は、より不良な予後に関連している大細胞/退形成変異体を除いて転帰を予測しなかった。[ 10 ][ 18 ][ 40 ]線維形成性髄芽腫(特にMBEN)を有する3歳以下の患者にみられる線維形成の組織学的所見は、古典的髄芽腫または大細胞型/退形成性髄芽腫の乳児および幼児の転帰と比べて予後が有意に良好なことを暗示することが数件の研究で観察されている。[ 18 ][ 28 ][ 29 ][ 30 ][ 41 ];[ 31 ][証拠レベル:2A]
他の胚芽腫については、組織学的な違いは多様な転帰と関連していない。
腫瘍細胞の生物学的/分子的特徴
新鮮凍結切片とホルマリン固定パラフィン包埋切片の両方でのゲノム解析(変異を同定するためのRNA遺伝子発現およびDNAメチル化プロファイルのほか、DNA塩基配列決定法を含む)により、髄芽腫の分子的サブタイプが同定されている。[ 3 ][ 4 ][ 5 ][ 6 ][ 16 ][ 17 ][ 42 ][ 43 ][ 44 ][ 45 ][ 46 ][ 47 ][ 48 ][ 49 ]これらのサブタイプには、WNT経路の活性化およびSHH経路の活性化を特徴とするもののほか、MYCやMYCNの変化など、ゲノムの変化を特徴とする追加のサブグループが含まれる。[ 3 ][ 4 ][ 5 ][ 6 ][ 16 ][ 17 ][ 42 ][ 43 ][ 44 ][ 45 ][ 46 ][ 47 ][ 48 ]腫瘍がWNT経路の活性化を示す髄芽腫の小児は通常、予後が非常に優れているのに対し、SHH経路が活性化した腫瘍を有する患者の予後はTP53変異の有無による影響を受ける(それぞれ、予後不良 vs 予後良好)。[ 50 ]残りの患者の転帰は、WNT経路の活性化を示す患者の転帰よりも不良である。髄芽腫における変異はサブタイプ特異的に観察されており、WNTサブタイプではCTNNB1変異が観察され、SHHサブタイプではPTCH1、SMO、およびSUFU変異が観察されている。反復性の変異の予後に対する意義は、そうした変異と関連する分子的サブタイプの予後に対する意義とぴったりと合致している。[ 4 ][ 51 ]再発時のサブタイプは、診断時の元の分子的サブタイプから変わらない。[ 52 ]
髄芽腫のサブタイプおよび特異的な分子的変化の予後的意義に関する詳しい情報については、本要約の髄芽腫の分子的サブタイプのセクションを参照のこと。
髄芽腫以外の胚芽腫に対する統合的ゲノム解析でもまた、さまざまな転帰を示す分子的サブタイプが同定されている。(詳細な情報については、本要約の髄芽腫以外の胚芽腫のサブタイプのセクションを参照のこと。)
治療後の追跡
胚芽腫の小児における再燃は、診断後最初の18ヵ月以内に起こる可能性が最も高い。[ 35 ][ 53 ]通常は、脳および脊椎に対する画像検査によるサーベイランスがルーチンの間隔で治療中および治療後に行われる(表2を参照のこと)。早期の無症状の状態で再発病変を検出できるようにデザインされたこうした画像検査の頻度は任意に決定されており、生存に明確に影響することは示されていない。[ 54 ][ 55 ][ 56 ][ 57 ]成長ホルモン補充療法が腫瘍再燃の可能性を高めることは示されていない。[ 30 ]
表2.髄芽腫および他の中枢神経系胚芽腫の治療中および治療後のサーベイランス検査 サーベイランス期間 サーベイランス期間中の受診頻度 検査 MRI = 磁気共鳴画像法。 a松果体芽腫については、診断から5年経過するまで6ヵ月ごとの脊髄の評価を継続する。これらの提唱は小規模のサンプルサイズに基づいているが、診断から5年経過するまで脊髄のサーベイランス検査を継続するための証拠がある。[ 58 ] 診断後最初の3年間 3ヵ月ごと 身体診察 最初の3年間は3ヵ月ごとの脳の画像検査、続いて次の2年間は6ヵ月ごと、その後は治療担当医の好みまたはプロトコルに従う;最初の2年間は3ヵ月ごとの脊髄のMRI、続いて1年間は6ヵ月ごと、その後は治療担当医の好みまたはプロトコルに従う。a 年1回の内分泌学の評価 1~2年ごとの神経心理学的検査 診断後3~5年間 6ヵ月ごと 身体診察 年1回の脳と脊椎の画像検査 年1回の内分泌学の評価 1~2年ごとの神経心理学的検査 診断から5年後以降 年1回 身体診察 年1回の脳の画像検査 年1回の内分泌学の評価 1~2年ごとの神経心理学的検査(任意) 小児および青年がん生存者には、治療から数ヵ月または数年経過後もがん療法の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。(小児および青年がん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)のPDQ要約を参照のこと。)
参考文献- Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Pfister S, Remke M, Benner A, et al.: Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27 (10): 1627-36, 2009.[PUBMED Abstract]
- Taylor MD, Northcott PA, Korshunov A, et al.: Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465-72, 2012.[PUBMED Abstract]
- Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088, 2008.[PUBMED Abstract]
- Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23 (31): 7951-7, 2005.[PUBMED Abstract]
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). Bethesda, Md: National Cancer Institute, 2012, Section 29. Also available online. Last accessed February 06, 2020.[PUBMED Abstract]
- Smoll NR, Drummond KJ: The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci 19 (11): 1541-4, 2012.[PUBMED Abstract]
- Chintagumpala MM, Paulino A, Panigrahy A, et al.: Embryonal and pineal region tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 671-99.[PUBMED Abstract]
- Packer RJ, Gajjar A, Vezina G, et al.: Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24 (25): 4202-8, 2006.[PUBMED Abstract]
- Bennett J, Ashmawy R, Ramaswamy V, et al.: The clinical significance of equivocal findings on spinal MRI in children with medulloblastoma. Pediatr Blood Cancer 64 (8): , 2017.[PUBMED Abstract]
- Warren KE, Vezina G, Poussaint TY, et al.: Response assessment in medulloblastoma and leptomeningeal seeding tumors: recommendations from the Response Assessment in Pediatric Neuro-Oncology committee. Neuro Oncol 20 (1): 13-23, 2018.[PUBMED Abstract]
- Fouladi M, Gajjar A, Boyett JM, et al.: Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol 17 (10): 3234-7, 1999.[PUBMED Abstract]
- Zeltzer PM, Boyett JM, Finlay JL, et al.: Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol 17 (3): 832-45, 1999.[PUBMED Abstract]
- Yao MS, Mehta MP, Boyett JM, et al.: The effect of M-stage on patterns of failure in posterior fossa primitive neuroectodermal tumors treated on CCG-921: a phase III study in a high-risk patient population. Int J Radiat Oncol Biol Phys 38 (3): 469-76, 1997.[PUBMED Abstract]
- Thompson MC, Fuller C, Hogg TL, et al.: Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24 (12): 1924-31, 2006.[PUBMED Abstract]
- Northcott PA, Korshunov A, Witt H, et al.: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 (11): 1408-14, 2011.[PUBMED Abstract]
- Rutkowski S, von Hoff K, Emser A, et al.: Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 28 (33): 4961-8, 2010.[PUBMED Abstract]
- Cohen BH, Zeltzer PM, Boyett JM, et al.: Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Childrens Cancer Group randomized trial. J Clin Oncol 13 (7): 1687-96, 1995.[PUBMED Abstract]
- Reddy AT, Janss AJ, Phillips PC, et al.: Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88 (9): 2189-93, 2000.[PUBMED Abstract]
- Timmermann B, Kortmann RD, Kühl J, et al.: Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol 20 (3): 842-9, 2002.[PUBMED Abstract]
- Jakacki RI, Zeltzer PM, Boyett JM, et al.: Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Childrens Cancer Group. J Clin Oncol 13 (6): 1377-83, 1995.[PUBMED Abstract]
- Hwang EI, Kool M, Burger PC, et al.: Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children's Oncology Group Randomized ACNS0332 Trial. J Clin Oncol : JCO2017764720, 2018.[PUBMED Abstract]
- Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.[PUBMED Abstract]
- Sharma MC, Mahapatra AK, Gaikwad S, et al.: Pigmented medulloepithelioma: report of a case and review of the literature. Childs Nerv Syst 14 (1-2): 74-8, 1998 Jan-Feb.[PUBMED Abstract]
- Müller K, Zwiener I, Welker H, et al.: Curative treatment for central nervous system medulloepithelioma despite residual disease after resection. Report of two cases treated according to the GPHO Protocol HIT 2000 and review of the literature. Strahlenther Onkol 187 (11): 757-62, 2011.[PUBMED Abstract]
- Horwitz M, Dufour C, Leblond P, et al.: Embryonal tumors with multilayered rosettes in children: the SFCE experience. Childs Nerv Syst 32 (2): 299-305, 2016.[PUBMED Abstract]
- Leary SE, Zhou T, Holmes E, et al.: Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 117 (14): 3262-7, 2011.[PUBMED Abstract]
- Giangaspero F, Perilongo G, Fondelli MP, et al.: Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91 (6): 971-7, 1999.[PUBMED Abstract]
- von Bueren AO, von Hoff K, Pietsch T, et al.: Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 13 (6): 669-79, 2011.[PUBMED Abstract]
- Rutkowski S, Gerber NU, von Hoff K, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol 11 (2): 201-10, 2009.[PUBMED Abstract]
- Rutkowski S, Bode U, Deinlein F, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352 (10): 978-86, 2005.[PUBMED Abstract]
- Albright AL, Wisoff JH, Zeltzer PM, et al.: Effects of medulloblastoma resections on outcome in children: a report from the Children's Cancer Group. Neurosurgery 38 (2): 265-71, 1996.[PUBMED Abstract]
- Taylor RE, Bailey CC, Robinson K, et al.: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol 21 (8): 1581-91, 2003.[PUBMED Abstract]
- Lannering B, Rutkowski S, Doz F, et al.: Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol 30 (26): 3187-93, 2012.[PUBMED Abstract]
- Thompson EM, Hielscher T, Bouffet E, et al.: Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol 17 (4): 484-95, 2016.[PUBMED Abstract]
- Jakacki RI, Burger PC, Kocak M, et al.: Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 776-83, 2015.[PUBMED Abstract]
- McManamy CS, Lamont JM, Taylor RE, et al.: Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62 (6): 627-32, 2003.[PUBMED Abstract]
- Massimino M, Antonelli M, Gandola L, et al.: Histological variants of medulloblastoma are the most powerful clinical prognostic indicators. Pediatr Blood Cancer 60 (2): 210-6, 2013.[PUBMED Abstract]
- Eberhart CG, Kratz J, Wang Y, et al.: Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol 63 (5): 441-9, 2004.[PUBMED Abstract]
- Garrè ML, Cama A, Bagnasco F, et al.: Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res 15 (7): 2463-71, 2009.[PUBMED Abstract]
- Tabori U, Baskin B, Shago M, et al.: Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28 (8): 1345-50, 2010.[PUBMED Abstract]
- Onvani S, Etame AB, Smith CA, et al.: Genetics of medulloblastoma: clues for novel therapies. Expert Rev Neurother 10 (5): 811-23, 2010.[PUBMED Abstract]
- Dubuc AM, Northcott PA, Mack S, et al.: The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10 (3): 215-23, 2010.[PUBMED Abstract]
- Giangaspero F, Wellek S, Masuoka J, et al.: Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112 (1): 5-12, 2006.[PUBMED Abstract]
