

ご利用について
医療専門家向けの本PDQがん情報要約では、神経芽腫の治療について、包括的な、専門家の査読を経た、そして根拠に基づいた情報を提供する。本要約は、患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 神経芽腫に関する一般情報
-
小児および青年期のがん患者の生存において、劇的な改善が達成されている。[ 1 ][ 2 ]1975年から2020年の間に小児がんの死亡率は50%以上低下した。[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ]神経芽腫患者の5年生存率は1975年から2020年の間に1歳未満の小児については86%から93%へ増加し、1~14歳の小児については34%から83%へ増加した。[ 2 ][ 3 ]
小児および青年期のがん生存者には、治療から数カ月または数年経過した後もがん治療の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。 小児期および青年期のがん生存者における晩期障害の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)の要約を参照のこと。
発生率および疫学
神経芽腫は小児において最も頻度の高い頭蓋外固形腫瘍である。米国では毎年650例以上の症例が診断される。[ 2 ][ 6 ][ 7 ][ 8 ]有病率は、およそ出生7,000当たり1例である。15歳未満の小児における発生率は、年間100万人当たり8.3例である。米国における神経芽腫症例の発生率は全体として安定している。[ 9 ]約37%の患者が乳児期に診断され、90%は診断時年齢が5歳未満であり、診断時年齢の中央値は生後17カ月である。[ 8 ][ 10 ] 診断時年齢に関するデータは、これが乳児期の疾患であり、生後1カ月以内での診断率が最も高いことを示している。[ 6 ][ 10 ][ 11 ]
複数の(神経芽腫の乳児に対するスクリーニングに関する)集団ベース研究によって、生後1年以内に臨床で発見されることなく自然退縮する神経芽腫が、臨床で発見される神経芽腫と少なくとも同程度に存在することが示されている。[ 12 ][ 13 ][ 14 ]
米国がん統計データベース(United States Cancer Statistics database)および米国国立がん登録プログラム生存データベース(National Program of Cancer Registries survival database)を用いて、2003年から2019年までの神経芽腫患者の発生率および転帰に関する疫学的傾向を記述した研究によると、 非ヒスパニック系の白人患者では、他のすべての人種および民族集団と比べて神経芽腫の発生リスクが高い。非ヒスパニック系白人患者と比較した相対リスクは、ヒスパニック系患者で0.54、非ヒスパニック系アジア人または太平洋諸島系患者で0.64、非ヒスパニック系アメリカンインディアンおよびアラスカ先住民患者で0.69、非ヒスパニック系黒人患者で0.73であった。[ 9 ]5年相対生存率は、非ヒスパニック系黒人患者(72.6%)と比較して、非ヒスパニック系白人患者(80.7%)とヒスパニック系患者(80.8%)で高かった。[ 9 ]
複数の疫学研究の知見からは、環境曝露やその他の曝露と神経芽腫の発生率増減との間に明確な関連は認められていない。[ 15 ]
神経芽腫のスクリーニング
家族性神経芽腫および遺伝的素因
家族性神経芽腫患者の希少コホートで生殖細胞系列DNAを解析した複数の研究から、腫瘍発生における複雑な遺伝学的根拠について洞察が得られている。神経芽腫患者の約1~2%に神経芽腫の家族歴がみられる。それらの患児は家族歴のない患児と比べて平均年齢が低く(診断時で生後9カ月)、約20%が原発性の神経芽腫を複数有している。
生殖細胞系列バリアント:以下のものを含むいくつかの生殖細胞系列バリアントについて、神経芽腫の遺伝的素因との関連が報告されている:
その他のがん素因症候群。他のがん素因症候群に関連する遺伝子異常を有する小児は、神経芽腫やその他の悪性疾患を発症するリスクが高い可能性がある。以下の症候群には古典的RAS経路の遺伝子が主に関係している:
さらに、神経芽腫は以下の症候群の患者でも報告されている:
シーケンシング技術の普及に伴い神経芽腫患者にみられる生殖細胞系列変異のスペクトラムは拡大している。例えば、ある研究では、SMARCA4の病的な生殖細胞系列バリアントを有する患者11例が同定された。[ 30 ]神経芽腫患者786人を対象とした別の研究では、対象患者の13.9%ががん素因遺伝子に病原性または病原性可能性の高い生殖細胞変異を有していた。生殖細胞系列で病原性変異が最も頻繁に観察された遺伝子は、BARD1、ERCC2、CHEK2、MSH3であった。神経芽腫の患者では、対照と比較して、BARD1、EZH2、ALK、PTCH1およびMSH3の病的生殖細胞系列バリアントが特異的に多く認められた。これらの変異を有する患者は、変異を持たない患者と比較して生存率が劣っていた。[ 31 ]別の研究でも、がん素因遺伝子における生殖細胞変異が予後不良と関連するという知見が再現された。さらに、従来のがん素因遺伝子を超える生殖細胞系列の機能的バリアントの負荷も予後因子であったことが示された。[ 32 ]SMARCA4遺伝子の詳細については、ラブドイド腫瘍素因症候群2型を参照のこと。
散発性の神経芽腫もまた、より弱い生殖細胞系列素因の結果として発生率が高まる可能性もある。ゲノムワイド関連解析により、神経芽腫の発生リスク増大と関連する、効果サイズの小さな共通のゲノム変異(一塩基多型)がいくつか同定されている。それらのゲノムリスク変異の大半には、神経芽腫の明確に異なる表現型(高リスク群 vs 低リスク群)との有意な関連が認められる。[ 33 ]
神経芽腫の素因およびサーベイランス
American Association for Cancer Research(AACR)によるスクリーニングに関する推奨が、2016年のChildhood Cancer Predisposition Workshopで公表された。AACRは、以下の対象者は10歳になるまでに腫瘍を早期発見するべく生化学検査とX線撮影によるサーベイランスを受けるべきと推奨している:[ 27 ]
サーベイランスは以下で構成される:[ 27 ]
サーベイランスは出生時または神経芽腫素因の診断時に開始し、6歳までは3カ月ごとに、その後10歳までは6カ月ごとに継続する。 コステロ症候群の患者はカテコールアミン分泌腫瘍が存在しない場合でも尿中カテコールアミンが高値となる場合があるため、値が高いか、有意な上昇がみられる場合に限り、超音波・胸部X線以外の検査を指示すべきである。[ 35 ]リー-フラウメニ症候群の患者には胸部X線を行うべきでない。[ 27 ]
ベックウィズ-ヴィーデマン症候群の小児の約5%は、CDKN1Cの活性を低下させるバリアントを有している。遺伝的亜型に分類されたベックウィズ-ヴィーデマン症候群に関するすべての大規模研究を対象としたレビューでは、CDKN1Cバリアントを有する小児が70人特定され、そのうち4.6%が神経芽腫を発症した。ウィルムス腫瘍および肝芽腫の症例は認められなかった。このような小児に対する神経芽腫スクリーニングの経験は乏しいため、一般に受け入れられているガイドラインはない。しかしながら、同研究の著者らは、尿中VMA/HVAによる4~6カ月毎のスクリーニングを提唱している。ベックウィズ-ヴィーデマン症候群の他の遺伝的亜型を有する患者では、神経芽腫の有病率は1%未満である。インプリンティング制御領域1(ICR1)にメチル化獲得遺伝子型がみられた123人の患児では、神経芽細胞群腫瘍は認められなかった。[ 36 ]
臨床像
小児の神経芽腫で最も頻度の高い徴候および症状は、腫瘍塊および転移によるものであり、具体的には以下のものがある:
青年期における神経芽腫の臨床像は、小児期のそれと類似する。 唯一の例外は骨髄転移の発生頻度が青年期では比較的低いことであり、肺や脳などのまれな部位への転移がより高頻度でみられる。[ 40 ]
オプソクローヌス/ミオクローヌス症候群
神経芽腫の小児ではまれに、小脳性運動失調やオプソクローヌス/ミオクローヌスなど、腫瘍随伴症状としての神経所見がみられる。[ 41 ] オプソクローヌス/ミオクローヌス症候群を呈する幼児のうち、約半数で神経芽腫が発見される。[ 42 ][ 43 ]英国における小児の年間発生率は、100万人当たり0.18例と推定されている。診断時年齢の平均は1.5~2歳である。[ 44 ]
通常の臨床像としては、神経芽腫が発見される数日前から進行性の神経機能障害で発症する。しかしながら、原発腫瘍の切除後長期間が経過してから神経症状が現れることもある。[ 42 ][ 45 ][ 46 ]オプソクローヌス/ミオクローヌス症候群を発症する神経芽腫患者は、しばしば神経芽腫に予後良好な生物学的特徴がみられ、生存率がきわめて良好であるが、腫瘍関連死の報告もある。[ 42 ]
オプソクローヌス/ミオクローヌス症候群は、まだ十分に解明されていない免疫機序によって引き起こされるようである。[ 42 ] 典型的には原発腫瘍にびまん性のリンパ球浸潤がみられる。[ 47 ]髄液ではB細胞数の増加がみられ、しばしばオリゴクローナルな免疫グロブリンバンドがみられる。ステロイド反応性のB細胞関連サイトカインの上昇もしばしばみられる。[ 48 ]
オプソクローヌス/ミオクローヌス症候群を伴う神経芽腫症例44例において、ゲノムコピー数プロファイルが解析された。 腫瘍の再発や疾患関連死が認められなかったため、全ゲノムプロファイルに予後予測上の意義は認められなかった。[ 49 ]
免疫学的介入や単純に神経芽腫の切除に対して鋭敏な神経学的反応がみられる患者もあるが、多くの症例では、改善は緩徐かつ部分的なものに留まる。 免疫療法により急性期の運動障害と運動失調は改善された一方、主に認知障害と行動障害で構成される長期的な神経心理学的障害に対する免疫療法の利益は明らかでない。治療で得られる速やかな改善の長期的な利益は、それが症状に対する治療であるか、基礎にある神経芽腫に対する治療であるかに関係なく、不明であるが、速やかな改善それ自体に価値があると考えられる。[ 46 ] [ 50 ]
急性症状に対して副腎皮質刺激ホルモンまたはコルチコステロイドによる治療が有効となる場合があるが、コルチコステロイドに反応しない患者もいる。[ 45 ][ 51 ] 種々の免疫調節薬、血漿交換療法、静注γグロブリン、およびリツキシマブによる他の治療法が選択された症例で有効であることが報告されている。[ 45 ][ 52 ][ 53 ][ 54 ][ 55 ]多剤併用の免疫抑制療法が検討されており、短期成績に改善がみられる。[ 56 ]化学療法で治療された患者では、短期の神経学的成績がより良好となることがあるが、これは化学療法の免疫抑制作用によるものである可能性がある。[ 41 ]
Children's Oncology Group(COG)は、オプソクローヌス/ミオクローヌス症候群の患者を対象とした初のランダム化オープンラベル第III相試験を完了した。[ 57 ] オプソクローヌス/ミオクローヌス運動失調症候群を併発した8歳未満の神経芽腫初発患者が、プレドニゾンと本腫瘍に対するリスク調整治療を単独または免疫グロブリン静注療法(IVIG)との併用で受ける2群にランダムに割り付けられた。[ 57 ]
診断
神経芽腫の診断的評価としては以下のものがある:
神経芽腫の診断には、小児腫瘍に精通した病理医の関与が必要である。 神経芽腫の中には、ヘマトキシリン・エオシン染色のみを用いる従来の光学顕微鏡検査では、リンパ腫やユーイング肉腫、横紋筋肉腫など小児の他のsmall round blue cell tumor(SRBCT)と形態学的に鑑別できないものがある。 そのような症例では、SRBCTをより具体的に診断するために、免疫組織化学的および細胞遺伝学的分析が必要となる。
国際合意により確立された神経芽腫診断の必要最低基準として、診断を下すには以下のいずれか1つを満たす必要がある:[ 64 ]
