

ご利用について
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood central nervous system atypical teratoid and rhabdoid tumor. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
CONTENTS
- General Information About Childhood Central Nervous System (CNS) Atypical Teratoid/Rhabdoid Tumor
-
Primary brain tumors, including atypical teratoid/rhabdoid tumors, are a diverse group of diseases that together constitute the most common solid tumor of childhood. The PDQ childhood brain tumor treatment summaries are primarily organized according to the World Health Organization classification of nervous system tumors.[ 1 ][ 2 ] Brain tumors are classified according to histology, but immunohistochemical analysis, cytogenetic and molecular genetic findings, and measures of mitotic activity are increasingly used in tumor diagnosis and classification. Tumor location and extent of spread are important factors that affect treatment and prognosis. For a full description of the classification of nervous system tumors and a link to the corresponding treatment summary for each type of brain tumor, refer to the PDQ summary on Childhood Brain and Spinal Cord Tumors Treatment Overview.
CNS atypical teratoid/rhabdoid tumor (AT/RT) is a rare, clinically aggressive tumor that most often affects children aged 3 years and younger but can occur in older children and adults. Approximately one-half of AT/RTs arise in the posterior fossa.[ 3 ] The diagnostic evaluation includes magnetic resonance imaging (MRI) of the neuraxis and lumbar cerebrospinal fluid examination. AT/RT has been linked to somatic and germline mutations of SMARCB1 and, less commonly, SMARCA4, both of which are tumor suppressor genes.[ 4 ] There is no current standard treatment for children with AT/RT. Multimodal treatment consisting of surgery, chemotherapy, and radiation therapy is under evaluation.
Based on present biologic understanding, AT/RT is part of a larger family of rhabdoid tumors. In this summary, the term AT/RT refers to CNS tumors only and the term rhabdoid tumor reflects the possibility of both CNS and non-CNS tumors. Unless specifically noted in the text, this summary is referring to CNS AT/RT.
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
Incidence
The exact incidence of childhood CNS AT/RT is difficult to determine because the tumor has only been recognized since 1996.[ 5 ]