- Jones DT, Jäger N, Kool M, et al.: Dissecting the genomic complexity underlying medulloblastoma. Nature 488 (7409): 100-5, 2012.[PUBMED Abstract]
- Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.[PUBMED Abstract]
- Kool M, Korshunov A, Remke M, et al.: Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473-84, 2012.[PUBMED Abstract]
- Schwalbe EC, Williamson D, Lindsey JC, et al.: DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125 (3): 359-71, 2013.[PUBMED Abstract]
- Goschzik T, Schwalbe EC, Hicks D, et al.: Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19 (12): 1602-1616, 2018.[PUBMED Abstract]
- Polkinghorn WR, Tarbell NJ: Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol 4 (5): 295-304, 2007.[PUBMED Abstract]
- Ramaswamy V, Remke M, Bouffet E, et al.: Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14 (12): 1200-7, 2013.[PUBMED Abstract]
- Packer RJ, Zhou T, Holmes E, et al.: Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children's Oncology Group trial A9961. Neuro Oncol 15 (1): 97-103, 2013.[PUBMED Abstract]
- Shaw DW, Geyer JR, Berger MS, et al.: Asymptomatic recurrence detection with surveillance scanning in children with medulloblastoma. J Clin Oncol 15 (5): 1811-3, 1997.[PUBMED Abstract]
- Torres CF, Rebsamen S, Silber JH, et al.: Surveillance scanning of children with medulloblastoma. N Engl J Med 330 (13): 892-5, 1994.[PUBMED Abstract]
- Kramer ED, Vezina LG, Packer RJ, et al.: Staging and surveillance of children with central nervous system neoplasms: recommendations of the Neurology and Tumor Imaging Committees of the Children's Cancer Group. Pediatr Neurosurg 20 (4): 254-62; discussion 262-3, 1994.[PUBMED Abstract]
- Sabel M, Fleischhack G, Tippelt S, et al.: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129 (3): 515-524, 2016.[PUBMED Abstract]
- Perreault S, Lober RM, Carret AS, et al.: Surveillance imaging in children with malignant CNS tumors: low yield of spine MRI. J Neurooncol 116 (3): 617-23, 2014.[PUBMED Abstract]
- 小児髄芽腫
-
髄芽腫に関連した遺伝性がん素因症候群
髄芽腫を含む脳腫瘍を有する小児のサブセットは、髄芽腫だけでなく他のがんも発症する素因となる生殖細胞変異を有することが次第に明らかになってきている。[ 1 ][ 2 ]これらの変異は、がんのサーベイランス、予防、診断、および管理に関して罹患した小児、同胞、両親のほか、潜在的に他の家系員に対して明らかな含意を有する。こうした変異はまた、腫瘍の特異的治療にも影響しうる。
髄芽腫は患者の約5%では遺伝性がん素因症候群の状況で発生する。[ 1 ][ 2 ]1,000人を超える患者の大規模研究により、髄芽腫を診断された全患者の約5%において生殖細胞変異が実証された。APC、BRCA2、PALB2、PTCH1、SUFU、およびTP53において、生殖細胞変異が同定された。[ 2 ]
髄芽腫に関連していることが知られている症候群には以下のものがある:
G蛋白共役受容体161(GPR161)においてヘテロ接合性の有害な生殖細胞変異が、SHH髄芽腫症例の約3%で同定された。[ 18 ]GPR161は、SHHシグナル伝達の阻害因子である。GPR161変異症例の診断時年齢中央値は1.5歳であった。GPR161遺伝子座におけるヘテロ接合性の消失(LOH)がすべての腫瘍で示され、患者6人中5人の腫瘍が染色体1q(GPR161が存在する)のコピー数の変化を伴わないLOHを示した。有害なGPR161変異を有する患者における髄芽腫以外のがんリスクは明らかになっていない。
ときに、髄芽腫はこれらの素因遺伝子において生殖細胞変異が存在する場合に最初の症状となる場合がある。以下のような状況では、生殖細胞検査を検討すべきである:
臨床像
定義上、髄芽腫は後頭蓋窩に発生する。[ 19 ][ 20 ]約80%の小児では、髄芽腫が第四脳室領域に発生する。初期の総体症状の大部分は、脳脊髄液(CSF)の閉塞とその結果として生じる、水頭症と呼ばれる脳内のCSFの蓄積に関係する。髄芽腫の小児は通常、症状の発現から2~3ヵ月以内に診断される。髄芽腫は、一般的に以下の徴候および症状を呈する:[ 21 ]
髄芽腫患者の20%では診断時に水頭症が見られず、最初は小脳の障害が現れることが多い。例えば、比較的側方に位置する小脳の髄芽腫は水頭症を引き起こさないことがあり、その位置のために側方性の小脳機能障害(四肢運動失調)を生じる可能性が高く、片側の距離測定障害や不安定さ、腫瘍と同側の第六および第七脳神経の衰弱として発現する。後に、腫瘍が正中線に向かって増殖し、CSFを閉塞するにつれて、水頭症に関連するより典型的な症状が明らかとなる。
脳神経の所見は、片側性または両側性の第六脳神経麻痺(通常は水頭症に関係する)を除いて、比較的まれである。[ 21 ]ときに、髄芽腫では腫瘍内の出血の結果、嗜眠と意識喪失の急性発症により、症状が急激に現れる。
乳児では、髄芽腫の発症は多様であり、以下が含まれる:
診察で、頭蓋内圧亢進による大泉門の隆起や、中脳視蓋の圧迫に続発して上方視が見られないことから下向きに偏向した眼球(いわゆる落陽現象の徴候)などの異常な眼球運動が見られることがある。
細胞分類および分子分類
髄芽腫は世界保健機関(WHO)の分類で以下の4つの組織型が認識されている:[ 20 ]
核サイズの増大、顕著な細胞学的多形性、多数の有糸分裂、およびアポトーシス小体を含む、退形成の特徴を示す髄芽腫はかなりの注目を集めている。[ 22 ][ 23 ]ほとんどの髄芽腫にはある程度の退形成が認められるため、退形成の基準の使用は主観的である。退形成の病巣は古典的髄芽腫と大細胞型髄芽腫のいずれの組織学的特徴を有する腫瘍にも発生する可能性があり、退形成性の多様体と大細胞型の多様体間でかなりの重複がみられ、これらはしばしば大細胞型/退形成性髄芽腫と呼ばれる。[ 22 ][ 23 ]慣習では、退形成がびまん性(腫瘍の50~80%に退形成が発生しているものとして広く定義されている)の場合に、髄芽腫が退形成性と考えられる。
小脳半球に最も一般的に生じる線維形成性/結節性の組織型変異を伴う髄芽腫の発生率は、乳児で高く、小児では低いが、青年および成人では再び高くなる。線維形成性/結節性の組織型変異はMBEN(小結節形成を伴う髄芽腫は広範な小葉構造を有する)とは異なる。MBENのサブタイプはほぼ例外なく乳児に発生し、予後は非常に優れている。[ 7 ][ 24 ]
髄芽腫の分子的サブタイプ
統合的分子解析によって複数の髄芽腫のサブタイプが同定されている。[ 25 ][ 26 ][ 27 ][ 28 ][ 29 ][ 30 ][ 31 ][ 32 ][ 33 ][ 34 ][ 35 ][ 36 ][ 37 ][ 38 ][ 39 ][ 40 ][ 41 ][ 42 ]2012年以降、髄芽腫は、WNT活性化、ソニック・ヘッジホッグ(SHH)活性化、グループ3、グループ4の髄芽腫など分子的に少なくとも4つの中心となるサブタイプに分類が可能であるという一般的なコンセンサスが得られている。ただし、同じ腫瘍の異なる領域には、他の本質的に異なる遺伝子変異が存在する可能性が高く、有効な分子標的療法を考案する上で複雑さが増している。[ 43 ]これらのサブタイプは原発部位と転移部位で安定したままである。[ 44 ][ 45 ]
2016年の世界保健機関(WHO)分類は、分子的に定義された髄芽腫に対して以下のカテゴリーを追加することで、このコンセンサスを支持している:[ 20 ]
これらのサブグループはさらなる下位分類が可能であり、こうした分類によりさらに多くの予後情報が得られる。[ 45 ][ 46 ][ 47 ]
髄芽腫、WNT活性化
WNT腫瘍はWNTシグナル伝達経路の異常を伴う髄芽腫で、すべての髄芽腫の約10%を占める。[ 46 ]WNT髄芽腫はWNTシグナル伝達遺伝子発現特性および免疫組織化学検査でβ-カテニン核染色を示す。[ 48 ]WNT腫瘍は通常、組織学的に古典的髄芽腫に分類され、大細胞型/退形成性の像を示すことはまれである。WNT髄芽腫は一般的に比較的年齢の高い患者(年齢中央値、10歳)に発生し、診断時に転移を来していることはまれである。
CTNNB1変異はWNT腫瘍症例の85~90%で観察され、CTNNB1変異が認められない症例の多くでAPC変異が発見される。腫瘍にAPC変異がみられるWNT腫瘍患者では、しばしばターコット症候群(すなわち、APC生殖細胞変異)が認められる。[ 47 ]WNT髄芽腫は、CTNNB1変異に加えて、症例の80~90%で6qの欠失(6モノソミー)を示す。診断時に18歳未満の髄芽腫患者のほとんどで6モノソミーが観察される一方、18歳以上の患者での頻度ははるかに低くなる(症例の約25%)ようである。[ 46 ][ 48 ]
WNTサブタイプは、主に年長児、青年、および成人にみられ、男性に多いとは言えない。一部は胚性菱脳唇領域からの脳幹由来であると考えられている。[ 49 ]WNT髄芽腫は小児、特に腫瘍にβ-カテニン核染色および証明された6qの欠失および/またはCTNNB1変異を有する個人において、非常に良好な転帰と関係している。[ 40 ][ 50 ][ 51 ]
髄芽腫、SHH活性化およびTP53変異、および髄芽腫、SHH活性化およびTP53野生型
SHH腫瘍はSHH経路の異常を伴う髄芽腫で、髄芽腫症例の約25%を占める。[ 46 ]SHH髄芽腫は、染色体9q欠失;線維形成性/結節性組織型;およびPTCH1、PTCH2、SMO、SUFU、およびGLI2を含むSHH経路遺伝子の変異を特徴とする。[ 48 ]
G蛋白共役受容体161(GPR161)においてヘテロ接合性の有害な生殖細胞変異が、SHH髄芽腫症例の約3%で同定された。[ 18 ]GPR161は、SHHシグナル伝達の阻害因子である。GPR161変異症例の診断時年齢中央値は1.5歳であった。GPR161遺伝子座におけるヘテロ接合性の消失(LOH)がすべての腫瘍で示され、患者6人中5人の腫瘍が染色体1q(GPR161が存在する)のコピー数の変化を伴わないLOHを示した。
U1スプライソソームの核内低分子RNA(snRNA)の第3ヌクレオチドにおける変異(r.3A>G)はSHH髄芽腫に非常に特異的である。[ 52 ][ 53 ]U1 snRNA r.3A>G変異は、成人におけるSHH髄芽腫の実質的にすべての症例、小児および青年における約1/3の症例で観察され、乳児症例では認められない。[ 53 ]U1 snRNA変異はRNAスプライシングを破綻させ、腫瘍抑制遺伝子(例、PTCH1)の不活性化およびがん遺伝子(例、GLI2)の活性化に至る。SHH髄芽腫の特異的サブタイプにおけるU1 snRNA r.3A>G変異の意義を以下に記述する。
SHH髄芽腫の発症年齢は二峰性分布を示し、主に3歳未満の小児および、より年齢の高い青年および成人にみられる。腫瘍は小脳外顆粒層から発生すると考えられている。発症時年齢の異質性は、以下のように、追加の分子生物学的な検討で同定される特異なサブセットを示す:
非転移性SHH髄芽腫を有する患者の転帰は、3歳未満の小児と成人で比較的良好である。[ 46 ]MBEN組織型を有する若年小児は特に予後良好である。[ 7 ][ 24 ][ 56 ][ 57 ][ 58 ]治療失敗のリスクが最も高いSHH髄芽腫患者は、腫瘍にTP53変異が認められ、しばしばGLI2またはMYCN増幅が共起し、大細胞型/退形成性組織型を伴う3歳より年長の小児である。[ 46 ][ 54 ][ 59 ]
予後不良な分子所見を有する患者の予後は悪く、従来の治療後に生存する患者は50%未満である。[ 41 ][ 54 ][ 59 ][ 60 ][ 61 ]
2016 WHO分類では、別個の疾患実体としてTP53変異を伴うSHH髄芽腫(髄芽腫、SHH活性化およびTP53変異)が同定されている。[ 20 ]SHH活性化髄芽腫症例の約25%にTP53変異が認められ、これらの症例ではまたTP53生殖細胞変異が高い割合で示されている(1件の研究では20例中9例)。これらの患者の年齢は一般的に5~18歳で、転帰不良である(5年全生存率、50%未満)。[ 61 ]この腫瘍はしばしば大細胞型/退形成性の組織像を示す。[ 61 ]
髄芽腫、非WNT/非SHH活性化
WHO分類では、一部にはこの区別について即時の臨床的影響が認められないことに基づいて、グループ3およびグループ4の髄芽腫が単一の疾患実体に統合されている。グループ3の髄芽腫は髄芽腫症例の約25%を占める一方、グループ4の髄芽腫は髄芽腫症例の約40%を占める。[ 46 ][ 48 ]グループ3およびグループ4の髄芽腫患者はいずれも主に男性である。[ 34 ][ 45 ]グループ3およびグループ4の髄芽腫は、遺伝子発現やDNAメチル化プロファイルなどの特徴に基づいてさらに細分できるが、こうした細分化の最適なアプローチは確立されていない。[ 46 ][ 47 ]
さまざまなゲノム変化がグループ3およびグループ4の髄芽腫において観察されている;しかしながら、症例の10%以上~20%では単一の変化も発生していない。ゲノム変化には、以下のものがある:
MYC遺伝子の増幅またはMYC遺伝子の過剰発現が認められるグループ3の患者の予後は不良で[ 45 ]、診断から5年後に生存しているのはこれらの患者の50%未満である。[ 46 ]この不良な予後は、特に診断時に4歳未満の小児に当てはまる。[ 41 ]しかしながら、MYC遺伝子の増幅が認められないグループ3の髄芽腫の3歳より年長の患者の予後は非WNT髄芽腫のほとんどの患者と同様であり、5年PFS率は70%を超える。[ 60 ][ 62 ]
グループ4の髄芽腫は乳児期および小児期を通じ、また成人期まで生じる。グループ4の髄芽腫患者の予後は他の非WNT髄芽腫患者の予後と同様であり、転移病変の有無、染色体11q欠失、および染色体17p欠失のような付加的因子による影響を受ける可能性がある。[ 38 ][ 39 ][ 46 ][ 59 ]1件の研究で、11番染色体の欠失または17番染色体の増加のいずれかを有したグループ4の患者は転移にもかかわらず低リスクであったことが明らかにされた。これらの細胞遺伝学的特徴の両方が認められない症例において、発症時の転移は高リスクおよび中リスク間で差異が生じた。[ 59 ]
グループ3およびグループ4の標準リスク患者(すなわち、MYC増幅または転移病変が認められない)について、染色体全体の増加または欠失は予後良好を暗示するようである。この知見は、SIOP-PNET-4(NCT01351870)臨床試験に登録された非WNT/非SHH髄芽腫患者91人のデータから得られ、1990年から2014年に治療された非WNT/非SHH髄芽腫の小児70人の独立した集団で確認された。[ 62 ]染色体異常として、以下のものが挙げられる:
髄芽腫を4つの主要なサブタイプに分けるという現在の分類法は、将来変更される可能性が高い。[ 46 ][ 47 ][ 63 ][ 64 ]各サブグループが分子的にさらに分析されるため、分子的特徴に基づいてサブグループ内でさらに細分される可能性が高いが、複数の独立した研究データが統合されるにつれて、研究がコンセンサスに至りつつある。一例として、補完バイオインフォマティクス(complementary bioinformatics)アプローチを用いて、公表されている複数の大規模コホート間の一致が解析され、さらに統一された亜型分類が記述された。グループ3およびグループ4の髄芽腫を有する小児について、8つの異なるサブグループがDNAメチル化クラスタリングにより決定された。特定のサブグループによって予後が異なっていた。[ 38 ][ 48 ][ 54 ][ 65 ]
髄芽腫の成人に対する分類が小児に対する分類において同様の予測能力を有するかどうかは不明である。[ 39 ][ 41 ]成人の髄芽腫に関する1件の研究では、MYCがん遺伝子の増幅はまれにしか認められず、6qの欠失およびWNTの活性化(β-カテニン核染色により同定)を示す腫瘍は小児の髄芽腫でみられる非常に優れた予後を共有していなかったが、別の研究では成人におけるWNT活性化腫瘍について非常に優れた予後が確認された。[ 39 ][ 41 ]
病期評価
歴史的に、病期分類は腫瘍の大きさと範囲に関する手術中の評価とともに、術後における脳および脊椎の神経画像検査とCSFの細胞学的評価に基づいていた(Chang病期分類システム)。腫瘍の範囲の術中評価は、診断前の脳脊髄幹の画像診断検査および原発部位の残存病変量を測定するための術後画像診断検査に取って代わられている。現在、以下の検査および手技が病期分類に用いられている:
腫瘍の範囲は以下のように決定される:
術後の残存腫瘍の程度は以下のように指定される:
1990年代以降のプロスペクティブ研究の実施においては、患者を平均リスクおよび高リスク髄芽腫のサブグループに分けるため、この病期分類システムが用いられている。[ 67 ][ 68 ][ 69 ]
(病理標本の50%以上を占める)びまん性の組織学的退形成の存在が、病期分類システムへの追加事項として組み込まれている。びまん性の退形成が認められた場合は、それ以外は平均リスク疾患の患者でも高リスク疾患へと病期が引き上げられる。
リスク層別化
リスク層別化は、播種性病変の神経放射線検査による評価、CSF細胞診、残存腫瘍量についての術後神経画像検査の評価、および患者の年齢に基づいて行われる。(詳しい情報については、本要約の病期評価のセクションを参照のこと。)3歳より年長の髄芽腫患者は、以下の2つのリスクグループに層別化されている:
若年の小児について、一部の研究では3歳未満の小児と4~5歳未満の小児に対して、平均リスク群(播種を認めず、残存腫瘍が1.5cm2以下)または高リスク群(播種性病変を認め、および/または残存腫瘍が1.5cm2を超える)への同様の分類が行われている。線維形成の組織学的所見もまた、より予後良好なリスクのサブグループ化(特にMBENのサブグループ)の暗示に用いられている。[ 70 ][ 71 ]
切除範囲および診断時の病変の範囲に基づくリスクグループの割り付けでは、治療成績を予測しない場合がある。分子遺伝学および組織学的因子の方が情報価値がある場合があるが、これらは患者の年齢、診断時の病変の範囲、受けた治療を考慮して評価する必要がある。[ 38 ][ 72 ]分子的細分類のリスク層別化は変更され、北米のプロスペクティブ研究(例、NCT01878617およびNCT02724579)において治療を割り付けるためのリスク層別化スキームに統合されつつある。[ 63 ]
小児髄芽腫に対する治療法選択肢の概要
表3では、新たに診断された、および再発した小児髄芽腫に対する標準治療法の選択肢について記述している。
表3.小児髄芽腫に対する標準治療法の選択肢 治療法群 標準治療法の選択肢 新たに診断された小児髄芽腫 3歳以下の小児 手術 補助化学療法 3歳より年長で平均リスク髄芽腫の小児 手術 補助放射線療法 補助化学療法 3歳より年長で高リスク髄芽腫の小児 手術 補助放射線療法 補助化学療法 再発した小児髄芽腫 標準治療法の選択肢は存在しない。(詳しい情報については、本要約の再発した小児髄芽腫およびその他のCNS胚芽腫の治療のセクションを参照のこと。) 手術
手術は腫瘍のタイプを組織学的に確認するため、また治療成績を改善する手段として治療の標準的要素と考えられている。安全に実施可能であれば、完全切除またはほぼ完全切除が最適と考えられる。[ 73 ][ 74 ]
手術後の小児は、術前の腫瘍関連脳損傷、水頭症、または手術関連脳損傷を原因とする重大な神経学的欠損を来している場合がある。[ 75 ][証拠レベル:3iC]かなりの数の髄芽腫患者が小脳無言症候群(後頭蓋窩症候群としても知られる)を発症する。小脳無言症候群の症状には以下のものがある:
小脳虫部の損傷および/または小脳皮質神経回路の破壊が無言症の原因ではないかと想定されているものの、小脳無言症候群の病因は依然として不明である。[ 76 ][ 77 ];[ 78 ][証拠レベル:3iC]平均リスクおよび高リスクの髄芽腫の小児を評価したChildren's Cancer Groupの2件の研究では、この症候群が患者の25%近くで同定された。[ 77 ][ 78 ][ 79 ];[ 80 ][証拠レベル:3iiiC]この症候群の患者の約50%に、長期の永続的な神経学的および神経認知的後遺症が現れる。[ 78 ][ 80 ]
放射線療法
原発腫瘍部位への放射線療法は通常、54Gy~55.8Gyの範囲で実施される。[ 81 ]ほとんどの場合、できれば原体照射法により、原発腫瘍部位の周囲に1~2cmのマージンを取って照射される。[ 81 ]小児腫瘍学グループ(COG)のACNS0331(NCT00085735)研究において、後頭蓋窩全体および腫瘍床 + マージンへのブースト量の低減は、平均リスク患者における治療成績を損なわなかった。[ 82 ][証拠レベル:1iiA]診断時に3~4歳より年長の小児では、すべての髄芽腫に対して診断時の病変の範囲などの危険因子に応じ、23.4~36Gyの範囲の線量で頭蓋脊髄照射が施行される。陽子線治療に関する1件のプロスペクティブ第II相毒性研究[ 83 ]および髄芽腫に対する陽子線と光子線を比較したレトロスペクティブな効力の報告[ 84 ]により、無増悪生存(PFS)、全生存(OS)、再燃パターン、および遅発性毒性作用について同等の転帰が実証された。陽子線治療(n = 38)または光子線治療(n = 46)のいずれかを受けた小児84人を対象にしたレトロスペクティブ研究により、陽子線治療を受けた小児では蝸牛への平均線量が低かったにもかかわらず、グレード3およびグレード4の耳毒性の割合は同程度であったことが実証されたため、他の因子(例、シスプラチン、内耳神経[第8脳神経]に関連する初発腫瘍の位置)が耳毒性の一因となることが示唆されている。[ 85 ]これらの治療技術について転帰の比較が研究段階にある。
化学療法は通常、放射線療法中および放射線療法後に実施される。
3歳未満の小児について、この年齢層では放射線の影響が大きいため、放射線療法を省略するか、遅らせる努力がなされる。小児はすべての年齢で、脳の発達に対する放射線の有害作用を受けやすい。特に低年齢の小児では、神経認知発達、成長、および内分泌機能が低下するという影響が頻繁に観察されている。[ 86 ][ 87 ][ 88 ][ 89 ][ 90 ]
小児髄芽腫の治療
3歳以下の小児の治療
髄芽腫の幼児に対する5年無病生存(DFS)率の範囲は30~70%となっている。長期生存者のほとんどは化学療法単独による治療が成功し、播種を認めず、線維形成の組織学的証拠を示した腫瘍が完全切除されている。[ 70 ][ 91 ][ 93 ];[ 94 ][証拠レベル:2A]
新たに髄芽腫と診断された3~4歳未満の小児の治療は絶えず進化している。未熟な神経系への有害な影響のために、複数の治療アプローチで頭蓋脊髄照射の延期、および症例によっては回避が試みられている。結果はさまざまで、使用された薬物レジメン、化学療法終了時または研究によっては患児が3歳に達する時期での頭蓋脊髄照射および局所への追加放射線療法の利用の違いによって、研究間の比較が困難となっている。
新たに診断された髄芽腫で3歳以下の小児に対する標準治療法の選択肢には以下がある:
手術
実施可能とみなされるなら、腫瘍の外科的完全切除が最適な治療である。線維形成性/結節性髄芽腫またはMBENの患者では古典的髄芽腫の患者より完全切除率が高いように、外科的切除可能性は組織型に関連している。[ 57 ][ 58 ]
補助化学療法
髄芽腫の若年小児に対する治療法には、シクロホスファミド、エトポシド、シスプラチン、およびビンクリスチンのような薬剤を含む多剤化学療法アプローチがあり、これに大量メトトレキサートの静脈内投与および/またはメトトレキサートまたはマフォスファミドの髄腔内投与、および/またはメトトレキサートの脳室内投与を併用する場合も併用しない場合もある。[ 58 ][ 70 ][ 91 ][ 93 ][ 95 ][ 96 ];[ 97 ][証拠レベル:2A];[ 98 ][証拠レベル:2B]
線維形成性髄芽腫またはMBEN患者にみられる線維形成の組織学的所見は、古典的髄芽腫または大細胞/退形成性髄芽腫の患者の転帰と比べて予後が有意に良好なことを暗示することが数件の研究で観察されている。[ 7 ][ 24 ][ 56 ][ 57 ][ 58 ];[ 71 ][証拠レベル:2A]
証拠(補助化学療法):
- German Hirntumor(HIT)2000多施設試験では、線維形成は良好なEFS率の独立予測因子であった。[ 58 ]
- COGの臨床試験CCG-9921でも、線維形成性髄芽腫(MBENを含む)の小児に良好な転帰が観察され、線維形成群ではEFS率が77%(±9%)、OS率が85%(±8%)であったのに対して、非線維形成群ではEFS率が17%(±5%)、OS率が29%(±6%)であった(EFSとOSのいずれの比較でもP < 0.0001)。[ 91 ]この研究では、線維形成性腫瘍の患者は増悪前に放射線療法を受けなかった。
- 現行の強化化学療法レジメンによる治療を受けた線維形成性髄芽腫またはMBENの小児と比較して、他の組織型サブタイプの小児は経過が不良である。
- 診断時に3歳未満の小児に対する別の治療法選択肢は、化学療法とその後に実施する自家幹細胞救助である。幹細胞救助によりサポートするより大量の骨髄除去的化学療法レジメンを用いた試験の結果でも、診断時に3歳未満の髄芽腫患児のサブグループでは、化学療法単独により治療可能なことが実証されている。[ 92 ][ 94 ][ 101 ][証拠レベル:2A]しかしながら、一部の研究では、化学療法後に原発腫瘍部位および/または頭蓋脊髄軸への放射線が追加されており、化学療法の効力の評価がより困難になっている。[ 100 ]
3歳より年長で平均リスク髄芽腫の小児の治療
新たに診断された平均リスク髄芽腫で3歳より年長の小児に対する標準治療法の選択肢には以下がある:
手術
実施可能とみなされれば、腫瘍の完全切除またはほぼ完全切除が最適と考えられる。[ 73 ]
補助放射線療法
放射線療法は通常、手術後に同時化学療法と併用して、または併用せずに開始される。[ 102 ][ 103 ][ 104 ]髄芽腫の小児で生存の最も優れた成績は、放射線療法を術後4~6週間以内に開始する場合に得られている。[ 103 ][ 104 ][ 105 ];[ 81 ][ 106 ][証拠レベル:1iA]
平均リスクの髄芽腫患者に対する放射線量は、後頭蓋窩または局所腫瘍床に対して54Gy、脳脊髄軸全体(すなわち、全脳および全脊椎)に対して23.4Gyで、頭蓋脊髄照射と呼ばれる。[ 102 ][ 103 ][ 104 ][ 107 ]
証拠(補助放射線療法):
- 非播種性病変を有する患者において、35Gyの頭蓋脊髄への照射線量と後頭蓋窩への55Gyのブースト照射を用いる放射線療法単独で治療された場合の5年EFS率は50~65%である。[ 69 ][ 103 ]
- 腫瘍の制御に必要となる最小の頭蓋脊髄照射線量は分かっていない。化学療法を実施せずに、頭蓋脊髄への放射線量を23.4Gyにまで低下させると、孤立性の軟髄膜の再発率が高くなった。[
107
]
COG研究(NCT00085735)において、比較的低線量の頭蓋脊髄照射が評価された。3~7歳の小児が、頭蓋脊髄への18Gyまたは23.4Gyの放射線量のほか、腫瘍床への標的容積を限定したブースト照射を受けるようにランダムに割り付けられた。[ 82 ][証拠レベル:1iiA]
分子的サブグループに応じた解析が待たれている。予後が最も良好な分子的サブグループであるWNT髄芽腫患者において頭蓋脊髄照射線量の18Gyへの縮小が現在研究されている(NCT02724579)。
- SIOP-PNET-4(NCT01351870)研究では、1日1回の放射線療法(脳脊髄軸への1.8Gy分割で23.4Gyおよび後頭蓋窩への30Gyのブースト照射)と1日2回の放射線療法(1Gy分割で36Gyおよび後頭蓋窩への24Gyのブースト照射)が比較された。[ 108 ]
- 放射線療法後に化学療法を追加する場合、23.4Gyの頭蓋脊髄放射線療法が有効な線量であることが示されている。[ 81 ][ 108 ][ 109 ][ 110 ]比較的低い線量の照射が現在評価されている。
- 髄芽腫における標準ブースト照射は後頭蓋窩全体を対象とするが、失敗データのパターンは後頭蓋窩全体の代わりに腫瘍床に放射線療法を実施した場合も同じく有効で、毒性低下とも関連していると明らかにしている。[ 111 ][ 112 ];[ 82 ][証拠レベル:1iiA]
補助化学療法
化学療法は現在、平均リスクの髄芽腫の小児に対する治療の標準的な構成要素である。
証拠(補助化学療法):
- 複数のプロスペクティブ・ランダム化試験および単一群試験により、放射線療法中および放射線療法後の補助化学療法が平均リスクの髄芽腫の小児におけるOSを改善することが示唆されている。[ 80 ][ 102 ][ 103 ][ 104 ][ 105 ][ 106 ]
- 脳脊髄軸に対する23.4Gyの低線量放射線療法を化学療法と合わせて施行すると、85%までの患者に疾患制御が得られることが示されており、長期の神経認知的後遺症の重症化が軽減される可能性がある。[ 81 ][ 109 ][ 110 ][ 114 ]
- シスプラチン、ロムスチン、ビンクリスチンの併用や、シスプラチン、シクロホスファミド、ビンクリスチンの併用など、さまざまな化学療法レジメンが使用され、成功を収めている。[
102
][
103
][
114
][
115
]これらの治療により、5年および10年EFS率およびOS率が増加しており、おそらく晩期再燃率が低下している。しかしながら、集学的治療で治療された長期生存者は、難聴、心合併症、二次性新生物などの晩期合併症(晩期障害)のリスクが高い。[
116
]
さらに、末梢血幹細胞救助でサポートするが、ビンクリスチンおよびシスプラチンの累積投与量を低減させる、放射線療法後の高用量シクロホスファミドでも同様の生存率がもたらされている。[ 51 ]
- 髄芽腫はしばしば化学療法に感受性があるが、平均リスクの髄芽腫患者における放射線療法前の化学療法が、放射線療法とその後の化学療法による治療と比較して生存率を改善することは示されていない。プロスペクティブ研究によっては、放射線療法前の化学療法がより不良な生存率と関係している。[ 103 ][ 104 ][ 105 ][ 106 ]
3歳より年長で高リスク髄芽腫の小児の治療
新たに診断された髄芽腫で、転移性病変を有するか、亜全切除しか受けられなかった3歳より年長の小児に対する標準治療法の選択肢には以下がある:
高リスク群の患者では、多くの研究によって、集学的治療は疾患制御期間および全DFS期間を改善することが実証されている。[ 51 ][ 117 ]いくつかの研究により、転移性病変を有する患者を含めて、高リスク病変がある患者の約50~70%が長期の疾患制御を得ることが示されている。[ 51 ][ 102 ][ 117 ][ 118 ][ 119 ];[ 120 ][証拠レベル:1iiA];[ 121 ][証拠レベル:2A];[ 122 ][証拠レベル:1iiA]
補助放射線療法
標準リスク群の治療とは対照的に、頭蓋脊髄への照射線量は一般に36Gyである。
補助化学療法
証拠(補助化学療法):
- 平均リスク群の小児に有用であることが明らかにされている薬物と同じ薬物が高リスク群の小児に広く使用されており、これにはシスプラチン、ロムスチン、シクロホスファミド、エトポシド、およびビンクリスチンが含まれる。[ 120 ]これらの治療により、5年および10年EFS率およびOS率が増加しており、おそらく晩期再燃率が低下している。しかしながら、集学的治療で治療された長期生存者は、難聴、心合併症、二次性新生物などの晩期合併症(晩期障害)のリスクが高い。[ 116 ]
- 放射線療法後、ビンクリスチンおよびシスプラチンの累積投与量の低減を伴う、末梢血幹細胞救助でサポートする大量骨髄非除去的化学療法も用いられており、約60%の5年PFS率がもたらされている。[ 51 ]
小児髄芽腫に対して臨床評価段階にある治療法の選択肢
選択された患者では、初期相の臨床試験が利用できる場合がある。これらの試験は、COGやPediatric Brain Tumor Consortiumなどの団体を介して利用できる。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は現在実施中の米国および/または施設の臨床試験の例である:
参考文献- Zhang J, Walsh MF, Wu G, et al.: Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 373 (24): 2336-46, 2015.[PUBMED Abstract]
- Waszak SM, Northcott PA, Buchhalter I, et al.: Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19 (6): 785-798, 2018.[PUBMED Abstract]
- Hamilton SR, Liu B, Parsons RE, et al.: The molecular basis of Turcot's syndrome. N Engl J Med 332 (13): 839-47, 1995.[PUBMED Abstract]
- Taylor MD, Mainprize TG, Rutka JT, et al.: Medulloblastoma in a child with Rubenstein-Taybi Syndrome: case report and review of the literature. Pediatr Neurosurg 35 (5): 235-8, 2001.[PUBMED Abstract]
- Miller RW, Rubinstein JH: Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 56 (1): 112-5, 1995.[PUBMED Abstract]
- Bourdeaut F, Miquel C, Richer W, et al.: Rubinstein-Taybi syndrome predisposing to non-WNT, non-SHH, group 3 medulloblastoma. Pediatr Blood Cancer 61 (2): 383-6, 2014.[PUBMED Abstract]
- Garrè ML, Cama A, Bagnasco F, et al.: Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res 15 (7): 2463-71, 2009.[PUBMED Abstract]
- Pastorino L, Ghiorzo P, Nasti S, et al.: Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A (7): 1539-43, 2009.[PUBMED Abstract]
- Brugières L, Remenieras A, Pierron G, et al.: High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J Clin Oncol 30 (17): 2087-93, 2012.[PUBMED Abstract]
- Evans DG, Farndon PA, Burnell LD, et al.: The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64 (5): 959-61, 1991.[PUBMED Abstract]
- Smith MJ, Beetz C, Williams SG, et al.: Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32 (36): 4155-61, 2014.[PUBMED Abstract]
- Li FP, Fraumeni JF: Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 43 (6): 1365-73, 1969.[PUBMED Abstract]
- Pearson AD, Craft AW, Ratcliffe JM, et al.: Two families with the Li-Fraumeni cancer family syndrome. J Med Genet 19 (5): 362-5, 1982.[PUBMED Abstract]
- de Chadarévian JP, Vekemans M, Bernstein M: Fanconi's anemia, medulloblastoma, Wilms' tumor, horseshoe kidney, and gonadal dysgenesis. Arch Pathol Lab Med 109 (4): 367-9, 1985.[PUBMED Abstract]
- Offit K, Levran O, Mullaney B, et al.: Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst 95 (20): 1548-51, 2003.[PUBMED Abstract]