- 光学顕微鏡検査(免疫組織検査または電子顕微鏡検査の併用の有無は問わない)により腫瘍組織から明確な病理診断が得られていること。
- 骨髄穿刺または骨髄生検により明確な腫瘍細胞(例、合胞体または免疫細胞学的に陽性の細胞集塊)が認められ、かつ尿中カテコールアミン代謝物値が高値を示していること。
胎児/新生児神経芽腫の経過観察と自然退縮
神経芽腫の乳児、特にINSS 4S/INRG MSパターンの転移がみられる乳児では、自然退縮現象がよく報告されている。[ 65 ]まれに、胎児超音波検査によって神経芽腫の疑いが出生前の段階で示されることもある。[ 66 ]自然退縮する可能性が高い神経芽腫腫瘍が疑われる生後6カ月以下の乳児における即時診断生検の必要性に関する管理上の推奨事項は現在も検討が続いている。INSS 4S/INRG MS例に関する詳しい情報については、原発巣および転移巣の評価のセクションを参照のこと。
自然退縮は一般に、以下の特徴を有する腫瘍でみられる:[ 67 ][ 68 ][ 69 ]
自然退縮に関連する別の特徴としては、テロメラーゼの発現がないこと[ 67 ][ 70 ]、H-Ras蛋白の発現[ 71 ]、神経成長因子受容体であるニューロトロフィン受容体TrkAの発現などがある。[ 72 ]
無症状の小さな早期副腎神経芽腫(スクリーニング、出生前超音波検査、または偶然行われた超音波検査で発見されたもの)を有するとみられる特定の乳児症例では、しばしば腫瘍の自然退縮がみられることが複数の研究により示唆されている。それらの患者は外科的介入や組織学的診断なしで安全に経過観察とすることができる。[ 73 ][ 74 ][ 75 ]
根拠(経過観察[自然退縮]):
- あるCOG研究において、画像検査によって1期の小さな副腎腫瘤(3.1 cm 以下)を検出された、厳選された6か月未満の乳児83名が生検なしで経過観察とされた。外科的介入の対象は、腫瘤の増大または進行がみられた患者と尿中カテコールアミン代謝物の高値がみられた患者とされた。[ 76 ]
- ドイツの臨床試験では、MYCN増幅を認めない限局性神経芽腫の乳児340人の成績が報告された。このうち190人が切除を受け、57人が化学療法で治療され、93人で肉眼的残存腫瘍が認められた。[ 77 ]
- カナダのケベック州とドイツで実施された神経芽腫のスクリーニング試験では、神経芽腫の発生率がスクリーニングを受けない集団で報告される値の2倍となり、この結果から、多くの神経芽腫が臨床的に診断されることなく、自然退縮していることが示唆される。[ 12 ][ 13 ][ 14 ]
予後因子
神経芽腫患者の予後には以下の因子が関連している:
治療方針の決定に役立てるため、これらの予後因子の一部を組み合わせたリスク群が考案されている。詳しい情報については、International Neuroblastoma Risk Group Staging System(INRGSS)およびChildren's Oncology Group(COG)Neuroblastoma Risk Groupingのセクションを参照のこと。
診断時年齢
乳児および小児
診断時年齢が5年生存率に及ぼす影響は顕著である。新規に神経芽腫と診断された患者4,832人を対象としたCOG ANBL00B1(NCT00904241)研究では、生後18カ月未満の患者の5年EFS率は82%、OS率は91%であった。対して、生後18カ月以上の患者では、5年EFS率が64%、OS率が74%であった。[ 78 ]
National Childhood Cancer Registry(NCCR)によると、2014年から2020年までの5年相対生存率は以下の通りであった:[ 2 ]
患者年齢が予後に及ぼす影響は、以下から明らかなように、臨床的および病理生物学的因子に強い影響を受ける:
青年期および若年成人
青年期および成人が神経芽腫を発症することはまれであり、全症例に占める割合は5%未満である。 この年齢層で神経芽腫が発生した場合、臨床経過はより若年の患者に発生した神経芽腫より緩徐であるが、しばしばde novoの化学療法抵抗性を示す。[ 81 ]青年期および若年成人における神経芽腫では、大きな腫瘍や両側性の副腎病変、褐色細胞腫様の特徴など、まれな臨床病理学的特徴がみられることがある。[ 82 ][エビデンスレベルC1]神経芽腫は10歳以上の青年または成人では、病期や部位にかかわらず、長期予後が比較的不良である。
青年期および若年成人患者でMYCN増幅が認められることはまれであるが(10~21歳で9%)、進行期の年長児では生存率が不良である。青年期および若年成人集団の腫瘍では分節性染色体異常がよくみられ、ALKおよびATRXバリアントの頻度が他よりはるかに高い。[ 83 ][ 84 ][ 85 ]青年期では、約40%の腫瘍にATRXの機能喪失型バリアントがみられるのに対して、このバリアントは比較的年齢の低い小児では20%未満、1歳未満の乳児では0%である。[ 81 ]一部の患者で複雑なDNAマイクロアレイ所見と新たな変異が報告されている。[ 82 ][エビデンスレベルC1]
青年期および若年成人患者(15~39歳)の5年OS率は38%である。[ 86 ][エビデンスレベルC1]10~21歳の患者の5年EFS率は32%であり、OS率は46%である。4期の患者では、10年EFS率が3%、OS率が5%である。[ 87 ]積極的な化学療法と手術により、これらの患者の50%以上で病状が最小限に抑えられることが示されている。[ 40 ][ 88 ]局所放射線療法や自家造血幹細胞移植、効果が確認されている薬剤の使用など、その他の治療法によって、青年期および成人における不良な予後が改善する可能性がある。[ 87 ][ 88 ]
成人
44人の患者(18~71歳)を対象とした単一施設の症例集積研究の報告によると、成人発症の神経芽腫は生物学的性質が小児または青年期の神経芽腫と異なるようである。[ 89 ]
上述のように、成人発症の神経芽腫では活性化型ALKバリアントが多く認められる。ある単一施設の後ろ向き研究では、ALK変異を有する再発神経芽腫の成人患者13人(年齢中央値34歳;範囲16~71歳)がロルラチニブによる治療を受けた。9人(69%)が完全または部分奏効を達成したが、うち5人には他のALK阻害薬による治療歴があった。ロルラチニブには、減量を要する有意な有害事象との関連が認められた。しかしながら、成人用の推奨用量を下回る用量で反応がみられた。[ 90 ]別の多施設共同試験では、ALK変異を有する再発または難治性神経芽腫の成人患者15人(18歳以上;年齢中央値24歳)がロルラチニブによる治療を受けた。奏効率(完全奏効、部分奏効、最小奏効)は67%であった。[ 91 ]
病期
1990年代までは、病期判定に画像所見と手術所見に基づくいくつかの分類システムが用いられていた。世界中で得られた結果を比較する試みとして、International Neuroblastoma Staging System(INSS)と呼ばれる外科病理学的病期分類システムが開発された。[ 64 ]このINSSにより、診断時の病期に基づき予後が予測されたが、生物学的要因との重要な交互作用の存在も明らかにされた。[ 3 ][ 4 ][ 11 ][ 64 ][ 79 ][ 80 ][ 92 ][ 93 ][ 94 ]しかしながら、外科的なアプローチは施設間で異なるため、局所病変のある患者のINSS病期にも大きな差が生じる可能性がある。診断時の進展範囲を画一的に定義するべく、International Neuroblastoma Risk Group Classification System用に手術前に適用するInternational Neuroblastoma Risk Group Staging System(INRGSS)が開発された。[ 95 ][ 96 ]INRGSSは現在、北米および欧州の共同グループ研究で用いられている。この病期分類システムは、局所領域リンパ節転移の影響を受けない。
ANBL00B1(NCT00904241)研究に登録された初発の神経芽腫患者において、INRGSSの病期別に見た5年EFS率およびOS率は以下の通りであった:[ 78 ]
詳しい情報については、以下のセクションを参照のこと:
腫瘍の組織型
神経芽腫の組織型は、予後およびリスク群の判定に有意な影響を及ぼす。詳しい情報については、神経芽腫群腫瘍の組織学的分類のセクションと表2を参照のこと。
初発の神経芽腫患者4,832人を対象としたANBL00B1(NCT00904241)研究では、International Neuroblastoma Pathology Classification(INPC)に従い、52%の腫瘍が予後良好、48%の腫瘍が予後不良に分類された。腫瘍が予後良好に分類された患者では、5年EFS率が88%、OS率が96%であった。腫瘍が予後不良に分類された患者では、5年EFS率が55%、5年OS率が66%であった(P < 0.0001)。[ 78 ]
予後良好と考えられる組織学的特徴としては以下のものがある:
有糸分裂/核崩壊指数の高さと未分化腫瘍細胞は予後不良の組織学的特徴と考えられているが、その予後予測上の価値は年齢に依存する。[ 100 ][ 101 ]
あるCOG研究(P9641[NCT00003119])では、複数の因子とともに組織型の治療成績に対する影響が検討された。MYCN増幅がない1期および2期神経芽腫の小児915人のうち、87%が初回手術と経過観察で治療された。症状が出現しているか、そうなるリスクがあるか、診断時に切除された腫瘍が50%未満であるか、手術単独の施行後に切除不能進行と判定された患者(13%)には、化学療法と手術による治療が行われた。組織学的特徴が良好な患者では、5年EFS率が90%~94%、OS率が99%~100%と報告された。予後不良の組織学的特徴を有する患者では、EFS率が80%~86%、OS率が89%~93%であった。[ 79 ]
神経芽腫の中間リスク群患者を対象としたCOG ANBL0531(NCT00499616)研究では、生物学的特徴に基づくアルゴリズムと反応に基づくアルゴリズムを用いて治療が割り付けられたが、これらのアルゴリズムには1p36および11q23のアレル状態が含まれていた。MYCN増幅がみられる患者は除外された。[ 102 ]
INRG Data Commonsのデータを用いた研究では、基礎にあるINPCの組織学的基準の予後予測上の価値が評価された。年齢、組織学的カテゴリー、核分裂-核崩壊指数(MKI)、および悪性度について、独立した予後予測能が示された。年齢に関連した4つの組織学的予後分類群が同定された(生後18カ月未満ではMKI低値 vs MKI高値、生後18カ月以上では高分化 vs 未分化/低分化)。確立されたCOGリスク基準を用いて作成された生存系統樹と比較して、INPCの代わりに個々の組織学的特徴を解析した場合、予後予測に意義のある別のサブグループが同定され、その妥当性が確認された。[ 104 ]INPCについては、神経芽腫群腫瘍の組織学的分類のセクションに記載されている。
生物学的特徴
詳しい情報については、神経芽腫のゲノムおよび生物学的特徴のセクションを参照のこと。
原発部位
神経芽腫の臨床的および生物学的特徴は、腫瘍の原発部位によって異なる。 国際リスクグループプロジェクト(International Risk Group Project)が複数の臨床試験データ8,389例を統合した研究では、以下の結果が観察され、臨床的・生物学的データが不完全な小規模先行研究の結果を裏付けた:[ 105 ]
Therapeutically Applicable Research to Generate Effect Treatments(TARGET)およびゲノムワイド関連解析のデータセットを用いた単一の研究において、副腎原発の神経芽腫(n = 646)のゲノムおよびエピゲノムデータが胸部交感神経節から発生した神経芽腫(n = 118)のデータと比較された。副腎原発の神経芽腫ではMYCN増幅などの構造的DNA異常が認められることが多かった一方、胸部腫瘍は結果として高二倍体となる有糸分裂チェックポイントの異常がみられた。 胸部腫瘍は副腎腫瘍と比べて、すべての症例(OR、1.89;P = 0.04)とMYCN増幅のない症例(OR、2.86;P = 0.003)でALKの機能獲得型異常を有する頻度が高かった。 胸部腫瘍は16%でALKバリアントがみられるため、この状況ではそれらのバリアントに対する塩基配列決定をルーチンに行うことを考慮すべきである。[ 106 ]