The incidence in older patients is unknown. However, in the Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry (AT/RT Registry), 12 of the 42 patients (29%) were older than 36 months at the time of diagnosis.[ 9 ]
Anatomy
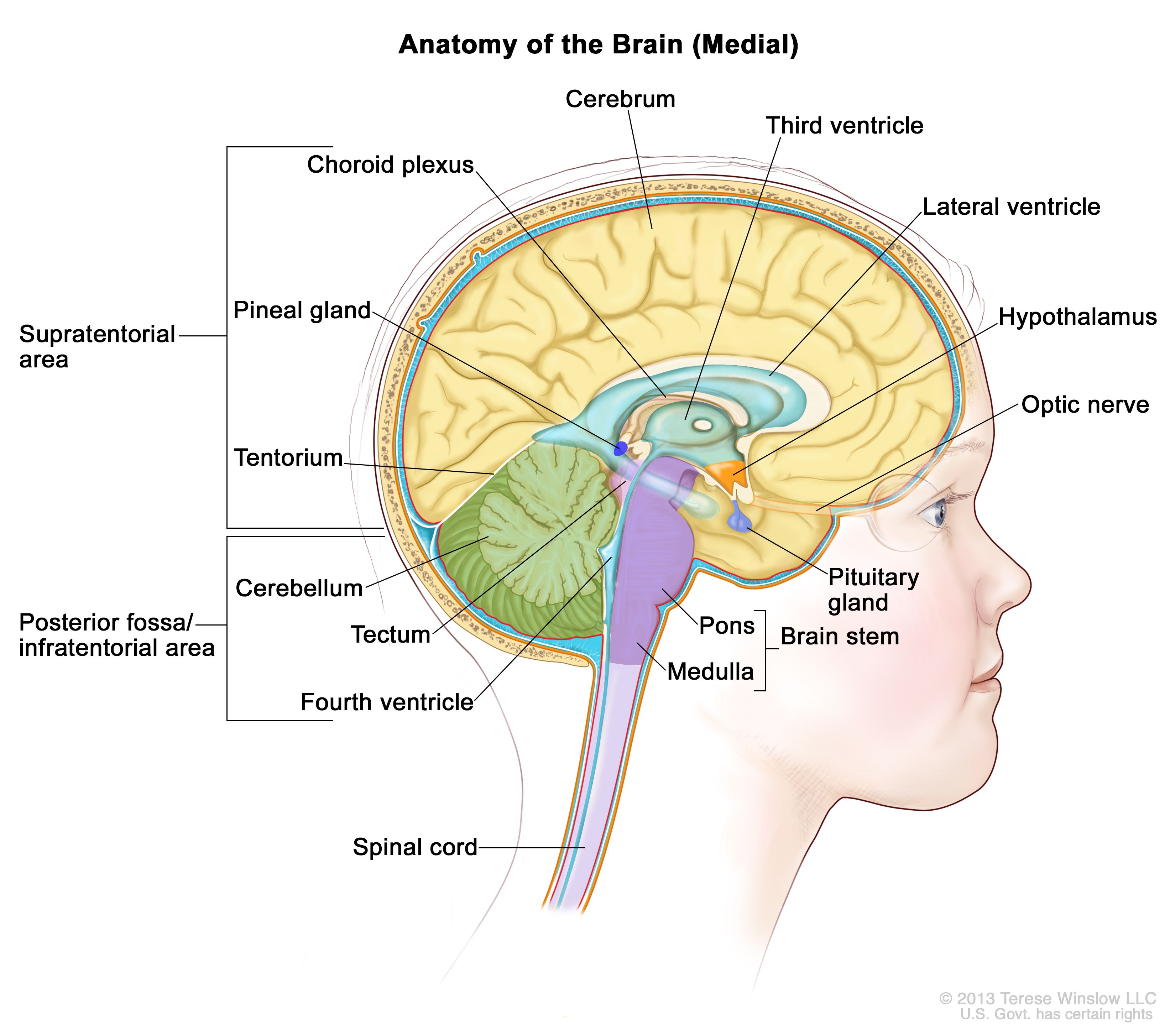
Anatomy of the inside of the brain, showing the pineal and pituitary glands, optic nerve, ventricles (with cerebrospinal fluid shown in blue), and other parts of the brain. The tentorium separates the cerebrum from the cerebellum. The infratentorium (posterior fossa) is the region below the tentorium that contains the brain stem, cerebellum, and fourth ventricle. The supratentorium is the region above the tentorium and denotes the region that contains the cerebrum. Clinical Presentation
Childhood AT/RT is a clinically aggressive tumor that primarily occurs in children younger than 3 years, but it also can occur in older children and has been reported in adults.[ 10 ][ 11 ]
Approximately one-half of all AT/RTs arise in the posterior fossa, although it can occur anywhere in the CNS.[ 3 ][ 6 ] Tumors of the posterior fossa may occur in the cerebellopontine angle or more midline. Involvement of individual cranial nerves has been noted.
Because AT/RT grows rapidly, patients typically have a fairly short history of progressive symptoms, measured in days to weeks. Signs and symptoms are dependent on tumor location. Young patients with posterior fossa tumors usually present with symptoms related to hydrocephalus, including the following:
They may also develop ataxia or regression of motor skills.
Registry data suggest that approximately 20% of patients present with disseminated disease.[ 9 ][ 12 ] Dissemination is typically through leptomeningeal pathways seeding the spine and other areas of the brain. Up to 35% of patients may present with germline mutations and be prone to synchronous, multifocal tumors. There are also rare reports of patients with synchronous renal rhabdoid tumor and CNS AT/RT.[ 13 ][ 14 ][ 15 ]
Diagnostic Evaluation
All patients with suspected childhood AT/RT should have MRI of the brain and spine. Unless medically contraindicated, patients should also have lumbar cerebrospinal fluid inspected for evidence of tumor. Patients may also undergo renal ultrasound to detect synchronous tumors.