- Dewire MD, Ellison DW, Patay Z, et al.: Fanconi anemia and biallelic BRCA2 mutation diagnosed in a young child with an embryonal CNS tumor. Pediatr Blood Cancer 53 (6): 1140-2, 2009.[PUBMED Abstract]
- Reid S, Renwick A, Seal S, et al.: Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 42 (2): 147-51, 2005.[PUBMED Abstract]
- Begemann M, Waszak SM, Robinson GW, et al.: Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J Clin Oncol 38 (1): 43-50, 2020.[PUBMED Abstract]
- Rorke LB: The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol 42 (1): 1-15, 1983.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Ramaswamy V, Remke M, Shih D, et al.: Duration of the pre-diagnostic interval in medulloblastoma is subgroup dependent. Pediatr Blood Cancer 61 (7): 1190-4, 2014.[PUBMED Abstract]
- McManamy CS, Lamont JM, Taylor RE, et al.: Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62 (6): 627-32, 2003.[PUBMED Abstract]
- Eberhart CG, Kratz J, Wang Y, et al.: Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. J Neuropathol Exp Neurol 63 (5): 441-9, 2004.[PUBMED Abstract]
- Giangaspero F, Perilongo G, Fondelli MP, et al.: Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91 (6): 971-7, 1999.[PUBMED Abstract]
- Onvani S, Etame AB, Smith CA, et al.: Genetics of medulloblastoma: clues for novel therapies. Expert Rev Neurother 10 (5): 811-23, 2010.[PUBMED Abstract]
- Dubuc AM, Northcott PA, Mack S, et al.: The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10 (3): 215-23, 2010.[PUBMED Abstract]
- Thompson MC, Fuller C, Hogg TL, et al.: Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24 (12): 1924-31, 2006.[PUBMED Abstract]
- Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088, 2008.[PUBMED Abstract]
- Tabori U, Baskin B, Shago M, et al.: Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28 (8): 1345-50, 2010.[PUBMED Abstract]
- Pfister S, Remke M, Benner A, et al.: Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27 (10): 1627-36, 2009.[PUBMED Abstract]
- Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23 (31): 7951-7, 2005.[PUBMED Abstract]
- Polkinghorn WR, Tarbell NJ: Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol 4 (5): 295-304, 2007.[PUBMED Abstract]
- Giangaspero F, Wellek S, Masuoka J, et al.: Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112 (1): 5-12, 2006.[PUBMED Abstract]
- Northcott PA, Korshunov A, Witt H, et al.: Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29 (11): 1408-14, 2011.[PUBMED Abstract]
- Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.[PUBMED Abstract]
- Jones DT, Jäger N, Kool M, et al.: Dissecting the genomic complexity underlying medulloblastoma. Nature 488 (7409): 100-5, 2012.[PUBMED Abstract]
- Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.[PUBMED Abstract]
- Taylor MD, Northcott PA, Korshunov A, et al.: Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465-72, 2012.[PUBMED Abstract]
- Kool M, Korshunov A, Remke M, et al.: Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473-84, 2012.[PUBMED Abstract]
- Pietsch T, Schmidt R, Remke M, et al.: Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol 128 (1): 137-49, 2014.[PUBMED Abstract]
- Cho YJ, Tsherniak A, Tamayo P, et al.: Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29 (11): 1424-30, 2011.[PUBMED Abstract]
- Gajjar A, Bowers DC, Karajannis MA, et al.: Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol 33 (27): 2986-98, 2015.[PUBMED Abstract]
- Morrissy AS, Cavalli FMG, Remke M, et al.: Spatial heterogeneity in medulloblastoma. Nat Genet 49 (5): 780-788, 2017.[PUBMED Abstract]
- Wang X, Dubuc AM, Ramaswamy V, et al.: Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol 129 (3): 449-57, 2015.[PUBMED Abstract]
- Schwalbe EC, Lindsey JC, Nakjang S, et al.: Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18 (7): 958-971, 2017.[PUBMED Abstract]
- Cavalli FMG, Remke M, Rampasek L, et al.: Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 31 (6): 737-754.e6, 2017.[PUBMED Abstract]
- Northcott PA, Buchhalter I, Morrissy AS, et al.: The whole-genome landscape of medulloblastoma subtypes. Nature 547 (7663): 311-317, 2017.[PUBMED Abstract]
- Northcott PA, Jones DT, Kool M, et al.: Medulloblastomics: the end of the beginning. Nat Rev Cancer 12 (12): 818-34, 2012.[PUBMED Abstract]
- Gibson P, Tong Y, Robinson G, et al.: Subtypes of medulloblastoma have distinct developmental origins. Nature 468 (7327): 1095-9, 2010.[PUBMED Abstract]
- Ellison DW, Dalton J, Kocak M, et al.: Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121 (3): 381-96, 2011.[PUBMED Abstract]
- Gajjar A, Chintagumpala M, Ashley D, et al.: Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7 (10): 813-20, 2006.[PUBMED Abstract]
- Shuai S, Suzuki H, Diaz-Navarro A, et al.: The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574 (7780): 712-716, 2019.[PUBMED Abstract]
- Suzuki H, Kumar SA, Shuai S, et al.: Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574 (7780): 707-711, 2019.[PUBMED Abstract]
- Kool M, Jones DT, Jäger N, et al.: Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25 (3): 393-405, 2014.[PUBMED Abstract]
- Robinson GW, Rudneva VA, Buchhalter I, et al.: Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19 (6): 768-784, 2018.[PUBMED Abstract]
- Leary SE, Zhou T, Holmes E, et al.: Histology predicts a favorable outcome in young children with desmoplastic medulloblastoma: a report from the children's oncology group. Cancer 117 (14): 3262-7, 2011.[PUBMED Abstract]
- Rutkowski S, von Hoff K, Emser A, et al.: Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 28 (33): 4961-8, 2010.[PUBMED Abstract]
- von Bueren AO, von Hoff K, Pietsch T, et al.: Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 13 (6): 669-79, 2011.[PUBMED Abstract]
- Shih DJ, Northcott PA, Remke M, et al.: Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32 (9): 886-96, 2014.[PUBMED Abstract]
- Schwalbe EC, Williamson D, Lindsey JC, et al.: DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol 125 (3): 359-71, 2013.[PUBMED Abstract]
- Zhukova N, Ramaswamy V, Remke M, et al.: Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31 (23): 2927-35, 2013.[PUBMED Abstract]
- Goschzik T, Schwalbe EC, Hicks D, et al.: Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19 (12): 1602-1616, 2018.[PUBMED Abstract]
- Gottardo NG, Hansford JR, McGlade JP, et al.: Medulloblastoma Down Under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol 127 (2): 189-201, 2014.[PUBMED Abstract]
- Louis DN, Perry A, Burger P, et al.: International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 24 (5): 429-35, 2014.[PUBMED Abstract]
- Sharma T, Schwalbe EC, Williamson D, et al.: Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138 (2): 309-326, 2019.[PUBMED Abstract]
- Fouladi M, Gajjar A, Boyett JM, et al.: Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol 17 (10): 3234-7, 1999.[PUBMED Abstract]
- Zeltzer PM, Boyett JM, Finlay JL, et al.: Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children's Cancer Group 921 randomized phase III study. J Clin Oncol 17 (3): 832-45, 1999.[PUBMED Abstract]
- Yao MS, Mehta MP, Boyett JM, et al.: The effect of M-stage on patterns of failure in posterior fossa primitive neuroectodermal tumors treated on CCG-921: a phase III study in a high-risk patient population. Int J Radiat Oncol Biol Phys 38 (3): 469-76, 1997.[PUBMED Abstract]
- Taylor RE, Bailey CC, Robinson K, et al.: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol 21 (8): 1581-91, 2003.[PUBMED Abstract]
- Rutkowski S, Bode U, Deinlein F, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352 (10): 978-86, 2005.[PUBMED Abstract]
- Rutkowski S, Gerber NU, von Hoff K, et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol 11 (2): 201-10, 2009.[PUBMED Abstract]
- Ramaswamy V, Remke M, Adamski J, et al.: Medulloblastoma subgroup-specific outcomes in irradiated children: who are the true high-risk patients? Neuro Oncol 18 (2): 291-7, 2016.[PUBMED Abstract]
- Albright AL, Wisoff JH, Zeltzer PM, et al.: Effects of medulloblastoma resections on outcome in children: a report from the Children's Cancer Group. Neurosurgery 38 (2): 265-71, 1996.[PUBMED Abstract]
- Thompson EM, Hielscher T, Bouffet E, et al.: Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol 17 (4): 484-95, 2016.[PUBMED Abstract]
- Stargatt R, Rosenfeld JV, Maixner W, et al.: Multiple factors contribute to neuropsychological outcome in children with posterior fossa tumors. Dev Neuropsychol 32 (2): 729-48, 2007.[PUBMED Abstract]
- Pollack IF, Polinko P, Albright AL, et al.: Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery 37 (5): 885-93, 1995.[PUBMED Abstract]