TARGETコホートでは、副腎原発の患者の70%と胸部原発の患者の51%が4期であった。MYCN増幅を考慮しないゲノムワイド関連解析では、副腎原発の患者の43%と胸部原発の患者の17%が4期であった。 多変量解析によると、MYCN増幅の有無、病期、および年齢(閾値は生後18カ月)で調整したとき、原発部位が副腎であることはゲノムワイド関連解析コホートにおいて予後不良の独立した予測因子となったが、TARGETコホートではそうならなかった。 副腎神経芽腫は、ゲノムワイド関連解析コホートまたはTARGETコホートを対象とした同様の多変量解析においても、EFS不良の独立した予測因子とならなかった。[ 106 ]
神経芽腫の原発部位が予後に及ぼす影響が発生部位と関連のある腫瘍の生物学的性質の差のみに依存するのかどうかは不明である。
多発性神経芽細胞腫の発生はまれであるが、通常は乳児にみられ、一般に予後良好である。[ 107 ]多発原発性神経芽腫の患者では、家族性神経芽腫と生殖細胞系列の病的ALKバリアントを考慮すべきである。
治療に対する反応
治療に対する反応に転帰との関連が報告されている。 COG ANBL0531(NCT00499616)研究で初回治療に対する反応が不良であった中間リスク群神経芽腫患者20人のうち、その後に6人が進行または再発を来し、1人が死亡した。[ 102 ]
高リスク群の患者では、寛解導入化学療法後の骨髄中における神経芽腫細胞の残存に予後不良との関連がみられる。微小残存病変を検出する高感度の手法を用いることで、予後を評価できる可能性がある。[ 108 ][ 109 ][ 110 ]例えば、高リスク群神経芽腫の小児における初回寛解導入化学療法後に(骨髄中の)神経芽腫細胞によって発現したRNA転写物の検出は、有意に不良なEFSおよびOSとの関連が報告されている。[ 111 ]
同様に、寛解導入化学療法完了後のCurieスコアが2を超えることを基準とするMIBG集積の持続は、MYCN増幅のない高リスク群患者において予後不良の予測因子である。腫瘍にMYCN増幅がある高リスク群患者では、寛解導入化学療法完了後の0を超えるCurieスコアに不良な転帰との関連がみられる。[ 112 ][ 113 ] タンデム移植を受けた北米患者を対象とした解析では、導入療法終了時のCurieスコアが0を超えていた患者はEFS率が低かったことが示された。[ 114 ]Curieスコアに関する詳しい情報については、Curieスコア法およびSIOPENスコア法のセクションを参照のこと。
連続した4つのCOG高リスク群試験の患者を対象とした解析では、導入療法終了時のInternational Neuroblastoma Response Criteria[ 64 ](1993年)に従った部分奏効(PR)以上の反応にEFS率およびOS率上昇との有意な関連が認められた。多変量解析(n = 407)では、11qのヘテロ接合性喪失(LOH)を認めないことが、依然としてPR以上の成績と有意な関連を示す唯一の因子であった(OR、1.962 vs 11q LOH;95%CI、1.104-3.487;P = 0.0216)。[ 115 ]
治療に関連した有糸分裂の減少と原発腫瘍の組織学的分化度の上昇も反応の予測因子である。[ 116 ]
原発腫瘍の縮小に基づく予後予測の正確性はあまり明確ではない。7つの大規模国際センターが実施した研究では、229人の高リスク群患者が様々な方法で治療された。具体的な治療法としては、化学療法、原発腫瘍の外科的切除、腫瘍床への放射線照射、高用量の骨髄破壊的治療 + 造血幹細胞移植のほか、大半の症例で選択されたサイトカインで強化した抗GD2抗体免疫療法とイソトレチノインの併用などがあった。導入化学療法後の原発腫瘍の反応が、最大径の30%以上の縮小、腫瘍体積の50%以上の縮小、腫瘍体積の65%以上の縮小(従来の放射線学的手法である3つの腫瘍径からの算出)という3つの尺度で測定された。測定は診断時と原発腫瘍切除前の導入化学療法後に実施された。導入化学療法終了時における原発腫瘍の反応は、いずれの尺度でも生存の予測因子にならなかった。[ 117 ]
LDHおよびフェリチンの測定値
1990年から2016年にかけて神経芽腫と診断された患者(n > 8,575)で構成される大規模国際コホートでは、LDHおよびフェリチンの血清中濃度が高いほど、5年EFS率およびOS率が不良であった。2009年以降の高リスク群神経芽腫患者では、LDHおよびフェリチンの血清中濃度が高いほど、3年EFS率およびOS率も不良であった。 診断時年齢、MYCN増幅の有無、およびINSS(4期)で調整した多変量解析においても、LDHおよびフェリチン値に独立した予後予測能が認められた(P < 0.0001)。[ 118 ][エビデンスレベルC1]
当初のINRG分類システムでは厳密な評価は行われなかったが、その後のINRG Data Commonsの解析により、2010年から2016年までの期間を含めて、すべての患者と高リスク群患者において、フェリチンおよびLDHの血清中濃度が統計学的に有意な独立した予後因子であったことが明確に示された。したがって、容易に得ることができるこれら2つの臨床検査値をINRGの予後分類システムに組み込むことが提案された。[ 118 ]
治療の実施時期
米国における神経芽腫の5年生存率は1975年から2020年までの間に、1歳未満の小児では86%から93%に、1~14歳の小児では34%から83%に上昇した。[ 2 ][ 3 ]神経芽腫のすべての乳児および小児患者の5年相対生存率は、1974年から1989年までの期間に診断された患者では46%であったのが、1999年から2005年までの期間に診断された患者では71%まで上昇した。[ 119 ]2014年から2020年までを対象としたより最近の推定では、15歳未満の乳児および小児の5年相対生存率が約85%とさらに高いことが示されている。[ 2 ]これらの統計値については、予後に年齢、病期、および生物学的特徴に応じた極端な不均一性がみられることから、解釈に注意が必要である。それでも、2000年から2019年までの期間に診断されて治療を受けた高リスク群患者の生存率は、1990年から1999年までの期間に診断された患者と比較して、有意に改善したことが複数の研究から示されている。[ 120 ][ 121 ]詳しい情報については、表1を参照のこと。同様に、COG ANBL0531(NCT00499616)研究でも、より早期に実施されたCOG-A3961(NCT00003093)研究と比較して、強度を大きく下げた化学療法で治療された中間リスク群患者の多くの部分集団において同等の成績が得られたことが明らかにされた。[ 102 ]
参考文献- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.[PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.[PUBMED Abstract]
- Gurney JG, Ross JA, Wall DA, et al.: Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992. J Pediatr Hematol Oncol 19 (5): 428-32, 1997 Sep-Oct.[PUBMED Abstract]
- United States Census Bureau: Age and Sex Composition in the United States: 2018. U.S. Census Bureau, 2018. Available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Mahapatra S, Challagundla KB: Neuroblastoma. Treasure Island, FL: StatPearls Publishing LLC, 2022. Available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Campbell K, Siegel DA, Umaretiya PJ, et al.: A comprehensive analysis of neuroblastoma incidence, survival, and racial and ethnic disparities from 2001 to 2019. Pediatr Blood Cancer 71 (1): e30732, 2024.[PUBMED Abstract]
- London WB, Castleberry RP, Matthay KK, et al.: Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol 23 (27): 6459-65, 2005.[PUBMED Abstract]
- Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012. Also available online. Last accessed May 22, 2024.[PUBMED Abstract]
- Takeuchi LA, Hachitanda Y, Woods WG, et al.: Screening for neuroblastoma in North America. Preliminary results of a pathology review from the Quebec Project. Cancer 76 (11): 2363-71, 1995.[PUBMED Abstract]
- Woods WG, Gao RN, Shuster JJ, et al.: Screening of infants and mortality due to neuroblastoma. N Engl J Med 346 (14): 1041-6, 2002.[PUBMED Abstract]
- Schilling FH, Spix C, Berthold F, et al.: Neuroblastoma screening at one year of age. N Engl J Med 346 (14): 1047-53, 2002.[PUBMED Abstract]
- Heck JE, Ritz B, Hung RJ, et al.: The epidemiology of neuroblastoma: a review. Paediatr Perinat Epidemiol 23 (2): 125-43, 2009.[PUBMED Abstract]
- Mossé YP, Laudenslager M, Longo L, et al.: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455 (7215): 930-5, 2008.[PUBMED Abstract]
- Mosse YP, Laudenslager M, Khazi D, et al.: Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 75 (4): 727-30, 2004.[PUBMED Abstract]
- Raabe EH, Laudenslager M, Winter C, et al.: Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 27 (4): 469-76, 2008.[PUBMED Abstract]
- van Limpt V, Schramm A, van Lakeman A, et al.: The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 23 (57): 9280-8, 2004.[PUBMED Abstract]