AT/RT cannot be reliably distinguished from other malignant brain tumors on the basis of clinical history or radiographic evaluation alone. Surgery is necessary to obtain tissue and confirm the diagnosis. Immunostaining for loss of SMARCB1 protein expression is used to confirm the diagnosis.[ 16 ][ 17 ]
Prognosis
Prognostic factors that affect survival for AT/RTs are not fully delineated.
Known factors associated with a poor outcome include the following:
Most published data on outcomes of patients with AT/RT are from small series and are retrospective in nature. Initial retrospective studies reported an average survival from diagnosis of only about 12 months.[ 5 ][ 6 ][ 10 ][ 20 ][ 21 ] In a retrospective report, 2-year overall survival was better for patients who underwent a gross-total resection than for those who had a subtotal resection. However, in this study, the effect of radiation therapy on survival was less clear.[ 20 ]
There are reports of long-term survivors.[ 22 ] Notably, improved survival has been reported for those receiving intensive multimodal therapy.[ 12 ][ 15 ]
参考文献- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. Lyon, France: IARC Press, 2016.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Dho YS, Kim SK, Cheon JE, et al.: Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst 31 (8): 1305-11, 2015.[PUBMED Abstract]
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.[PUBMED Abstract]
- Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.[PUBMED Abstract]
- Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.[PUBMED Abstract]
- Ho DM, Hsu CY, Wong TT, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99 (5): 482-8, 2000.[PUBMED Abstract]
- Woehrer A, Slavc I, Waldhoer T, et al.: Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116 (24): 5725-32, 2010.[PUBMED Abstract]
- Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.[PUBMED Abstract]
- Burger PC, Yu IT, Tihan T, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22 (9): 1083-92, 1998.[PUBMED Abstract]
- Lutterbach J, Liegibel J, Koch D, et al.: Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol 52 (1): 49-56, 2001.[PUBMED Abstract]
- Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.[PUBMED Abstract]
- Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.[PUBMED Abstract]
- Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.[PUBMED Abstract]
- Seeringer A, Reinhard H, Hasselblatt M, et al.: Synchronous congenital malignant rhabdoid tumor of the orbit and atypical teratoid/rhabdoid tumor--feasibility and efficacy of multimodal therapy in a long-term survivor. Cancer Genet 207 (9): 429-33, 2014.[PUBMED Abstract]
- Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.[PUBMED Abstract]
- Margol AS, Judkins AR: Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet 207 (9): 358-64, 2014.[PUBMED Abstract]
- Kordes U, Gesk S, Frühwald MC, et al.: Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer 49 (2): 176-81, 2010.[PUBMED Abstract]
- Dufour C, Beaugrand A, Le Deley MC, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118 (15): 3812-21, 2012.[PUBMED Abstract]
- Lafay-Cousin L, Hawkins C, Carret AS, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48 (3): 353-9, 2012.[PUBMED Abstract]
- Athale UH, Duckworth J, Odame I, et al.: Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 31 (9): 651-63, 2009.[PUBMED Abstract]
- Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.[PUBMED Abstract]
- Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.[PUBMED Abstract]
- Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.[PUBMED Abstract]
- Tumor Biology of Childhood CNS Atypical Teratoid/Rhabdoid Tumor
-
Childhood central nervous system (CNS) atypical teratoid/rhabdoid tumor (AT/RT) was first described as a discrete clinical entity in 1987 [ 1 ] on the basis of its distinctive pathologic and genetic characteristics. Before then, it was most often classified as a medulloblastoma, CNS primitive neuroectodermal tumor (CNS PNET), or choroid plexus carcinoma. The World Health Organization (WHO) began classifying AT/RT as an embryonal grade IV neoplasm in 1993.[ 2 ]
Histologically, AT/RT is morphologically heterogeneous, typically containing sheets of large epithelioid cells with abundant eosinophilic cytoplasm and scattered rhabdoid cells, most often with accompanying components of primitive neuroectodermal cells (small round blue cells), mesenchymal cells, and/or glial cells.[ 3 ]
Immunohistochemical staining for epithelial markers (cytokeratin or epithelial membrane antigen), glial fibrillary acidic protein, synaptophysin (or neurofilament), and smooth muscle (desmin) may help to identify the heterogeneity of differentiation, but will vary depending on the cellular composition.[ 4 ] Rhabdoid cells, while not present in all AT/RTs, will express vimentin, epithelial membrane antigen, and smooth muscle actin.