- Robertson PL, Muraszko KM, Holmes EJ, et al.: Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children's Oncology Group. J Neurosurg 105 (6): 444-51, 2006.[PUBMED Abstract]
- Wells EM, Khademian ZP, Walsh KS, et al.: Postoperative cerebellar mutism syndrome following treatment of medulloblastoma: neuroradiographic features and origin. J Neurosurg Pediatr 5 (4): 329-34, 2010.[PUBMED Abstract]
- Gudrunardottir T, Sehested A, Juhler M, et al.: Cerebellar mutism: review of the literature. Childs Nerv Syst 27 (3): 355-63, 2011.[PUBMED Abstract]
- Wolfe-Christensen C, Mullins LL, Scott JG, et al.: Persistent psychosocial problems in children who develop posterior fossa syndrome after medulloblastoma resection. Pediatr Blood Cancer 49 (5): 723-6, 2007.[PUBMED Abstract]
- Packer RJ, Gajjar A, Vezina G, et al.: Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24 (25): 4202-8, 2006.[PUBMED Abstract]
- Michalski JM, Janss A, Vezina G, et al.: Results of COG ACNS0331: a phase III trial of involved-field radiotherapy (IFRT) and low dose craniospinal irradiation (LD-CSI) with chemotherapy in average-risk medulloblastoma: a report from the Children's Oncology Group. [Abstract] Int J Radiat Oncol Biol Phys 96 (5): A-LBA-2, 937-8, 2016. Also available online. Last accessed February 06, 2020.[PUBMED Abstract]
- Yock TI, Yeap BY, Ebb DH, et al.: Long-term toxic effects of proton radiotherapy for paediatric medulloblastoma: a phase 2 single-arm study. Lancet Oncol 17 (3): 287-98, 2016.[PUBMED Abstract]
- Eaton BR, Esiashvili N, Kim S, et al.: Clinical Outcomes Among Children With Standard-Risk Medulloblastoma Treated With Proton and Photon Radiation Therapy: A Comparison of Disease Control and Overall Survival. Int J Radiat Oncol Biol Phys 94 (1): 133-8, 2016.[PUBMED Abstract]
- Paulino AC, Mahajan A, Ye R, et al.: Ototoxicity and cochlear sparing in children with medulloblastoma: Proton vs. photon radiotherapy. Radiother Oncol 128 (1): 128-132, 2018.[PUBMED Abstract]
- Ris MD, Packer R, Goldwein J, et al.: Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children's Cancer Group study. J Clin Oncol 19 (15): 3470-6, 2001.[PUBMED Abstract]
- Packer RJ, Sutton LN, Atkins TE, et al.: A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. J Neurosurg 70 (5): 707-13, 1989.[PUBMED Abstract]
- Johnson DL, McCabe MA, Nicholson HS, et al.: Quality of long-term survival in young children with medulloblastoma. J Neurosurg 80 (6): 1004-10, 1994.[PUBMED Abstract]
- Walter AW, Mulhern RK, Gajjar A, et al.: Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3720-8, 1999.[PUBMED Abstract]
- Laughton SJ, Merchant TE, Sklar CA, et al.: Endocrine outcomes for children with embryonal brain tumors after risk-adapted craniospinal and conformal primary-site irradiation and high-dose chemotherapy with stem-cell rescue on the SJMB-96 trial. J Clin Oncol 26 (7): 1112-8, 2008.[PUBMED Abstract]
- Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.[PUBMED Abstract]
- Chi SN, Gardner SL, Levy AS, et al.: Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol 22 (24): 4881-7, 2004.[PUBMED Abstract]
- Grill J, Sainte-Rose C, Jouvet A, et al.: Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6 (8): 573-80, 2005.[PUBMED Abstract]
- Cohen BH, Geyer JR, Miller DC, et al.: Pilot Study of Intensive Chemotherapy With Peripheral Hematopoietic Cell Support for Children Less Than 3 Years of Age With Malignant Brain Tumors, the CCG-99703 Phase I/II Study. A Report From the Children's Oncology Group. Pediatr Neurol 53 (1): 31-46, 2015.[PUBMED Abstract]
- Duffner PK, Horowitz ME, Krischer JP, et al.: Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328 (24): 1725-31, 1993.[PUBMED Abstract]
- Ater JL, van Eys J, Woo SY, et al.: MOPP chemotherapy without irradiation as primary postsurgical therapy for brain tumors in infants and young children. J Neurooncol 32 (3): 243-52, 1997.[PUBMED Abstract]
- Grundy RG, Wilne SH, Robinson KJ, et al.: Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer 46 (1): 120-33, 2010.[PUBMED Abstract]
- Blaney SM, Kocak M, Gajjar A, et al.: Pilot study of systemic and intrathecal mafosfamide followed by conformal radiation for infants with intracranial central nervous system tumors: a pediatric brain tumor consortium study (PBTC-001). J Neurooncol 109 (3): 565-71, 2012.[PUBMED Abstract]
- Lafay-Cousin L, Bouffet E, Strother D, et al.: Phase II Study of Nonmetastatic Desmoplastic Medulloblastoma in Children Younger Than 4 Years of Age: A Report of the Children's Oncology Group (ACNS1221). J Clin Oncol 38 (3): 223-231, 2020.[PUBMED Abstract]
- Lafay-Cousin L, Smith A, Chi SN, et al.: Clinical, Pathological, and Molecular Characterization of Infant Medulloblastomas Treated with Sequential High-Dose Chemotherapy. Pediatr Blood Cancer 63 (9): 1527-34, 2016.[PUBMED Abstract]
- Dhall G, Grodman H, Ji L, et al.: Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the "Head Start" I and II protocols. Pediatr Blood Cancer 50 (6): 1169-75, 2008.[PUBMED Abstract]
- Packer RJ, Sutton LN, Elterman R, et al.: Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg 81 (5): 690-8, 1994.[PUBMED Abstract]
- Bailey CC, Gnekow A, Wellek S, et al.: Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med Pediatr Oncol 25 (3): 166-78, 1995.[PUBMED Abstract]
- Kortmann RD, Kühl J, Timmermann B, et al.: Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT '91. Int J Radiat Oncol Biol Phys 46 (2): 269-79, 2000.[PUBMED Abstract]
- Taylor RE, Bailey CC, Robinson KJ, et al.: Impact of radiotherapy parameters on outcome in the International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 study of preradiotherapy chemotherapy for M0-M1 medulloblastoma. Int J Radiat Oncol Biol Phys 58 (4): 1184-93, 2004.[PUBMED Abstract]
- von Hoff K, Hinkes B, Gerber NU, et al.: Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT'91. Eur J Cancer 45 (7): 1209-17, 2009.[PUBMED Abstract]
- Thomas PR, Deutsch M, Kepner JL, et al.: Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol 18 (16): 3004-11, 2000.[PUBMED Abstract]
- Sabel M, Fleischhack G, Tippelt S, et al.: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129 (3): 515-524, 2016.[PUBMED Abstract]
- Oyharcabal-Bourden V, Kalifa C, Gentet JC, et al.: Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23 (21): 4726-34, 2005.[PUBMED Abstract]
- Merchant TE, Kun LE, Krasin MJ, et al.: Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys 70 (3): 782-7, 2008.[PUBMED Abstract]
- Fukunaga-Johnson N, Sandler HM, Marsh R, et al.: The use of 3D conformal radiotherapy (3D CRT) to spare the cochlea in patients with medulloblastoma. Int J Radiat Oncol Biol Phys 41 (1): 77-82, 1998.[PUBMED Abstract]
- Huang E, Teh BS, Strother DR, et al.: Intensity-modulated radiation therapy for pediatric medulloblastoma: early report on the reduction of ototoxicity. Int J Radiat Oncol Biol Phys 52 (3): 599-605, 2002.[PUBMED Abstract]
- Carrie C, Grill J, Figarella-Branger D, et al.: Online quality control, hyperfractionated radiotherapy alone and reduced boost volume for standard risk medulloblastoma: long-term results of MSFOP 98. J Clin Oncol 27 (11): 1879-83, 2009.[PUBMED Abstract]
- Packer RJ, Goldwein J, Nicholson HS, et al.: Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children's Cancer Group Study. J Clin Oncol 17 (7): 2127-36, 1999.[PUBMED Abstract]
- Nageswara Rao AA, Wallace DJ, Billups C, et al.: Cumulative cisplatin dose is not associated with event-free or overall survival in children with newly diagnosed average-risk medulloblastoma treated with cisplatin based adjuvant chemotherapy: report from the Children's Oncology Group. Pediatr Blood Cancer 61 (1): 102-6, 2014.[PUBMED Abstract]
- Salloum R, Chen Y, Yasui Y, et al.: Late Morbidity and Mortality Among Medulloblastoma Survivors Diagnosed Across Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 37 (9): 731-740, 2019.[PUBMED Abstract]
- Verlooy J, Mosseri V, Bracard S, et al.: Treatment of high risk medulloblastomas in children above the age of 3 years: a SFOP study. Eur J Cancer 42 (17): 3004-14, 2006.[PUBMED Abstract]
- Gandola L, Massimino M, Cefalo G, et al.: Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J Clin Oncol 27 (4): 566-71, 2009.[PUBMED Abstract]
- Evans AE, Jenkin RD, Sposto R, et al.: The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg 72 (4): 572-82, 1990.[PUBMED Abstract]
- Jakacki RI, Burger PC, Zhou T, et al.: Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children's Oncology Group Phase I/II study. J Clin Oncol 30 (21): 2648-53, 2012.[PUBMED Abstract]
- von Bueren AO, Kortmann RD, von Hoff K, et al.: Treatment of Children and Adolescents With Metastatic Medulloblastoma and Prognostic Relevance of Clinical and Biologic Parameters. J Clin Oncol 34 (34): 4151-4160, 2016.[PUBMED Abstract]