- Serra A, Häberle B, König IR, et al.: Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol 30 (10): 728-32, 2008.[PUBMED Abstract]
- Satgé D, Moore SW, Stiller CA, et al.: Abnormal constitutional karyotypes in patients with neuroblastoma: a report of four new cases and review of 47 others in the literature. Cancer Genet Cytogenet 147 (2): 89-98, 2003.[PUBMED Abstract]
- Mosse Y, Greshock J, King A, et al.: Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol 4 (12): 769-71, 2003.[PUBMED Abstract]
- Gillani R, Collins RL, Crowdis J, et al.: Rare germline structural variants increase risk for pediatric solid tumors. Science 387 (6729): eadq0071, 2025.[PUBMED Abstract]
- Moroni I, Bedeschi F, Luksch R, et al.: Costello syndrome: a cancer predisposing syndrome? Clin Dysmorphol 9 (4): 265-8, 2000.[PUBMED Abstract]
- Cotton JL, Williams RG: Noonan syndrome and neuroblastoma. Arch Pediatr Adolesc Med 149 (11): 1280-1, 1995.[PUBMED Abstract]
- Gutmann DH, Ferner RE, Listernick RH, et al.: Neurofibromatosis type 1. Nat Rev Dis Primers 3: 17004, 2017.[PUBMED Abstract]
- Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.[PUBMED Abstract]
- Bougnères P, Pantalone L, Linglart A, et al.: Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab 93 (10): 3971-80, 2008.[PUBMED Abstract]
- Maas SM, Vansenne F, Kadouch DJ, et al.: Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 170 (9): 2248-60, 2016.[PUBMED Abstract]
- Witkowski L, Nichols KE, Jongmans M, et al.: Germline pathogenic SMARCA4 variants in neuroblastoma. J Med Genet 60 (10): 987-992, 2023.[PUBMED Abstract]
- Kim J, Vaksman Z, Egolf LE, et al.: Germline pathogenic variants in neuroblastoma patients are enriched in BARD1 and predict worse survival. J Natl Cancer Inst 116 (1): 149-159, 2024.[PUBMED Abstract]
- Seo ES, Lee JW, Lim J, et al.: Germline functional variants contribute to somatic mutation and outcomes in neuroblastoma. Nat Commun 15 (1): 8360, 2024.[PUBMED Abstract]
- Tolbert VP, Coggins GE, Maris JM: Genetic susceptibility to neuroblastoma. Curr Opin Genet Dev 42: 81-90, 2017.[PUBMED Abstract]
- Matser YAH, Verly IRN, van der Ham M, et al.: Optimising urinary catecholamine metabolite diagnostics for neuroblastoma. Pediatr Blood Cancer 70 (6): e30289, 2023.[PUBMED Abstract]
- Kratz CP, Rapisuwon S, Reed H, et al.: Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 157 (2): 83-9, 2011.[PUBMED Abstract]
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.[PUBMED Abstract]
- Citak C, Karadeniz C, Dalgic B, et al.: Intestinal lymphangiectasia as a first manifestation of neuroblastoma. Pediatr Blood Cancer 46 (1): 105-7, 2006.[PUBMED Abstract]
- Bourdeaut F, de Carli E, Timsit S, et al.: VIP hypersecretion as primary or secondary syndrome in neuroblastoma: A retrospective study by the Société Française des Cancers de l'Enfant (SFCE). Pediatr Blood Cancer 52 (5): 585-90, 2009.[PUBMED Abstract]
- Mahoney NR, Liu GT, Menacker SJ, et al.: Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol 142 (4): 651-9, 2006.[PUBMED Abstract]
- Conte M, Parodi S, De Bernardi B, et al.: Neuroblastoma in adolescents: the Italian experience. Cancer 106 (6): 1409-17, 2006.[PUBMED Abstract]
- Matthay KK, Blaes F, Hero B, et al.: Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer Lett 228 (1-2): 275-82, 2005.[PUBMED Abstract]
- Rudnick E, Khakoo Y, Antunes NL, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children's Cancer Group Study. Med Pediatr Oncol 36 (6): 612-22, 2001.[PUBMED Abstract]
- Antunes NL, Khakoo Y, Matthay KK, et al.: Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol 22 (4): 315-20, 2000 Jul-Aug.[PUBMED Abstract]
- Pang KK, de Sousa C, Lang B, et al.: A prospective study of the presentation and management of dancing eye syndrome/opsoclonus-myoclonus syndrome in the United Kingdom. Eur J Paediatr Neurol 14 (2): 156-61, 2010.[PUBMED Abstract]
- Pranzatelli MR: The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol 15 (3): 186-228, 1992.[PUBMED Abstract]
- Mitchell WG, Davalos-Gonzalez Y, Brumm VL, et al.: Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and neurologic sequelae. Pediatrics 109 (1): 86-98, 2002.[PUBMED Abstract]
- Cooper R, Khakoo Y, Matthay KK, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: histopathologic features-a report from the Children's Cancer Group. Med Pediatr Oncol 36 (6): 623-9, 2001.[PUBMED Abstract]
- Pranzatelli MR, Tate ED, McGee NR: Demographic, Clinical, and Immunologic Features of 389 Children with Opsoclonus-Myoclonus Syndrome: A Cross-sectional Study. Front Neurol 8: 468, 2017.[PUBMED Abstract]
- Hero B, Clement N, Øra I, et al.: Genomic Profiles of Neuroblastoma Associated With Opsoclonus Myoclonus Syndrome. J Pediatr Hematol Oncol 40 (2): 93-98, 2018.[PUBMED Abstract]
- Catsman-Berrevoets CE, Aarsen FK, van Hemsbergen ML, et al.: Improvement of neurological status and quality of life in children with opsoclonus myoclonus syndrome at long-term follow-up. Pediatr Blood Cancer 53 (6): 1048-53, 2009.[PUBMED Abstract]
- Connolly AM, Pestronk A, Mehta S, et al.: Serum autoantibodies in childhood opsoclonus-myoclonus syndrome: an analysis of antigenic targets in neural tissues. J Pediatr 130 (6): 878-84, 1997.[PUBMED Abstract]