Immunohistochemistry for the SMARCB1 protein is useful in establishing the diagnosis of AT/RT. A loss of SMARCB1 staining is noted in neoplastic cells, but staining is retained in non-neoplastic cells (e.g., vascular endothelial cells).[ 5 ][ 6 ][ 7 ]
AT/RT is a rapidly growing tumor that can have an MIB-1 labeling index of 50% to 100%.[ 8 ]
Genomics of CNS Atypical Teratoid/Rhabdoid Tumor (AT/RT)
SMARCB1 gene
AT/RT was the first primary pediatric brain tumor in which a candidate tumor suppressor gene, SMARCB1 (previously known as INI1 and hSNF5), was identified.[ 9 ] SMARCB1 is genomically altered in most rhabdoid tumors, including CNS, renal, and extrarenal rhabdoid malignancies.[ 9 ] Loss of SMARCB1/SMARCA4 staining is a defining marker for AT/RT. Additional genomic alterations (mutations and gains/losses) in other genes are very uncommon in patients with SMARCB1-associated AT/RT. Less commonly, SMARCA4-negative (with retained SMARCB1) tumors have been described.[ 10 ] No other genes are recurrently mutated in AT/RT.[ 11 ][ 12 ][ 13 ]
SMARCB1 is a component of a switch (SWI) and sucrose non-fermenting (SNF) adenosine triphosphate–dependent chromatin-remodeling complex.[ 14 ] Rare familial cases of rhabdoid tumors expressing SMARCB1 and lacking SMARCB1 mutations have also been associated with germline mutations of SMARCA4/BRG1, another member of the SWI/SNF chromatin-remodeling complex.[ 7 ][ 15 ]
The 2016 WHO classification defines AT/RT by the presence of either SMARCB1 or SMARCA4 alterations. Tumors with histological features of AT/RT that lack these genomic alterations are termed CNS embryonal tumor with rhabdoid features.[ 2 ]
Despite the absence of recurring genomic alterations beyond SMARCB1 (and, more rarely, other SWI/SNF complex members), biologically distinctive subsets of AT/RT have been identified.[ 16 ][ 17 ] The following three distinctive subsets of AT/RT were identified through the use of DNA methylation arrays for 150 AT/RT tumors and gene expression arrays for 67 AT/RT tumors:[ 17 ]
In addition to somatic mutations, germline mutations in SMARCB1 have been reported in a substantial subset of AT/RT patients.[ 9 ][ 19 ] A study of 65 children with rhabdoid tumors found that 23 (35%) had germline mutations and/or deletions of SMARCB1.[ 5 ] Children with germline alterations in SMARCB1 presented at an earlier age than did sporadic cases (median age, approximately 5 months vs. 18 months) and were more likely to present with synchronous, multifocal tumors.[ 5 ] One parent was found to be a carrier of the SMARCB1 germline abnormality in 7 of 22 evaluated cases showing germline alterations, with four of the carrier parents being unaffected by SMARCB1-associated cancers.[ 5 ] This indicates that AT/RT shows an autosomal dominant inheritance pattern with incomplete penetrance.
Gonadal mosaicism has also been observed, as evidenced by families in which multiple siblings are affected by AT/RT and have identical SMARCB1 alterations, but both parents lack a SMARCB1 mutation/deletion.[ 5 ][ 6 ] Screening for germline SMARCB1 mutations in children diagnosed with AT/RT may provide useful information for counseling families on the genetic implications of their child’s AT/RT diagnosis.[ 5 ]
Loss of SMARCB1 or SMARCA4 protein expression has therapeutic significance, because this loss creates a dependence of the cancer cells on EZH2 activity.[ 20 ] Preclinical studies have shown that some AT/RT xenograft lines with SMARCB1 loss respond to EZH2 inhibitors with tumor growth inhibition and occasional tumor regression.[ 21 ][ 22 ] In a study of the EZH2 inhibitor tazemetostat, objective responses were observed in adult patients whose tumors had either SMARCB1 or SMARCA4 loss (non-CNS malignant rhabdoid tumors and epithelioid sarcoma).[ 23 ] (Refer to the Treatment of Recurrent Childhood CNS Atypical Teratoid/Rhabdoid Tumor section of this summary for more information.)