- Tarbell NJ, Friedman H, Polkinghorn WR, et al.: High-risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol 31 (23): 2936-41, 2013.[PUBMED Abstract]
- 髄芽腫以外の小児胚芽腫
-
中枢神経系(CNS)原発腫瘍の最新の世界保健機関(WHO)分類では、他のCNS腫瘍を定義する特異的な病理組織学的特徴または分子的変化が認められない神経外胚葉由来の髄芽腫以外の腫瘍はすべて、CNS胚芽腫と分類される。CNS胚芽腫には次の腫瘍が含まれる:多層性ロゼットを有する胎児性腫瘍(ETMR)、C19MC変異型;髄上皮腫;CNS胚芽腫、他に特定されない(NOS);非定型奇形腫様/ラブドイド腫瘍(AT/RT);およびラブドイドの特徴を示すCNS胚芽腫。[ 1 ]これらの腫瘍は、AT/RTを除いて、本セクションで考察する(詳しい情報については、小児CNS AT/RTの治療に関するPDQ要約を参照のこと)。松果体芽腫は胚芽腫と組織学的特徴を共有しており、慣習的に同じ方法で治療されるため、松果体芽腫については本要約内で考察する(詳しい情報については、本要約の小児松果体芽腫のセクションを参照のこと)。
臨床像
髄芽腫以外の胚芽腫の発症もまた比較的急速で、神経系における腫瘍の位置によって異なる。胚芽腫は増殖が速い傾向があり、通常は最初の症状の発現から3ヵ月以内に診断される。
髄芽腫以外の胚芽腫はCNSのいずれの部位にも発生し、症状は不定である。通常は、嗜眠および嘔吐に関連した重篤な神経機能障害がみられる。テント上胚芽腫(図1を参照のこと)は、大脳皮質における病変の部分に応じて片側不全麻痺や視野欠損といった限局性の神経学的欠損を引き起こす。痙攣発作および鈍麻を引き起こすこともある。
細胞分類および分子分類
CNS腫瘍のWHO分類では、ETMRおよびラブドイドの特徴を示す非定型奇形腫様腫瘍は例外であるものの、主に組織学的および免疫組織学的特徴によって髄芽腫以外の胚芽腫を分類する。[ 1 ]定義上、これらの腫瘍は大脳半球、脳幹、または脊髄に発生し、多彩な分化形態を示すことのある未分化または低分化神経上皮細胞からなる。この分類は、腫瘍の病理組織学的特徴と腫瘍の位置に基づいて、以下のようになる:
異なる領域の神経細胞分化を示すCNS胚芽腫は大脳神経芽腫と呼ばれ、神経節細胞が存在する場合には神経節芽細胞腫と呼ばれる。同様に、髄上皮腫は特異的な組織学的パターンを有し、別の疾患実体として残っている。[ 1 ][ 2 ]
胚芽腫および松果体芽腫のゲノムの分子キャラクタリゼーションにより、これらの腫瘍間でかなりの不均一性が実証されている。これらの腫瘍はまた、分子的に髄芽腫と異なっている。[ 3 ][ 4 ]
WHO分類システムではまだ、(ETMR、C19MC変異型およびSMARCB1欠失を伴うAT/RTを除いて)髄芽腫以外の胚芽腫の分類に分子所見が採用されていないが、将来は組織学的所見と分子所見の両方、さらにはおそらく神経系の発生部位に基づいて分類される可能性が非常に高い。
髄芽腫以外の胚芽腫のサブタイプ
髄芽腫以外の胚芽腫の分子的サブタイプ
323例の髄芽腫以外の胚芽腫についてDNAメチル化パターンの教師なしクラスタリングを実施した研究では、髄芽腫以外の胚芽腫と診断されたこれらの腫瘍の約半数が、他の既知の小児脳腫瘍(例、高悪性度グリオーマおよび非定型奇形腫様/ラブドイド腫瘍[AT/RT])に特徴的な分子プロファイルを示したことが明らかにされた。[ 4 ]この観察は、このクラスの腫瘍を、生物学に基づく適切な診断に割り付けるために分子キャラクタリゼーションが有用であることを強調している。
髄芽腫以外の胚芽腫として診断された同じ集まりの323例の腫瘍では、分子キャラクタリゼーションにより、ゲノム的および生物学的に異なるサブタイプが同定されており、これには以下のものがある:
CNSのテント上原始神経外胚葉性腫瘍(CNS-PNET)および松果体芽腫を有する患者を対象にした1件の臨床試験において、テント上胚芽腫を正確に診断する上でDNAメチル化プロファイル解析の寄与が実証された。[ 14 ]松果体芽腫症例では、メチル化プロファイル解析により下された診断と中央病理診断により下された診断との間で高い一致がみられた(29例中26例)。しかしながら、残りの31人の患者について、メチル化プロファイル解析により下された診断は、18人の患者で高悪性度グリオーマ、2人の患者でAT/RT、および2人の患者でRELA融合陽性上衣腫であった。中央病理診断により下された診断とメチル化プロファイル解析により下された診断間の不一致の判定では、再検査された10例でメチル化プロファイル解析が支持された。
髄上皮腫
古典的なC19MC増幅を伴う髄上皮腫はETMR、C19MC変異型と考えられる(上述のETMRの情報を参照のこと)。しかしながら、腫瘍に髄上皮腫の組織学的特徴があるが、C19MC増幅を伴わない場合、WHO分類システム内で組織学的に別個の腫瘍として識別され、髄上皮腫と呼ばれる。[ 15 ][ 16 ]髄上皮腫はまれで、乳児および幼児に最も多く発生する傾向がみられる。組織学的に胚神経管を再現する髄上皮腫は、主に脳室内のテント上に発生する傾向があるが、神経根に沿ってテント下の馬尾および神経外でも発生する場合がある。[ 15 ][ 16 ]
病期評価
髄芽腫以外および髄上皮腫以外の胚芽腫患者は、髄芽腫の小児に用いられるものと同様に病期分類される;ただし、治療を目的とした平均リスクおよび高リスクのサブグループへの患者の割り付けは行われない(詳しい情報については、本要約の髄芽腫の病期評価のセクションを参照のこと)。
髄上皮腫は頻繁に脳脊髄幹に播種する。[ 19 ]髄上皮腫の病期分類は髄芽腫と同じ方法で実施される;ただし、治療を目的とした平均リスクおよび高リスクのサブグループへの患者の割り付けは行われない(詳しい情報については、本要約の髄芽腫の病期評価のセクションを参照のこと)。
髄芽腫以外の小児胚芽腫に対する治療法選択肢の概要
表4では、新たに診断された、および再発した髄芽腫以外および髄上皮腫以外の小児胚芽腫および髄上皮腫に対する標準治療法の選択肢について記述している。
髄芽腫以外の小児胚芽腫の治療
(CNS非定型奇形腫様/ラブドイド腫瘍の治療に関する詳しい情報については、小児中枢神経系非定型奇形腫様/ラブドイド腫瘍の治療に関するPDQ要約を参照のこと。)
(髄上皮腫の治療に関する情報については、本要約の小児の多層性ロゼットを有する胎児性腫瘍または髄上皮腫の治療のセクションを参照のこと。)
3歳以下の小児の治療
新たに診断された髄芽腫以外および髄上皮腫以外の胚芽腫で3歳以下の小児に対する標準治療法の選択肢には以下のものがある:
- 手術。
- 補助化学療法。
3歳以下の髄芽腫以外および髄上皮腫以外の胚芽腫の小児の治療は、3歳以下の髄芽腫の小児について概説したものと同様である。(詳しい情報については、本要約の髄芽腫の3歳以下の小児の治療のセクションを参照のこと)。
化学療法のみの使用による治療成績はさまざまで、5年生存率は0~50%の範囲であった。[ 20 ][ 21 ][ 22 ];[ 23 ][証拠レベル:2Di]化学療法ベースのレジメンに頭蓋脊髄照射を追加することで一部の小児では治療が成功する可能性があるが、神経発達の低下が予想される。[ 24 ][証拠レベル:2A]
3歳より年長の小児の治療
新たに診断された髄芽腫以外および髄上皮腫以外の胚芽腫で3歳より年長の小児に対する標準治療法の選択肢には以下のものがある:
手術
証拠(手術):
- 積極的な外科的切除の試みは、新たに診断された髄芽腫以外の胚芽腫の管理における第一段階である。これまでの研究では、切除の程度が転帰の予測因子であることは示されていないが[ 25 ][ 26 ][ 27 ]、1件の研究で腫瘍が完全切除された場合に生存の改善が示されている。[ 28 ][証拠レベル:2A]分子的に分類された髄芽腫以外の胚芽腫について発表された1件の研究(COG-ACNS0332[NCT00392327])では、残存腫瘍が1.5cm2未満であった患者に対する全生存(OS)が、残存腫瘍が1.5cm2を超えた患者と比較して改善されたことが明らかにされた。[ 14 ][証拠レベル:1iiA]
- 髄芽腫以外の胚芽腫はしばしば切除可能である;報告されているケースシリーズでは、患者の50~75%で完全切除またはほぼ完全切除が実施された。[ 25 ][ 26 ];[ 14 ][証拠レベル:1iiA]
補助放射線療法
手術後の髄芽腫以外の胚芽腫の小児は、通常、高リスク髄芽腫の小児が受ける治療と同様の治療を受ける。
患者は慣習的に、髄芽腫の場合と同様に、局所への追加放射線療法を実施する脳脊髄軸全体への放射線で治療される。[ 27 ]しかしながら、局所への追加放射線療法は腫瘍の大きさおよび大脳皮質におけるその位置のために問題となることがある。また、非播種性病変を有する小児では頭蓋脊髄照射が原発腫瘍部位への放射線単独よりも優れているという決定的な証拠はない。[ 25 ][ 26 ][ 27 ]
補助化学療法
放射線療法中およびその後の化学療法のアプローチは、高リスク髄芽腫の小児に用いられているものとほぼ同じである。25~50%の3~5年OS率が報告されている。[ 25 ][ 26 ][ 27 ];[ 28 ][ 29 ][証拠レベル:2A];[ 30 ][証拠レベル:3iiiB]
従来の病理検査でCNS原始神経外胚葉性腫瘍(PNET)と診断されていた松果体以外の腫瘍について発表された1件の研究において、これらの症例の71%がDNAメチル化検査では膠芽腫または他の診断であることが明らかにされた。髄芽腫以外の胚芽腫患者(n = 36)(松果体芽腫、n = 26を含む)の5年OS率は78.5%(95%信頼区間[CI]、62.2%-94.8%)であった。対照的に、膠芽腫患者の5年OS率は12%(95%CI、0%-24.7%)であった。この研究では、カルボプラチンまたはイソトレチノインを受けた小児に対する有益性は示されなかった。[ 14 ][証拠レベル:1iiA]この研究により、伝統的にCNS-PNETと呼ばれる腫瘍の分子分類の重要性が強調されている。[ 4 ]
小児の多層性ロゼットを有する胎児性腫瘍または髄上皮腫の治療
新たに診断された髄上皮腫および多層性ロゼットを有する胎児性腫瘍(ETMR)に対する治療の基礎となるデータはほとんどない。治療法の考慮は通常、高リスク髄芽腫の小児および診断時年齢が3歳以下の他の胚芽腫の小児に対するものと同じである。(詳しい情報については、本要約の3歳より年長で高リスク髄芽腫の小児の治療および3歳以下の小児の治療のセクションを参照のこと。)
参考文献- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Benesch M, Sperl D, von Bueren AO, et al.: Primary central nervous system primitive neuroectodermal tumors (CNS-PNETs) of the spinal cord in children: four cases from the German HIT database with a critical review of the literature. J Neurooncol 104 (1): 279-86, 2011.[PUBMED Abstract]
- Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.[PUBMED Abstract]
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.[PUBMED Abstract]
- Korshunov A, Sturm D, Ryzhova M, et al.: Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128 (2): 279-89, 2014.[PUBMED Abstract]
- Picard D, Miller S, Hawkins CE, et al.: Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol 13 (8): 838-48, 2012.[PUBMED Abstract]
- Spence T, Sin-Chan P, Picard D, et al.: CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 128 (2): 291-303, 2014.[PUBMED Abstract]
- Kleinman CL, Gerges N, Papillon-Cavanagh S, et al.: Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46 (1): 39-44, 2014.[PUBMED Abstract]
- Li M, Lee KF, Lu Y, et al.: Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16 (6): 533-46, 2009.[PUBMED Abstract]
- Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.[PUBMED Abstract]
- Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.[PUBMED Abstract]
- Ueno-Yokohata H, Okita H, Nakasato K, et al.: Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 47 (8): 861-3, 2015.[PUBMED Abstract]
- Roy A, Kumar V, Zorman B, et al.: Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 6: 8891, 2015.[PUBMED Abstract]
- Hwang EI, Kool M, Burger PC, et al.: Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children's Oncology Group Randomized ACNS0332 Trial. J Clin Oncol : JCO2017764720, 2018.[PUBMED Abstract]
- Louis DN, Ohgaki H, Wiestler OD, et al.: The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114 (2): 97-109, 2007.[PUBMED Abstract]
- Sharma MC, Mahapatra AK, Gaikwad S, et al.: Pigmented medulloepithelioma: report of a case and review of the literature. Childs Nerv Syst 14 (1-2): 74-8, 1998 Jan-Feb.[PUBMED Abstract]
- Jakobiec FA, Kool M, Stagner AM, et al.: Intraocular Medulloepitheliomas and Embryonal Tumors With Multilayered Rosettes of the Brain: Comparative Roles of LIN28A and C19MC. Am J Ophthalmol 159 (6): 1065-1074.e1, 2015.[PUBMED Abstract]
- Korshunov A, Jakobiec FA, Eberhart CG, et al.: Comparative integrated molecular analysis of intraocular medulloepitheliomas and central nervous system embryonal tumors with multilayered rosettes confirms that they are distinct nosologic entities. Neuropathology 35 (6): 538-44, 2015.[PUBMED Abstract]
- Müller K, Zwiener I, Welker H, et al.: Curative treatment for central nervous system medulloepithelioma despite residual disease after resection. Report of two cases treated according to the GPHO Protocol HIT 2000 and review of the literature. Strahlenther Onkol 187 (11): 757-62, 2011.[PUBMED Abstract]
- Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.[PUBMED Abstract]
- Marec-Berard P, Jouvet A, Thiesse P, et al.: Supratentorial embryonal tumors in children under 5 years of age: an SFOP study of treatment with postoperative chemotherapy alone. Med Pediatr Oncol 38 (2): 83-90, 2002.[PUBMED Abstract]
- Grill J, Sainte-Rose C, Jouvet A, et al.: Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6 (8): 573-80, 2005.[PUBMED Abstract]
- Fangusaro J, Finlay J, Sposto R, et al.: Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs): report of the Head Start I and II experience. Pediatr Blood Cancer 50 (2): 312-8, 2008.[PUBMED Abstract]
- Friedrich C, von Bueren AO, von Hoff K, et al.: Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro Oncol 15 (2): 224-34, 2013.[PUBMED Abstract]
- Cohen BH, Zeltzer PM, Boyett JM, et al.: Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Childrens Cancer Group randomized trial. J Clin Oncol 13 (7): 1687-96, 1995.[PUBMED Abstract]
- Reddy AT, Janss AJ, Phillips PC, et al.: Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer 88 (9): 2189-93, 2000.[PUBMED Abstract]