- Bell J, Moran C, Blatt J: Response to rituximab in a child with neuroblastoma and opsoclonus-myoclonus. Pediatr Blood Cancer 50 (2): 370-1, 2008.[PUBMED Abstract]
- Corapcioglu F, Mutlu H, Kara B, et al.: Response to rituximab and prednisolone for opsoclonus-myoclonus-ataxia syndrome in a child with ganglioneuroblastoma. Pediatr Hematol Oncol 25 (8): 756-61, 2008.[PUBMED Abstract]
- Pranzatelli MR, Tate ED, Travelstead AL, et al.: Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol 28 (9): 585-93, 2006.[PUBMED Abstract]
- Ertle F, Behnisch W, Al Mulla NA, et al.: Treatment of neuroblastoma-related opsoclonus-myoclonus-ataxia syndrome with high-dose dexamethasone pulses. Pediatr Blood Cancer 50 (3): 683-7, 2008.[PUBMED Abstract]
- Pranzatelli MR, Tate ED: Dexamethasone, Intravenous Immunoglobulin, and Rituximab Combination Immunotherapy for Pediatric Opsoclonus-Myoclonus Syndrome. Pediatr Neurol 73: 48-56, 2017.[PUBMED Abstract]
- de Alarcon PA, Matthay KK, London WB, et al.: Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Health 2 (1): 25-34, 2018.[PUBMED Abstract]
- Kumar P, Willard VW, Embry L, et al.: Late cognitive and adaptive outcomes of patients with neuroblastoma-associated opsoclonus-myoclonus-ataxia-syndrome: A report from the Children's Oncology Group. Pediatr Blood Cancer 71 (7): e31039, 2024.[PUBMED Abstract]
- Vik TA, Pfluger T, Kadota R, et al.: (123)I-mIBG scintigraphy in patients with known or suspected neuroblastoma: Results from a prospective multicenter trial. Pediatr Blood Cancer 52 (7): 784-90, 2009.[PUBMED Abstract]
- Yang J, Codreanu I, Servaes S, et al.: I-131 MIBG post-therapy scan is more sensitive than I-123 MIBG pretherapy scan in the evaluation of metastatic neuroblastoma. Nucl Med Commun 33 (11): 1134-7, 2012.[PUBMED Abstract]
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.[PUBMED Abstract]
- Pio L, Brisse HJ, Alaggio R, et al.: Image-guided core-needle or surgical biopsy for neuroblastoma diagnosis in children: A systematic review and meta-analysis from the International Society of Pediatric Surgical Oncology (IPSO). Pediatr Blood Cancer 71 (2): e30789, 2024.[PUBMED Abstract]
- Schoeman S, Bagatell R, Cahill AM, et al.: Percutaneous biopsy for the diagnosis, risk stratification, and molecular profiling of neuroblastoma: A single-center retrospective study. Pediatr Blood Cancer 71 (4): e30887, 2024.[PUBMED Abstract]
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.[PUBMED Abstract]
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.[PUBMED Abstract]
- Jennings RW, LaQuaglia MP, Leong K, et al.: Fetal neuroblastoma: prenatal diagnosis and natural history. J Pediatr Surg 28 (9): 1168-74, 1993.[PUBMED Abstract]
- Brodeur GM: Spontaneous regression of neuroblastoma. Cell Tissue Res 372 (2): 277-286, 2018.[PUBMED Abstract]
- Guan J, Hallberg B, Palmer RH: Chromosome Imbalances in Neuroblastoma-Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers (Basel) 13 (23): , 2021.[PUBMED Abstract]
- Schneiderman J, London WB, Brodeur GM, et al.: Clinical significance of MYCN amplification and ploidy in favorable-stage neuroblastoma: a report from the Children's Oncology Group. J Clin Oncol 26 (6): 913-8, 2008.[PUBMED Abstract]
- Hiyama E, Hiyama K, Yokoyama T, et al.: Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1 (3): 249-55, 1995.[PUBMED Abstract]
- Kitanaka C, Kato K, Ijiri R, et al.: Increased Ras expression and caspase-independent neuroblastoma cell death: possible mechanism of spontaneous neuroblastoma regression. J Natl Cancer Inst 94 (5): 358-68, 2002.[PUBMED Abstract]
- Brodeur GM, Minturn JE, Ho R, et al.: Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res 15 (10): 3244-50, 2009.[PUBMED Abstract]
- Yamamoto K, Ohta S, Ito E, et al.: Marginal decrease in mortality and marked increase in incidence as a result of neuroblastoma screening at 6 months of age: cohort study in seven prefectures in Japan. J Clin Oncol 20 (5): 1209-14, 2002.[PUBMED Abstract]
- Okazaki T, Kohno S, Mimaya J, et al.: Neuroblastoma detected by mass screening: the Tumor Board's role in its treatment. Pediatr Surg Int 20 (1): 27-32, 2004.[PUBMED Abstract]
- Fritsch P, Kerbl R, Lackner H, et al.: "Wait and see" strategy in localized neuroblastoma in infants: an option not only for cases detected by mass screening. Pediatr Blood Cancer 43 (6): 679-82, 2004.[PUBMED Abstract]
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.[PUBMED Abstract]
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.[PUBMED Abstract]
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.[PUBMED Abstract]
- McCarthy LC, Chastain K, Flatt TG, et al.: Neuroblastoma in Adolescents and Children Older than 10 Years: Unusual Clinicopathologic and Biologic Features. J Pediatr Hematol Oncol 41 (8): 586-595, 2019.[PUBMED Abstract]
- Mazzocco K, Defferrari R, Sementa AR, et al.: Genetic abnormalities in adolescents and young adults with neuroblastoma: A report from the Italian Neuroblastoma group. Pediatr Blood Cancer 62 (10): 1725-32, 2015.[PUBMED Abstract]
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.[PUBMED Abstract]
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.[PUBMED Abstract]
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.[PUBMED Abstract]
- Mossé YP, Deyell RJ, Berthold F, et al.: Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer 61 (4): 627-35, 2014.[PUBMED Abstract]