Rhabdoid Tumor Predisposition Syndrome (RTPS)
RTPS, related primarily to germline SMARCB1 alterations, has been clearly defined.[ 9 ][ 19 ] RTPS is highly suggested in patients with synchronous occurrence of extracranial malignant rhabdoid tumor (kidney or soft tissue) and AT/RT, bilateral malignant rhabdoid tumors of the kidney, or malignant rhabdoid tumors in two or more siblings.
This syndrome is manifested by a marked predisposition to the development of malignant rhabdoid tumors in infancy and early childhood. Up to one-third of AT/RTs are thought to arise in the setting of RTPS, and most of these occur within the first year of life. The most common non-CNS malignancy of RTPS is malignant rhabdoid tumor of the kidney, which is also noted in infancy.
(Refer to the Rhabdoid predisposition syndrome section in the PDQ summary on Wilms Tumor and Other Childhood Kidney Tumors Treatment for more information about RTPS.)
Cribriform Neuroepithelial Tumor
Cribriform neuroepithelial tumor is histologically and clinically distinct from AT/RT, but it has genomic and epigenomic characteristics that are very similar to AT/RT TYR.[ 18 ] Like AT/RT, cribriform neuroepithelial tumor occurs in young children (median age, 1–2 years) and tumor cells lack SMARCB1 expression. Histologically, cribriform neuroepithelial tumor is characterized by the presence of cribriform strands and ribbons, but there is an absence of rhabdoid tumor cells with abundant eosinophilic cytoplasm. Like AT/RT TYR, tyrosinase expression is commonly observed. The outcome of patients with cribriform neuroepithelial tumor is more favorable than the outcome of patients with AT/RT TYR, with only one death reported among ten children with cribriform neuroepithelial tumor.[ 18 ]
参考文献- Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. Lyon, France: IARC Press, 2016.[PUBMED Abstract]
- McLendon RE, Adekunle A, Rajaram V, et al.: Embryonal central nervous system neoplasms arising in infants and young children: a pediatric brain tumor consortium study. Arch Pathol Lab Med 135 (8): 984-93, 2011.[PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.[PUBMED Abstract]
- Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.[PUBMED Abstract]
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.[PUBMED Abstract]
- Kleihues P, Louis DN, Scheithauer BW, et al.: The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 61 (3): 215-25; discussion 226-9, 2002.[PUBMED Abstract]
- Biegel JA, Tan L, Zhang F, et al.: Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8 (11): 3461-7, 2002.[PUBMED Abstract]
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.[PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.[PUBMED Abstract]
- Kieran MW, Roberts CW, Chi SN, et al.: Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer 59 (7): 1155-7, 2012.[PUBMED Abstract]
- Hasselblatt M, Isken S, Linge A, et al.: High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 52 (2): 185-90, 2013.[PUBMED Abstract]
- Biegel JA, Kalpana G, Knudsen ES, et al.: The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62 (1): 323-8, 2002.[PUBMED Abstract]
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.[PUBMED Abstract]
- Torchia J, Picard D, Lafay-Cousin L, et al.: Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16 (5): 569-82, 2015.[PUBMED Abstract]
- Johann PD, Erkek S, Zapatka M, et al.: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3): 379-93, 2016.[PUBMED Abstract]
- Johann PD, Hovestadt V, Thomas C, et al.: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 27 (4): 411-418, 2017.[PUBMED Abstract]
- Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.[PUBMED Abstract]
- Wilson BG, Wang X, Shen X, et al.: Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 (4): 316-28, 2010.[PUBMED Abstract]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.[PUBMED Abstract]
- Kurmasheva RT, Sammons M, Favours E, et al.: Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 64 (3): , 2017.[PUBMED Abstract]
- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.[PUBMED Abstract]
- Stage Information for Childhood CNS Atypical Teratoid/Rhabdoid Tumor
There is no defined staging system for childhood central nervous system atypical teratoid/rhabdoid tumor. For treatment purposes, patients are classified as having newly diagnosed or recurrent disease with or without neuraxis dissemination.