- Timmermann B, Kortmann RD, Kühl J, et al.: Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol 20 (3): 842-9, 2002.[PUBMED Abstract]
- Jakacki RI, Burger PC, Kocak M, et al.: Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 776-83, 2015.[PUBMED Abstract]
- Chintagumpala M, Hassall T, Palmer S, et al.: A pilot study of risk-adapted radiotherapy and chemotherapy in patients with supratentorial PNET. Neuro Oncol 11 (1): 33-40, 2009.[PUBMED Abstract]
- Johnston DL, Keene DL, Lafay-Cousin L, et al.: Supratentorial primitive neuroectodermal tumors: a Canadian pediatric brain tumor consortium report. J Neurooncol 86 (1): 101-8, 2008.[PUBMED Abstract]
- 小児松果体芽腫
-
世界保健機関(WHO)は、松果体芽腫を松果体腫瘍のグループに分類している。しかしながら、松果体芽腫は他の胚芽腫と組織学的特徴を共有しており、慣習的に他の胚芽腫として治療されるため、松果体芽腫は本要約内で検討されている。[ 1 ]
臨床像
松果体芽腫は、しばしば第三脳室レベルでの脳脊髄液閉塞による水頭症および視蓋領域では脳幹後部への圧迫に関係した他の症状を引き起こす。症状には一連の眼球運動の異常(パリノー症候群)が含まれ、瞳孔の対光反射は不良であるが良好な遠近調節、上方視が見られないこと、眼球後退または輻輳眼振、眼瞼後退により明らかにされる。これらの腫瘍はまた、増殖するにつれて片側不全麻痺および運動失調を引き起こすことがある。[ 2 ]
細胞分類および分子分類
松果体芽腫は組織学的に髄芽腫に類似しており、胚芽腫と組織学的特徴を共有する;しかしながら、WHO分類により、松果体芽腫の組織発生は松果体細胞(pineocyte)と関連しており、別個に分類されている。[ 1 ]この分類では、これらの腫瘍の分子遺伝学的性質が考慮されていない。[ 1 ]
胚芽腫および松果体芽腫のゲノムの分子キャラクタリゼーションにより、これらの腫瘍間でかなりの不均一性が実証されている。これらの腫瘍はまた、分子的に髄芽腫と異なっている。[ 3 ][ 4 ]
WHO分類システムではまだ、髄芽腫以外の胚芽腫の分類に分子所見が採用されていないが、将来は組織学的所見と分子所見の両方、さらにはおそらく神経系の発生部位に基づいて分類される可能性が非常に高い。
以前は慣習的に胚芽腫とともに分類されていた松果体芽腫は現在では、世界保健機関(WHO)により松果体実質細胞腫瘍として分類されている。松果体芽腫に対する治療法が胚芽腫に用いられている治療法に非常に類似していることを考慮して、本要約でも松果体芽腫を中枢神経系(CNS)胚芽腫とともに含める以前の慣習に従う。松果体芽腫は、後述のようにRB1遺伝子およびDICER1遺伝子の両方における生殖細胞変異に関連している:
病期評価
診断時の播種は松果体芽腫患者の10~30%で起こっている。[ 10 ]腫瘍の位置のため、完全切除はまれであり、ほとんどの患者は術後治療の前には生検のみまたは亜全切除しか受けられない。[ 10 ][ 11 ]松果体芽腫の小児に対する病期分類は髄芽腫の小児と同じ方法で実施される;ただし、治療を目的とした平均リスクおよび高リスクのサブグループへの患者の割り付けは行われない(詳しい情報については、本要約の髄芽腫の病期分類のセクションを参照のこと)。[ 10 ]
小児松果体芽腫に対する治療法選択肢の概要
表5では、新たに診断された、および再発した小児松果体芽腫に対する標準治療法の選択肢について記述している。
表5.小児松果体芽腫に対する標準治療法の選択肢 治療法群 標準治療法の選択肢 新たに診断された小児松果体芽腫 3歳以下の小児 生検(診断のため)または亜全切除 補助化学療法 自家骨髄救助または末梢血幹細胞救助を併用する大量骨髄除去的化学療法 3歳より年長の小児 手術 補助放射線療法 補助化学療法 再発した小児松果体芽腫 標準治療法の選択肢は存在しない。(詳しい情報については、本要約の再発した小児髄芽腫およびその他のCNS胚芽腫の治療のセクションを参照のこと。) 小児松果体芽腫の治療
3歳以下の小児の治療
松果体芽腫で3歳以下の小児に対する標準治療法の選択肢には以下がある:
生検
松果体芽腫の診断には通常、生検が実施される。
補助化学療法
3歳以下の松果体芽腫の幼児では、放射線療法が避けられない場合、それが必要となる時期が遅くなることを期待して、通常は最初に化学療法を用いて治療する。[ 12 ]この小児群の全般的な予後は依然としてきわめて不良である。2件の連続的多施設プロスペクティブ臨床試験で化学療法による治療を受けた3歳未満の5人の小児は全員死亡した。[ 13 ][証拠レベル:2A]化学療法が奏効した小児において、化学療法後に必要となる放射線療法の時期および線量は不明である。化学療法ベースのレジメンに頭蓋脊髄照射を追加することで一部の小児では治療が成功する可能性があるが、神経発達の低下が予想される。[ 14 ][証拠レベル:2A]
自家骨髄救助または末梢血幹細胞救助を併用する大量骨髄除去的化学療法
自家骨髄救助または末梢血幹細胞救助を併用する高用量の骨髄除去的化学療法が使用されており、幼児においてある程度の成功を収めている。[ 15 ][証拠レベル:2Di]
3歳より年長の小児の治療
新たに診断された松果体芽腫で3歳より年長の小児に対する標準治療法の選択肢には以下のものがある:
手術
松果体芽腫の患者に対する初期治療は通常、腫瘍を診断するための手術である。[ 16 ]松果体芽腫患者では完全切除およびほぼ完全切除が行えることはまれであり、切除の程度が治療成績に及ぼす影響は不明である。[ 10 ][ 11 ]
補助放射線療法
松果体芽腫患者に対する通常の術後治療は放射線療法で開始されるが、一部の試験では放射線療法前の化学療法が用いられている。腫瘍部位への放射線の総線量は通常分割照射法を用いて54~55.8Gyである。[ 10 ][ 11 ]
この腫瘍はくも膜下腔全体に播種する傾向があるため、23.4~36Gyの線量による頭蓋脊髄の放射線照射も推奨されている。[ 10 ][ 11 ]
補助化学療法
化学療法は通常、診断時に播種が認められない小児における高リスク髄芽腫について概説されたものと同じ方法で用いられる。(詳しい情報については、本要約の3歳より年長で高リスク髄芽腫の小児の治療のセクションを参照のこと。)
診断時に限局性疾患で、積極的切除を受けた小児の5年無病生存率は50%を超える。[ 10 ][ 11 ][ 17 ][ 18 ][証拠レベル:1iiA]髄芽腫以外の胚芽腫患者36人(松果体芽腫患者26人を含む)を対象にした小児腫瘍学グループ(COG)のCOG-ACNS0332(NCT00392327)研究により、78.5%(95%信頼区間、62.2%-94.8%)の5年全生存(OS)率が報告された。[ 18 ][証拠レベル:1iiA]
診断時に播種性病変が認められる患者の生存は、かなり不良である。[ 10 ][ 11 ]COG-ACNS0332(NCT00392327)研究において、転移状態によるイベントフリー生存またはOSにおける有意差は認められなかった。
小児松果体芽腫に対して臨床評価段階にある治療法の選択肢
松果体芽腫の患者については、末梢血幹細胞救助でサポートする放射線療法後の比較的高用量の化学療法の使用、および放射線療法中の化学療法の使用を含む、多様な治療アプローチが評価段階にある。
選択された患者では、初期相の臨床試験が利用できる場合がある。これらの試験は、COGやPediatric Brain Tumor Consortiumなどの団体を介して利用できる。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
参考文献- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Chintagumpala MM, Paulino A, Panigrahy A, et al.: Embryonal and pineal region tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 671-99.[PUBMED Abstract]
- Pomeroy SL, Tamayo P, Gaasenbeek M, et al.: Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870): 436-42, 2002.[PUBMED Abstract]
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.[PUBMED Abstract]
- de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.[PUBMED Abstract]
- Ramasubramanian A, Kytasty C, Meadows AT, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (4): 825-9, 2013.[PUBMED Abstract]
- Abramson DH, Dunkel IJ, Marr BP, et al.: Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156 (6): 1319-20, 2013.[PUBMED Abstract]
- Turaka K, Shields CL, Meadows AT, et al.: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 59 (1): 121-5, 2012.[PUBMED Abstract]
- de Kock L, Sabbaghian N, Druker H, et al.: Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 128 (4): 583-95, 2014.[PUBMED Abstract]
- Jakacki RI, Zeltzer PM, Boyett JM, et al.: Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Childrens Cancer Group. J Clin Oncol 13 (6): 1377-83, 1995.[PUBMED Abstract]
- Timmermann B, Kortmann RD, Kühl J, et al.: Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol 20 (3): 842-9, 2002.[PUBMED Abstract]
- Mason WP, Grovas A, Halpern S, et al.: Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol 16 (1): 210-21, 1998.[PUBMED Abstract]
- Hinkes BG, von Hoff K, Deinlein F, et al.: Childhood pineoblastoma: experiences from the prospective multicenter trials HIT-SKK87, HIT-SKK92 and HIT91. J Neurooncol 81 (2): 217-23, 2007.[PUBMED Abstract]
- Friedrich C, von Bueren AO, von Hoff K, et al.: Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro Oncol 15 (2): 224-34, 2013.[PUBMED Abstract]
- Fangusaro J, Finlay J, Sposto R, et al.: Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs): report of the Head Start I and II experience. Pediatr Blood Cancer 50 (2): 312-8, 2008.[PUBMED Abstract]
- Jakacki RI, Burger PC, Kocak M, et al.: Outcome and prognostic factors for children with supratentorial primitive neuroectodermal tumors treated with carboplatin during radiotherapy: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 776-83, 2015.[PUBMED Abstract]
- Gururangan S, McLaughlin C, Quinn J, et al.: High-dose chemotherapy with autologous stem-cell rescue in children and adults with newly diagnosed pineoblastomas. J Clin Oncol 21 (11): 2187-91, 2003.[PUBMED Abstract]
- Hwang EI, Kool M, Burger PC, et al.: Extensive Molecular and Clinical Heterogeneity in Patients With Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children's Oncology Group Randomized ACNS0332 Trial. J Clin Oncol : JCO2017764720, 2018.[PUBMED Abstract]
- 再発した小児髄芽腫およびその他のCNS胚芽腫の治療
-
中枢神経系(CNS)胚芽腫はすべての型で再発は珍しくなく、通常は治療の36ヵ月以内に起こる。しかしながら、初期治療から何年も経過後に腫瘍が再発することもある。[ 1 ][ 2 ][ 3 ]こうした晩期再燃、特に診断から5年以上経過後に起こる再燃では、高悪性度グリオーマといった二次腫瘍との鑑別が困難な場合があり、組織学的確認が推奨され、通常は必要とされる。原発部位に再発することもあれば、再燃時に播種していることもある。非近接の再燃部位としては脊髄軟膜、頭蓋内、脳脊髄液などがあり、これらの部位の1つの場合もあれば複数の場合もあり、原発腫瘍の再燃に関連していることもある。[ 1 ][ 2 ][ 4 ]神経外病変再燃がみられる場合があるが、まれであり、主にみられるのは放射線療法単独による治療を受けた患者である。[ 5 ][証拠レベル:3iiiA]
諸研究では、診断時に播種が認められない患者においてさえも、放射線療法の線量または化学療法の種類に関係なく、約1/3の患者が原発腫瘍部位のみに再燃し、1/3が原発腫瘍部位および遠隔部位に再燃し、1/3が原発部位の再燃を伴わずに遠隔部位に再燃することが認められている。[ 1 ][ 2 ][ 4 ]
治療法選択肢
再発した小児CNS胚芽腫に対する標準治療法の選択肢は存在しない。(詳しい情報については、小児中枢神経系非定型奇形腫様/ラブドイド腫瘍の治療に関するPDQ要約の再発小児CNS非定型奇形腫様/ラブドイド腫瘍に対する治療のセクションを参照のこと。)
ほとんどの小児では治療は症状緩和目的で行われ、放射線療法および化学療法による治療歴のある患者で疾患制御は一時的であり、80%超が2年以内に増悪する。[ 3 ];[ 6 ][証拠レベル:3iB]幼児(主に放射線療法が未治療で診断時年齢が3歳未満の患児)については、再手術、放射線療法、および化学療法により比較的長期の疾患制御が可能である。[ 4 ][ 7 ][ 8 ][ 9 ]
治療アプローチには以下がある:
- 手術。
- 放射線療法。
- 化学療法。
- 幹細胞救助を併用する大量化学療法。
- 分子標的療法。
手術
再燃時には、すべての胚芽腫に対して再発の範囲を確認するための十分な評価が適応とされる。二次性腫瘍および治療に関連する脳壊死などの病変は、臨床的には腫瘍再発と鑑別することができないため、再燃の確認には生検または外科的切除術が必要とされる。手術の必要性は、初発腫瘍のタイプ、初期治療から病変の再発までの期間、および臨床的な症候論に基づいて、個別に判断しなければならない。
放射線療法
既に放射線療法および化学療法を受けている再発胚芽腫の患者は、以前の放射線療法の部位および線量に応じ、原発腫瘍部位への再照射、播種性病変がある部位に対する局所領域の放射線療法、まれに頭蓋脊髄放射線療法など、追加の放射線療法の候補となりうる。[ 10 ]ほとんどの症例で、こうした治療は症状緩和目的である。定位放射線療法および/または救助化学療法もまた使用できる(詳しい情報については、本要約の化学療法のサブセクションを参照のこと)。[ 11 ]
化学療法
再発CNS胚芽腫は、シクロホスファミド、シスプラチン、カルボプラチン、ロムスチン、エトポシド、トポテカン、テモゾロミドなどの化学療法薬の単独使用または併用および抗血管新生薬の規則正しい治療法に反応する可能性がある。[ 7 ][ 12 ][ 13 ][ 14 ][ 15 ][ 16 ][ 17 ][ 18 ][ 19 ][ 20 ][ 21 ];[ 22 ][ 23 ][証拠レベル:2A]これらの患者の約30~50%は、従来の化学療法に対して客観的奏効を示すが、長期の疾患制御はまれである。
選択された再発髄芽腫の患者で、主に診断時に化学療法単独による治療を受け、局所再発を来した乳児および幼児では、化学療法と局所放射線療法の併用による追加治療後に長期の疾患制御が得られる可能性がある;この可能性は再発病変の完全切除を受けることができる患者で最大となりうる。[ 24 ][証拠レベル:2A];[ 25 ][証拠レベル:3iiiA]
St. Jude Children's Research Hospitalの研究(SJYC07[NCT00602667])では、進行性疾患を有する29人の患者が頭蓋脊髄照射(線量中央値、36Gy;四分位範囲、36~36)を受けた。頭蓋脊髄照射を受けた29人の患者中、発表時に生存していた患者は18人(62%)であったのに対し、頭蓋脊髄照射を受けなかった25人の患者では6人(24%)であった。[ 26 ][証拠レベル:2Di]
幹細胞救助を併用する大量化学療法
過去に放射線療法を受けた患者では、自家骨髄救助または末梢血幹細胞救助によりサポートされる大量化学療法レジメンが使用されており、その成績はさまざまである。[ 8 ][ 9 ][ 27 ][ 28 ][ 29 ][ 30 ][証拠レベル:2A];[ 31 ][証拠レベル:3iiB];[ 32 ][ 33 ][証拠レベル:3iiiA]
- こうしたレジメンにより、客観的奏効が頻繁にみられ、患者の50~75%に得られている;しかしながら、長期的な疾患制御が得られるのは患者の30%未満であり、主に初回再燃の患者および再燃時の病変が限局性のみの患者で認められる。[ 9 ];[ 30 ][証拠レベル:2A];[ 31 ][証拠レベル:3iiB]