- Kushner BH, Kramer K, LaQuaglia MP, et al.: Neuroblastoma in adolescents and adults: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 41 (6): 508-15, 2003.[PUBMED Abstract]
- Suzuki M, Kushner BH, Kramer K, et al.: Treatment and outcome of adult-onset neuroblastoma. Int J Cancer 143 (5): 1249-1258, 2018.[PUBMED Abstract]
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.[PUBMED Abstract]
- Goldsmith KC, Park JR, Kayser K, et al.: Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med 29 (5): 1092-1102, 2023.[PUBMED Abstract]
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.[PUBMED Abstract]
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.[PUBMED Abstract]
- Campbell K, Gastier-Foster JM, Mann M, et al.: Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children's Oncology Group. Cancer 123 (21): 4224-4235, 2017.[PUBMED Abstract]
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.[PUBMED Abstract]
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.[PUBMED Abstract]
- Kubota M, Suita S, Tajiri T, et al.: Analysis of the prognostic factors relating to better clinical outcome in ganglioneuroblastoma. J Pediatr Surg 35 (1): 92-5, 2000.[PUBMED Abstract]
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.[PUBMED Abstract]
- Isaacs H: Fetal and neonatal neuroblastoma: retrospective review of 271 cases. Fetal Pediatr Pathol 26 (4): 177-84, 2007 Jul-Aug.[PUBMED Abstract]
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.[PUBMED Abstract]
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.[PUBMED Abstract]
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.[PUBMED Abstract]
- Vo KT, Matthay KK, Neuhaus J, et al.: Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol 32 (28): 3169-76, 2014.[PUBMED Abstract]
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.[PUBMED Abstract]
- Hiyama E, Yokoyama T, Hiyama K, et al.: Multifocal neuroblastoma: biologic behavior and surgical aspects. Cancer 88 (8): 1955-63, 2000.[PUBMED Abstract]
- Burchill SA, Lewis IJ, Abrams KR, et al.: Circulating neuroblastoma cells detected by reverse transcriptase polymerase chain reaction for tyrosine hydroxylase mRNA are an independent poor prognostic indicator in stage 4 neuroblastoma in children over 1 year. J Clin Oncol 19 (6): 1795-801, 2001.[PUBMED Abstract]
- Seeger RC, Reynolds CP, Gallego R, et al.: Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 18 (24): 4067-76, 2000.[PUBMED Abstract]
- Bochennek K, Esser R, Lehrnbecher T, et al.: Impact of minimal residual disease detection prior to autologous stem cell transplantation for post-transplant outcome in high risk neuroblastoma. Klin Padiatr 224 (3): 139-42, 2012.[PUBMED Abstract]
- van Zogchel LMJ, Decarolis B, van Wezel EM, et al.: Sensitive liquid biopsy monitoring correlates with outcome in the prospective international GPOH-DCOG high-risk neuroblastoma RT-qPCR validation study. J Exp Clin Cancer Res 43 (1): 331, 2024.[PUBMED Abstract]
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.[PUBMED Abstract]
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.[PUBMED Abstract]
- Streby KA, Parisi MT, Shulkin BL, et al.: Impact of diagnostic and end-of-induction Curie scores with tandem high-dose chemotherapy and autologous transplants for metastatic high-risk neuroblastoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 70 (8): e30418, 2023.[PUBMED Abstract]
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.[PUBMED Abstract]
- George RE, Perez-Atayde AR, Yao X, et al.: Tumor histology during induction therapy in patients with high-risk neuroblastoma. Pediatr Blood Cancer 59 (3): 506-10, 2012.[PUBMED Abstract]
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.[PUBMED Abstract]
- Moroz V, Machin D, Hero B, et al.: The prognostic strength of serum LDH and serum ferritin in children with neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 67 (8): e28359, 2020.[PUBMED Abstract]
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.[PUBMED Abstract]
- Bagatell R, DuBois SG, Naranjo A, et al.: Children's Oncology Group's 2023 blueprint for research: Neuroblastoma. Pediatr Blood Cancer 70 Suppl 6 (Suppl 6): e30572, 2023.[PUBMED Abstract]
- 神経芽腫群腫瘍の組織学的分類
-
神経芽腫は、小児期のsmall round blue cell tumor(SRBCT)の一種に分類されている。神経芽腫は、成熟した神経節腫から成熟度の低い神経節芽腫や未熟な神経芽腫まで、多様な分化度を示す細胞集合体で構成される不均一な腫瘍群である。このような差には、この種の腫瘍の悪性度の多様性が反映されている。[ 1 ]
International Neuroblastoma Pathology Classification(INPC)
INPCシステムは、当初のShimada分類で得られた経験から導き出された分類であり、この2つの分類システムの比較を表1に示す。INPCの判定では、治療開始前に採取した腫瘍検体を以下の形態学的特徴について組織学的に評価する:[ 2 ][ 3 ][ 4 ][ 5 ][ 6 ]
これらの組織学的パラメータと患者の年齢に基づいて、予後良好と予後不良が定義されている。 この分類システムの予後予測上の意義は、同様の基準を採用した他の分類システムのそれと併せて、いくつかの研究で確認されている(表1を参照のこと)。[ 2 ][ 3 ][ 4 ][ 6 ]