- Treatment of Newly Diagnosed Childhood CNS Atypical Teratoid/Rhabdoid Tumor
-
A standard treatment for children with central nervous system (CNS) atypical teratoid/rhabdoid tumor (AT/RT) has not yet been defined. Given the highly aggressive nature of the tumor, most patients have been treated with intensive multimodal therapy. However, the young age of most patients limits the extent of treatment, particularly for radiation therapy.
Treatment options for newly diagnosed CNS AT/RT include the following:
Surgery, Chemotherapy, and Radiation Therapy (Multimodal Therapy)
The extent of the surgical resection may affect survival. Data from the Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry (AT/RT Registry) suggest that patients who have had a complete resection may have a longer median survival, although complete surgical resection is often difficult because of the invasive nature of the tumor.[ 1 ]
Chemotherapy has been the main adjuvant therapy for very young children with AT/RT. Cooperative group studies that included children younger than 36 months demonstrated poor survival when treated with standard chemotherapeutic regimens alone.[ 2 ] The Children’s Cancer Group reported a 2-year event-free survival (EFS) of 14% for 28 children younger than 36 months treated with multiagent chemotherapy.[ 3 ]
Intensive regimens that utilize varying combinations of high-dose chemotherapy,[ 4 ][Level of evidence: 3iA]; [ 5 ][ 6 ][Level of evidence: 3iiiDi] intrathecal chemotherapy, and radiation therapy have led to prolonged survival for some patients. Thirteen patients in the AT/RT Registry were treated with high-dose chemotherapy with hematopoietic stem cell rescue as part of initial therapy.[ 1 ] Four of these patients, two of whom also received radiation, were alive without progressive disease 21.5 to 90 months after diagnosis at last report. Of 15 evaluable children (all younger than 32 months at diagnosis) who were on a chemotherapy Head Start III protocol, 2 survived for more than 47 months.[ 7 ][Level of evidence: 3iA]
Radiation therapy appears to have a positive impact on survival for AT/RT patients.
Evidence (radiation therapy):
- Of the 42 patients in the AT/RT Registry, 13 patients (31%) received radiation therapy in addition to chemotherapy as part of their primary therapy.[ 1 ] The radiation field was to the primary tumor bed in nine children, and the tumor bed and the craniospinal axis in four children. The median survival of these patients was 48 months, while the median survival of all patients on the registry was 16.75 months.
- In a retrospective series of 31 patients with AT/RT from St. Jude Children's Research Hospital, the 2-year EFS for patients older than 3 years was 78%, which was considerably better than the EFS (11%) for patients younger than 3 years.[ 8 ] All but one of the surviving patients (seven of eight) in the older group received craniospinal irradiation and intensive chemotherapy with hematopoietic stem cell transplant; only 3 of the 22 younger patients received any form of radiation therapy, two of whom are disease free.
- In a Surveillance, Epidemiology, and End Results registry review, radiation therapy was associated with improved survival in children younger than 3 years.[ 9 ]
- In the European Registry for rhabdoid tumors series, radiation therapy was also associated with an improved survival, with a 6-year overall survival (OS) rate of 66% (± 0.1%) in irradiated patients.[ 10 ][Level of evidence: 3iA]
Evidence (multimodal therapy):
- The Third Intergroup Rhabdomyosarcoma Study (IRS-III) utilized radiation therapy, intrathecal methotrexate, cytarabine, hydrocortisone, and systemic multiagent chemotherapy. The results of small retrospective series were encouraging,[ 11 ][ 12 ] leading to the first prospective study of multimodality treatment in this group of patients.