- さらに、再燃した髄芽腫に対する治療計画の一部として移植を意図すると規定した全国的試験の結果では、修復療法を開始して、この戦略により長期の無病生存を達成した患者はわずか約5%であったことが示された。[ 30 ][ 34 ]そのため、移植時点から報告される研究は、移植をベースとしたアプローチが再燃患者の全集団にもたらす有益性を過大評価する。
- 播種性病変を有する患者では、長期的な疾患制御はまれである。[ 35 ][証拠レベル:3iA]
分子標的療法
髄芽腫の異なるサブタイプに関連する分子的および遺伝的変化に関する知識の増加を受け、再発病変を有する小児において、precision therapyとも呼ばれる分子標的療法の研究が盛んに行われている。
再発ソニック・ヘッジホッグ(SHH)の髄芽腫患者では、SHH PTCH1阻害薬であるビスモデギブが小児期患者12人中3人で放射線学的奏効を示しており、2人の奏効の持続期間は2ヵ月未満、1人は6ヵ月超であった。PTCH1またはSMO段階のSHH経路の上流の変異を有する患者のみが奏効を生じた。[ 36 ]しかしながら、成長板の不可逆的な融合が発生するため、ビスモデギブの使用は骨格が成熟した小児に制限される。[ 37 ]
再発した小児髄芽腫およびその他のCNS胚芽腫に対して臨床評価段階にある治療法の選択肢
選択された患者では、初期相の臨床試験が利用できる場合がある。これらの試験は、小児腫瘍学グループ(COG)やPediatric Brain Tumor Consortiumなどの団体を介して利用できる。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は現在実施中の米国および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も、入手することができる。
参考文献- Taylor RE, Bailey CC, Robinson K, et al.: Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET-3 Study. J Clin Oncol 21 (8): 1581-91, 2003.[PUBMED Abstract]
- Packer RJ, Goldwein J, Nicholson HS, et al.: Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children's Cancer Group Study. J Clin Oncol 17 (7): 2127-36, 1999.[PUBMED Abstract]
- Sabel M, Fleischhack G, Tippelt S, et al.: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129 (3): 515-524, 2016.[PUBMED Abstract]
- Oyharcabal-Bourden V, Kalifa C, Gentet JC, et al.: Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23 (21): 4726-34, 2005.[PUBMED Abstract]
- Mazloom A, Zangeneh AH, Paulino AC: Prognostic factors after extraneural metastasis of medulloblastoma. Int J Radiat Oncol Biol Phys 78 (1): 72-8, 2010.[PUBMED Abstract]
- Johnston DL, Keene D, Strother D, et al.: Survival Following Tumor Recurrence in Children With Medulloblastoma. J Pediatr Hematol Oncol 40 (3): e159-e163, 2018.[PUBMED Abstract]
- Gentet JC, Doz F, Bouffet E, et al.: Carboplatin and VP 16 in medulloblastoma: a phase II Study of the French Society of Pediatric Oncology (SFOP). Med Pediatr Oncol 23 (5): 422-7, 1994.[PUBMED Abstract]
- Kadota RP, Mahoney DH, Doyle J, et al.: Dose intensive melphalan and cyclophosphamide with autologous hematopoietic stem cells for recurrent medulloblastoma or germinoma. Pediatr Blood Cancer 51 (5): 675-8, 2008.[PUBMED Abstract]
- Butturini AM, Jacob M, Aguajo J, et al.: High-dose chemotherapy and autologous hematopoietic progenitor cell rescue in children with recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors: the impact of prior radiotherapy on outcome. Cancer 115 (13): 2956-63, 2009.[PUBMED Abstract]
- Wetmore C, Herington D, Lin T, et al.: Reirradiation of recurrent medulloblastoma: does clinical benefit outweigh risk for toxicity? Cancer 120 (23): 3731-7, 2014.[PUBMED Abstract]
- Abe M, Tokumaru S, Tabuchi K, et al.: Stereotactic radiation therapy with chemotherapy in the management of recurrent medulloblastomas. Pediatr Neurosurg 42 (2): 81-8, 2006.[PUBMED Abstract]
- Friedman HS, Oakes WJ: The chemotherapy of posterior fossa tumors in childhood. J Neurooncol 5 (3): 217-29, 1987.[PUBMED Abstract]
- Needle MN, Molloy PT, Geyer JR, et al.: Phase II study of daily oral etoposide in children with recurrent brain tumors and other solid tumors. Med Pediatr Oncol 29 (1): 28-32, 1997.[PUBMED Abstract]
- Gaynon PS, Ettinger LJ, Baum ES, et al.: Carboplatin in childhood brain tumors. A Children's Cancer Study Group Phase II trial. Cancer 66 (12): 2465-9, 1990.[PUBMED Abstract]
- Allen JC, Walker R, Luks E, et al.: Carboplatin and recurrent childhood brain tumors. J Clin Oncol 5 (3): 459-63, 1987.[PUBMED Abstract]
- Ashley DM, Longee D, Tien R, et al.: Treatment of patients with pineoblastoma with high dose cyclophosphamide. Med Pediatr Oncol 26 (6): 387-92, 1996.[PUBMED Abstract]
- Lefkowitz IB, Packer RJ, Siegel KR, et al.: Results of treatment of children with recurrent medulloblastoma/primitive neuroectodermal tumors with lomustine, cisplatin, and vincristine. Cancer 65 (3): 412-7, 1990.[PUBMED Abstract]
- Friedman HS, Mahaley MS, Schold SC, et al.: Efficacy of vincristine and cyclophosphamide in the therapy of recurrent medulloblastoma. Neurosurgery 18 (3): 335-40, 1986.[PUBMED Abstract]
- Castello MA, Clerico A, Deb G, et al.: High-dose carboplatin in combination with etoposide (JET regimen) for childhood brain tumors. Am J Pediatr Hematol Oncol 12 (3): 297-300, 1990.[PUBMED Abstract]
- Cefalo G, Massimino M, Ruggiero A, et al.: Temozolomide is an active agent in children with recurrent medulloblastoma/primitive neuroectodermal tumor: an Italian multi-institutional phase II trial. Neuro Oncol 16 (5): 748-53, 2014.[PUBMED Abstract]
- Le Teuff G, Castaneda-Heredia A, Dufour C, et al.: Phase II study of temozolomide and topotecan (TOTEM) in children with relapsed or refractory extracranial and central nervous system tumors including medulloblastoma with post hoc Bayesian analysis: A European ITCC study. Pediatr Blood Cancer 67 (1): e28032, 2020.[PUBMED Abstract]
- Minturn JE, Janss AJ, Fisher PG, et al.: A phase II study of metronomic oral topotecan for recurrent childhood brain tumors. Pediatr Blood Cancer 56 (1): 39-44, 2011.[PUBMED Abstract]
- Peyrl A, Chocholous M, Kieran MW, et al.: Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59 (3): 511-7, 2012.[PUBMED Abstract]
- Ridola V, Grill J, Doz F, et al.: High-dose chemotherapy with autologous stem cell rescue followed by posterior fossa irradiation for local medulloblastoma recurrence or progression after conventional chemotherapy. Cancer 110 (1): 156-63, 2007.[PUBMED Abstract]
- Bakst RL, Dunkel IJ, Gilheeney S, et al.: Reirradiation for recurrent medulloblastoma. Cancer 117 (21): 4977-82, 2011.[PUBMED Abstract]
- Robinson GW, Rudneva VA, Buchhalter I, et al.: Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19 (6): 768-784, 2018.[PUBMED Abstract]
- Dunkel IJ, Boyett JM, Yates A, et al.: High-dose carboplatin, thiotepa, and etoposide with autologous stem-cell rescue for patients with recurrent medulloblastoma. Children's Cancer Group. J Clin Oncol 16 (1): 222-8, 1998.[PUBMED Abstract]
- Park JE, Kang J, Yoo KH, et al.: Efficacy of high-dose chemotherapy and autologous stem cell transplantation in patients with relapsed medulloblastoma: a report on the Korean Society for Pediatric Neuro-Oncology (KSPNO)-S-053 study. J Korean Med Sci 25 (8): 1160-6, 2010.[PUBMED Abstract]
- Gilman AL, Jacobsen C, Bunin N, et al.: Phase I study of tandem high-dose chemotherapy with autologous peripheral blood stem cell rescue for children with recurrent brain tumors: a Pediatric Blood and MarrowTransplant Consortium study. Pediatr Blood Cancer 57 (3): 506-13, 2011.[PUBMED Abstract]
- Pizer B, Donachie PH, Robinson K, et al.: Treatment of recurrent central nervous system primitive neuroectodermal tumours in children and adolescents: results of a Children's Cancer and Leukaemia Group study. Eur J Cancer 47 (9): 1389-97, 2011.[PUBMED Abstract]
- Massimino M, Gandola L, Spreafico F, et al.: No salvage using high-dose chemotherapy plus/minus reirradiation for relapsing previously irradiated medulloblastoma. Int J Radiat Oncol Biol Phys 73 (5): 1358-63, 2009.[PUBMED Abstract]
- Gururangan S, Krauser J, Watral MA, et al.: Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro Oncol 10 (5): 745-51, 2008.[PUBMED Abstract]
- Dunkel IJ, Gardner SL, Garvin JH, et al.: High-dose carboplatin, thiotepa, and etoposide with autologous stem cell rescue for patients with previously irradiated recurrent medulloblastoma. Neuro Oncol 12 (3): 297-303, 2010.[PUBMED Abstract]
- Gajjar A, Pizer B: Role of high-dose chemotherapy for recurrent medulloblastoma and other CNS primitive neuroectodermal tumors. Pediatr Blood Cancer 54 (4): 649-51, 2010.[PUBMED Abstract]
- Bowers DC, Gargan L, Weprin BE, et al.: Impact of site of tumor recurrence upon survival for children with recurrent or progressive medulloblastoma. J Neurosurg 107 (1 Suppl): 5-10, 2007.[PUBMED Abstract]
- Robinson GW, Orr BA, Wu G, et al.: Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol 33 (24): 2646-54, 2015.[PUBMED Abstract]
- Robinson GW, Kaste SC, Chemaitilly W, et al.: Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8 (41): 69295-69302, 2017.[PUBMED Abstract]
- 本要約の変更点(05/28/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
本要約には編集上の変更がなされた。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児髄芽腫およびその他の中枢神経系胚芽腫の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Childhood Medulloblastoma and Other Central Nervous System Embryonal Tumors Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/brain/hp/child-cns-embryonal-treatment-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389418]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。