表1. International Neuroblastoma Pathology Classification(Shimada分類)に従った神経芽腫群腫瘍の予後評価a International Neuroblastoma Pathology Classification 原著のShimada分類 予後群 MKI = 核分裂-核崩壊指数。 a許可を得て転載。Copyright © 1999 American Cancer Society. All rights reserved.[ 2 ] Hiroyuki Shimada, Inge M. Ambros, Louis P. Dehner, Jun-ichi Hata, Vijay V. Joshi, Borghild Roald, Daniel O. Stram, Robert B. Gerbing, John N. Lukens, Katherine K. Matthay, Robert P. Castleberry, The International Neuroblastoma Pathology Classification (the Shimada System), Cancer, volume 86, issue 2, pages 364–72. b神経芽腫の亜型については別の箇所で詳述している。[ 7 ] cまれな亜型であり、この年齢群で特に多く診断される。さらなる調査と分析が必要である。 dこれらの腫瘍カテゴリーに対する予後分類に年齢との関係は認められない。 神経芽腫: (シュワンストローマ減少型)b ストローマ減少型 予後良好: 予後良好 予後良好 1.5歳未満 低分化または分化かつMKI低値または中等値の腫瘍 1.5~5歳 高分化かつMKI低値の腫瘍 予後不良: 予後不良 予後不良 1.5歳未満 a)未分化腫瘍c b)MKI高値の腫瘍 1.5~5歳 a)未分化または低分化腫瘍 b)MKI中等値または高値の腫瘍 5歳以上 すべての腫瘍 神経節芽腫、混在型 (シュワンストローマ豊富型) ストローマ豊富混在型(予後良好) 予後良好d 神経節腫: (シュワンストローマ優位型) 成熟途中型 高分化(予後良好) 予後良好d 成熟型 神経節腫 神経節芽腫、結節型 (シュワンストローマ豊富型/ストローマ優位型およびストローマ減少型の複合) ストローマ豊富結節型(予後不良) 予後不良d MYCN増幅のある神経芽腫は、その大半がINPCで予後不良とされるが、7%ほどは予後良好と判定される。それらの腫瘍は一般に、たとえ遺伝子増幅があってもMYCNを発現せず、そのような患者はMYCN増幅があってMYCNが過剰発現している患者と比べて予後良好である。[ 8 ]
INRG Data Commons(18,865例)のINPCデータの構成要素が個別に解析され、診断時年齢、組織学的カテゴリー、MKI、および分化度について、独立した予後予測能の妥当性が確認された。 組織学的に判定可能な4つの予後分類群が同定された(生後18カ月未満でMKI低値 vs MKI高値、生後18カ月以上で高分化 vs 未分化/低分化)。また、この解析では、年齢とINPCの交絡が排除されたリスクスキーマを用いることで、MYCN増幅がなく、MKIが中等度または高度で二倍体の1期または2期腫瘍を有する生後547日以上の患者が新たな予後不良の患者群として同定され、そのイベントフリー生存率(EFS)は46%と非常に不良であった。[ 9 ][エビデンスレベルC1]
一部の症例では、生検所見に実際の神経芽腫群腫瘍の型が十分に反映されていない場合がある。例えば、生検で神経節腫または神経節芽腫と診断された患者125人を対象とした研究では、混在型に対して外科的切除が行われた。39%の症例で病理診断が変更され、そのうち14例(12%)では神経芽腫または結節性神経節芽腫に変更された。[ 10 ]
参考文献- Joshi VV, Silverman JF: Pathology of neuroblastic tumors. Semin Diagn Pathol 11 (2): 107-17, 1994.[PUBMED Abstract]
- Shimada H, Ambros IM, Dehner LP, et al.: The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86 (2): 364-72, 1999.[PUBMED Abstract]
- Shimada H, Umehara S, Monobe Y, et al.: International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (9): 2451-61, 2001.[PUBMED Abstract]
- Goto S, Umehara S, Gerbing RB, et al.: Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (10): 2699-708, 2001.[PUBMED Abstract]
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.[PUBMED Abstract]
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.[PUBMED Abstract]
- Shimada H, Ambros IM, Dehner LP, et al.: Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer 86 (2): 349-63, 1999.[PUBMED Abstract]
- Suganuma R, Wang LL, Sano H, et al.: Peripheral neuroblastic tumors with genotype-phenotype discordance: a report from the Children's Oncology Group and the International Neuroblastoma Pathology Committee. Pediatr Blood Cancer 60 (3): 363-70, 2013.[PUBMED Abstract]
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.[PUBMED Abstract]
- Burnand KM, Neville J, Budzanowski A, et al.: Management of Ganglioneuroma and Ganglioneuroblastoma Intermixed: A United Kingdom Children's Cancer and Leukaemia Group (UK CCLG) Nationwide Study Report. Pediatr Blood Cancer 72 (2): e31445, 2025.[PUBMED Abstract]
- 神経芽腫群腫瘍の病期分類およびリスク分類システム
-
International Neuroblastoma Staging System(INSS)
INSSは1986年にChildren's Oncology Group(COG)により策定・採用され、1993年に欧州と日本の共同研究グループにも採用された。INSSは術後病期分類システムであり、局所領域進展の腫瘍がINSSの1期、2A期、2B期、3期のいずれであるかを判定するために、正中線上の組織、リンパ節の状態、および重要なこととして最初の治療としての外科的切除の範囲の観点で分類した腫瘍の位置を用いる。[ 1 ][ 2 ]このシステムは、病期分類およびリスク層別化の世界的な調和の第一段階となった。 神経芽腫の生物学的および遺伝学的性質に関する理解がさらに進んだ結果、COG研究のリスク群と治療割り付けを容易にするため、INSS病期に加えて臨床的および生物学的因子を組み込んだリスク分類システムが開発された。[ 1 ][ 2 ][ 3 ][ 4 ]COGがINSSを最後に使用した研究は中間リスク群患者を対象としたANBL0531(NCT00499616)研究で、この研究は2014年に終了した。
International Neuroblastoma Risk Group Staging System(INRGSS)
外科的切除の範囲とは独立した病期分類システムを構築するべく、画像所見に基づく危険因子(image-defined risk factor:IDRF)を用いて局所領域腫瘍をL1期(IDRFなし)、L2期(IDRFあり)、M期(遠隔転移あり)、M(INSS 4S期に相当)のいずれかに分類するINRGSSが2005年に開発された。 例えば、脊髄圧迫がある症例では、横断面で脊柱管の3分の1を超えて浸潤を認める場合、クモ膜下腔が描出不可能な場合、または脊髄の磁気共鳴信号強度が異常である場合に、IDRFが1つあるとみなされる。 INRGSSに関する詳しい情報については、表2とIDRFの一覧(原著のIDRFおよびCOG IDRF)を参照のこと。
多くの研究において、IDRFの存在に術中合併症の増加、不完全な腫瘍切除、および生存率の低下との関連が報告されている。[ 5 ][ 6 ][ 7 ]2014年以来、COGとInternational Society of Paediatric Oncology Europe Neuroblastoma(SIOPEN)の臨床試験では、INSSの代わりに、International Neuroblastoma Risk Group(INRG)分類(表2を参照)のために特別に開発された術前病期分類であるINRGSSが用いられている。
表2. International Neuroblastoma Risk Group Staging Systema 病期 記述 IDRF = 画像所見に基づく危険因子;INSS = International Neuroblastoma Staging System。 aMonclair et al.[ 5 ]から改変; [ 6 ] L1 IDRFの一覧aで定義された重要組織への進展が認められない限局性腫瘍で、1つの身体区画内に限局している。 L2 IDRFが認められる局所領域腫瘍。a M 遠隔転移あり(MS期を除く)。 MS 生後18カ月未満の小児で遠隔転移がみられるが、転移巣が皮膚、肝臓、骨髄に限定されている。原発巣はINSS 1期、2期、3期のいずれかである。 原著で定義されたIDRFとしては以下のものがある:[ 5 ][ 7 ]
COGのIDRFでは、解剖学的な位置決定アプローチが用いられており、具体的には以下のものがある:[ 6 ][ 8 ];[ 7 ][エビデンスレベルC1]
外科的切除可能性の評価にIDRFを含めるべきである。 IDRFが多いほど、手術の合併症発生率が高く、完全切除の可能性が低くなる。IDRFが2つ以上みられる場合は、外科的切除ではなく化学療法で治療を開始することの議論を診断時から進めるべきである。合併症の発生率を抑えるため、浸潤性の腫瘍に対して外科的切除の先行を避けることが極めて重要である。ある国際研究の解析では、診断時または手術前に特定のIDRFがみられる患者では、原発腫瘍の90%を超える切除を達成できる可能性が低くなる可能性があることが示された。[ 9 ]中間リスク群を対象としたCOG試験では、施設の試験責任医師と中央評価者によるIDRF評価結果の一致度が評価されたが、一致症例の割合はわずか51.9%であった。[ 10 ]
術前補助化学療法はIDRFの排除に必ずしも有効であるとは限らない。2001年から2006年にかけて実施されたEuropean Unresectable Neuroblastoma試験の後ろ向き研究では、MYCN増幅のない1歳以上のINSS 3期神経芽腫患者143人のデータが検討された。切除不能とみなされる外科的危険因子がすべての患者に認められた。中央判定されたあるサブセットでは、53%の患者がInternational Neuroblastoma Pathology Classificationで予後不良の組織学的特徴ありと判定された。診断時点で、228のIDRFが確認された。[ 8 ];[ 11 ][エビデンスレベルC1]
病期分類システムであるINRGSSは、INRG分類に採用された予後マーカーの一つである。[ 12 ]詳しい情報については、表4を参照のこと。
INRGSSでは病期を4つ(L1、L2、M、MS)に分類する。 限局性の腫瘍は、20あるIDRFのうち1つでも認めるか否かという基準に基づき、L1期またはL2期に分類される。[ 5 ]
INRG Task Forceはまた、診断時と治療後の両方で骨髄中の神経芽腫細胞を発見して定量化するためのコンセンサスを得た手法を報告している。骨髄転移病変の定量化は、治療効果判定の精度改善につながる可能性があり[ 13 ]、現行の効果判定基準であるInternational Neuroblastoma Response Criteriaに組み込まれている。[ 14 ]
MS期に4S期とは異なる定義を採用するというINRG Task Forceの決定は、L2期の原発巣と4Sパターンの転移巣を有する少数の乳児(生後12~18カ月を含む)では治療成績が良好との報告に基づくものであった。[ 5 ][ 15 ]INRGのデータを解析したその後の研究では、診断時に生後12~18カ月であったMS期患者では、いくつかの生物学的特徴から予後不良が予測されたことが示された。しかしながら、生物学的に予後良好とされた生後12~18カ月のMS期神経芽腫患者の長期成績は、生後12カ月未満のINSS 4S期神経芽腫の患者と同程度であった。[ 15 ]