- On the basis of the previous pilot series, a prospective multi-institutional trial was conducted for children with newly diagnosed CNS AT/RT. Treatment was divided into five phases: preirradiation, chemoradiation, consolidation, maintenance, and continuation therapy. Intrathecal chemotherapy was administered, alternating intralumbar and intraventricular routes. Radiation therapy was either focal (54 Gy) or craniospinal (36 Gy, plus primary boost), depending on the child's age and extent of disease at diagnosis.[ 13 ]
- A report using the National Cancer Database described the outcome of 361 children with CNS AT/RT who were aged 0 to 18 years at diagnosis and treated in two periods (2004–2008 and 2009–2012). The report confirmed the increased use of trimodal therapy during the study period and an improvement in outcome.[ 14 ]
Because AT/RT is responsive to radiation therapy, this modality is incorporated into many treatment protocols.[ 15 ]
Prospective cooperative group clinical trials for AT/RT are greatly needed to better understand how age and extent of therapy affect survival.
Treatment Options Under Clinical Evaluation
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the Children’s Oncology Group, the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
参考文献- Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.[PUBMED Abstract]
- Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.[PUBMED Abstract]
- Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.[PUBMED Abstract]
- Nicolaides T, Tihan T, Horn B, et al.: High-dose chemotherapy and autologous stem cell rescue for atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 98 (1): 117-23, 2010.[PUBMED Abstract]
- Gardner SL, Asgharzadeh S, Green A, et al.: Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer 51 (2): 235-40, 2008.[PUBMED Abstract]
- Finkelstein-Shechter T, Gassas A, Mabbott D, et al.: Atypical teratoid or rhabdoid tumors: improved outcome with high-dose chemotherapy. J Pediatr Hematol Oncol 32 (5): e182-6, 2010.[PUBMED Abstract]
- Zaky W, Dhall G, Ji L, et al.: Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 61 (1): 95-101, 2014.[PUBMED Abstract]
- Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.[PUBMED Abstract]
- Buscariollo DL, Park HS, Roberts KB, et al.: Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118 (17): 4212-9, 2012.[PUBMED Abstract]
- Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.[PUBMED Abstract]
- Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.[PUBMED Abstract]
- Zimmerman MA, Goumnerova LC, Proctor M, et al.: Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol 72 (1): 77-84, 2005.[PUBMED Abstract]
- Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.[PUBMED Abstract]
- Fischer-Valuck BW, Chen I, Srivastava AJ, et al.: Assessment of the treatment approach and survival outcomes in a modern cohort of patients with atypical teratoid rhabdoid tumors using the National Cancer Database. Cancer 123 (4): 682-687, 2017.[PUBMED Abstract]
- De Amorim Bernstein K, Sethi R, Trofimov A, et al.: Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys 86 (1): 114-20, 2013.[PUBMED Abstract]
- Treatment of Recurrent Childhood CNS Atypical Teratoid/Rhabdoid Tumor
-
There is no standard treatment for children with recurrent atypical teratoid/rhabdoid tumor (AT/RT). Trials of molecularly targeted therapy are ongoing. In a study of the EZH2 inhibitor tazemetostat in adult patients with epithelioid sarcoma and non–central nervous system (CNS) malignant rhabdoid tumors with SMARCB1 or SMARCA4 loss, prolonged stable disease and objective responses were observed.[ 1 ] The activity of tazemetostat in children with AT/RT is under clinical evaluation.
Patients or families who desire additional disease-directed therapy should consider entering trials of novel therapeutic approaches because no standard agents have demonstrated clinically significant activity.
Regardless of whether a decision is made to pursue disease-directed therapy at the time of progression, palliative care remains a central focus of management. This ensures that quality of life is maximized while attempting to reduce symptoms and stress related to the terminal illness.
Treatment Options Under Clinical Evaluation
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the Children’s Oncology Group (COG), the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
参考文献- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.[PUBMED Abstract]
- Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
- Changes to This Summary (12/17/2019)
-
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Recurrent Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor
Added text to state that patients or families who desire additional disease-directed therapy should consider entering trials of novel therapeutic approaches because no standard agents have demonstrated clinically significant activity.
Added text to state that regardless of whether a decision is made to pursue disease-directed therapy at the time of progression, palliative care remains a central focus of management. This ensures that quality of life is maximized while attempting to reduce symptoms and stress related to the terminal illness.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
- About This PDQ Summary
-
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood central nervous system atypical teratoid and rhabdoid tumor. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/brain/hp/child-cns-atrt-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389426]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.