INRGSS、年齢、および生物学的因子を組み合わせることにより、個々の患者をリスクに基づく適切な治療アプローチの指針となる予後予測に有用なINRGリスク群に割り当てる。 INRGSSの妥当性は、MYCN増幅を認めないINSS病期判定済みの限局性神経芽腫を対象とした以下の後ろ向き研究において検討された:
国際的なプロトコルの大半が、リスク層別化と治療法の割り付けに用いられるINRG病期の定義に、IDRFの収集と使用を組み込んでいる。[ 17 ][ 18 ]COGは2006年からINRGSSのデータを収集して評価している。2014年に開始されたCOG試験では、外科医からの情報とともにINRGSSを用いて、L1期、L2期、およびMS期患者を含めた高リスク群以外の患者集団に対する治療法が決定された(ANBL1232[NCT02176967]、募集は締め切られた)。なお、INSSでは生後12カ月未満の患者しか4S期に分類できないのに対し、INRGSSでは生後18カ月未満の患者までMS期に分類できるという点に注意すること。INSS 4S期の原発巣はINSS 1期または2期でなければならないが、MS期の原発巣はL1またはL2(INSS 1期、2期、3期を含む)であればよい。INRGSSは進行中のCOG研究で使用されており、切除範囲には依存せず、年齢および生物学的変数と組み合わせた治療前の画像所見に依存する。標準化された命名体系を用いることが、より統一的な病期分類に大きく寄与し、世界のさまざまな地域で実施された臨床試験の比較を容易にすると期待される。
Children's Oncology Group(COG)Neuroblastoma Risk Grouping
COG ANBL00B1(NCT00904241)研究のデータは、2000年から2023年までリスク分類および臨床試験の適格判定に用いられた臨床的および生物学的予後マーカーの情報を迅速かつ信頼性高く入手するための基盤となった。現在では、リスク群分類を促進するためにAPEC14B1試験のデータが用いられている。COGリスク分類に関する詳しい情報については、表3を参照のこと。
COGは、2007年から2017年にかけてANBL00B1研究に登録された患者4,832人のデータに基づいて、リスク分類を更新した。[ 19 ]臨床的および生物学的因子に基づき、患者を低リスク群、中間リスク群、高リスク群に分類すると定義されている(表3を参照)。
表3. Children's Oncology Group Committeeが用いているリスク分類 高リスク群 1.M期、生後18カ月以上、他の特徴は問わない 2.M期、生後18カ月未満、MYCN増幅あり 3.MS期またはL2期、MYCN増幅あり 4.L2期、予後不良の組織学的特徴あり、生後18カ月以上 5.M期またはMS期、生後12~18カ月以上、予後不良の特徴を少なくとも1つ認める: —予後不良の組織学的特徴 —分節性染色体異常 —二倍体腫瘍 6.L1期、不完全切除、MYCN増幅あり 低リスク群 1.L1期、MYCN増幅なし、他の特徴は問わない 2.L1期、完全切除、MYCN増幅なし 3.MS期、生後12カ月未満、予後良好の特徴をすべて認める: —無症状 —予後良好の組織型 —分節性染色体異常を有さない —高二倍体腫瘍 中間リスク群 高リスク群および低リスク群の定義を満たさない他のすべての患者a a完全な分類については、Irwin MS et al.を参照のこと。[ 19 ] International Neuroblastoma Risk Group(INRG)
患者のリスク層別化と治療割り付けのための評価には、予後因子(臨床的および生物学的特徴)の組合せが何十年にもわたり用いられてきた。[ 12 ]スキーマは国際共同グループ間で異なる。INRGタスクフォースは、以下に概説するような、病期分類および治療前リスク分類のための統一されたアプローチを開発する努力を主導してきた。[ 20 ]これらの因子を用いてリスクを判定するアルゴリズムは複雑であり、新たな知識に応じてわずかに変化する。 INRG Classification Systemは、さまざまな臨床試験から集めた8,800人以上の神経芽腫群腫瘍患者を対象とした35の予後因子に関するsurvival-tree analysisの結果に基づいて設計された。この解析では、以下に示すINPC(Shimada分類)の基礎的な組織学的特徴も対象に含められた;[ 20 ][ 21 ]
INRGの分類スキームでは神経芽腫群腫瘍患者を、INRG病期、年齢、組織学的カテゴリー、腫瘍の分化度、MYCN増幅、11qの異常(検討された唯一の分節性染色体異常)、および倍数性に基づいて、16の治療前リスク群のいずれかに分類する。臨床試験(表4を参照)に登録されていたことから質の高いデータがあった患者8,800人の転帰に従って、4段階のリスクが定義された。
全体的なリスク分類において、組織学的カテゴリーはL1期およびL2期の全腫瘍についての重要なリスク決定因子であり、分化度は生後18カ月以上の患児における神経芽腫および結節型神経節芽腫の予後因子である。INRGの目標は、国際的な協力関係を強化し、世界中で実施された臨床試験の結果を相互比較できるよう患者を一様に分類することである。[ 20 ]
表4. International Neuroblastoma Risk Group(INRG)の治療前分類スキームa INRG病期 組織学的カテゴリー 腫瘍分化度 11qの異常 倍数性 治療前リスク群 GN = 神経節腫;GNB = 神経節芽腫;NA = 増幅なし。 a許可を得て転載。 © (2015) American Society of Clinical Oncology. All rights reserved. Pinto N et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma, J Clin Oncol 33 (27), 2015: 3008–3017.[ 12 ] L1/L2 GN成熟途中型、GNB混在型 A(超低リスク) L1 GN成熟途中型とGNB混在型を除くすべて NA B(超低リスク) 増幅あり K(高リスク) L2 生後18カ月未満 GN成熟途中型とGNB混在型を除くすべて NA なし D(低リスク) あり G(中間リスク) 生後18カ月以上 GNB、結節型神経芽腫 高分化 NA なし E(低リスク) あり H(中間リスク) 低分化または未分化 NA H(中間リスク) 増幅あり N(高リスク) M 生後18カ月未満 NA 高二倍体 F(低リスク) 生後12カ月未満 NA 二倍体 I(中間リスク) 生後12カ月~18カ月未満 NA 二倍体 J(中間リスク) 生後18カ月未満 増幅あり O(高リスク) 生後18カ月以上 P(高リスク) MS 生後18カ月未満 NA なし C(超低リスク) あり Q(高リスク) 増幅あり R(高リスク) 患者の年齢はすべてのリスク層別化システムに採用されているため、細胞分類システムとしては年齢を用いないものが望ましく、INRG Classification Systemのための予後判定基準を選択するためのsurvival-tree analysisでは、INPCやShimada分類ではなく、基本的な組織学的基準が用いられた。表5に示すように、2つの患者サブセットにおいて予後は組織学的所見によって最も明確に区別された。
表5. International Neuroblastoma Risk Groupに基づく神経芽腫群腫瘍患者サブセットの組織学的識別a INSS病期/組織学的亜型 症例数 EFS(%) OS(%) EFS = イベントフリー生存率;GN = 神経節腫;GNB = 神経節芽腫;INSS = International Neuroblastoma Staging System;NB = 神経芽腫;OS = 全生存期間。 a出典:Cohn et al.[ 20 ] INSS 1期、2期、3期、4S期 5,131 83 ± 1 91 ± 1 GN、成熟途中型 162 97 ± 2 98 ± 2 GNB、混在型 NB 4,970 83 ± 1 90 ± 1 GNB、結節型 INSS 2期、3期;日齢547以上 260 69 ± 3 81 ± 2 11q正常かつ高分化 16 80 ± 16 100 11q異常または未分化 49 61 ± 11 73 ± 11 INRG Risk Classification Schemaには、INRGに基づく組織学的サブセットが組み込まれている。
原発巣および転移巣の評価
神経芽腫では約70%の患者に診断時から遠隔転移がみられる。治療開始前に転移巣に対する徹底的な評価が行われる。 典型的には以下の検査が行われる:[ 1 ]
CTおよびMRI
メタヨードベンジルグアニジン(MIBG)シンチグラフィ
転移病変の範囲は、軟部組織、骨髄、および皮質骨を含めて、あらゆる部位の病変に適用可能なMIBGシンチグラフィを用いて評価される。 神経芽腫の約90%でMIBGの集積が認められる。MIBGシンチグラフィの感度および特異度は90~99%であり、MIBGの集積は原発部位と転移部位で等しく分布する。[ 23 ]ヨウ素123(123I)の半減期は比較的短いが、放射線量が低く、画像品質が優れており、甲状腺毒性が低く、費用も安価であるため、131Iよりも好ましい。
軟部組織および骨転移を同定するには、123I-MIBGによる撮像が最適である。ある前向き比較研究では、PET-CTよりも優れていることが示された。[ 24 ]132人の神経芽腫小児を対象とした後ろ向きレビューでは、99mテクネチウム(Tc)-メチレンジホスホン酸(99mTc-MDP)による骨シンチグラフィの施行は、123I-MIBGシンチグラフィまたはPETを用いて決定された病期または臨床的管理の変更につながるような新たな転移部位を同定できなかった。骨シンチグラフィは神経芽腫の標準的な病期診断検査としては用いられない。[ 25 ]
診断時に行うベースラインのMIBGシンチグラフィは、疾患の反応をモニタリングし、治療後サーベイランスを実施する上で非常に優れた方法となる。[ 26 ]神経芽腫の初発患者60人を対象とした123I-MIBGとPETの画像ペアの後ろ向き解析により、INSS 1期および2期患者における原発巣の進展範囲の特定においてはPETの方が優れており、残存腫瘤検出の感度も高いことが示された。対照的に、4期患者に対する123I-MIBG画像検査は、骨髄および骨転移の検出に優れていた。[ 27 ]
Curieスコア法およびSIOPENスコア法
腫瘍の進展範囲および予後予測上の価値を評価するための半定量的なスコアリング法が、複数の研究グループによって検討されている。 腫瘍の進展範囲および治療効果の評価に最も一般的に用いられているスコアリング方法は、Curie法とSIOPEN法である。
German Pediatric Oncology Groupは、1歳以上の4期神経芽腫患者58人を対象とした後ろ向き研究において、Curieスコア法とSIOPENスコア法を予後予測上の価値について比較した。 この研究では、診断時および導入化学療法後における(これら2つの方法の)予後予測上の価値に一致が認められた。診断時点では、Curieスコア2点以下とSIOPENスコア4点以下(最良のカットオフ)に、より高いスコアとの比較において、EFS率および全生存(OS)率が有意に改善した。 4サイクルの導入化学療法後にSIOPENおよびCurieスコアにより完全奏効と判定された患者は、転移部位に集積が残存していた患者と比べて、予後良好であった。しかしながら、その後に化学療法の第4~6サイクルにみられたMIBG陽性転移の消失は予後に影響を及ぼさなかった。[ 32 ]
引用された臨床試験には、移植および免疫療法終了後のCurieまたはSIOPENスコアの寛解導入後の評価結果が含まれていなかった。それらの評価に関連したカットオフ値や成績は、寛解導入前および寛解導入後のスコアと異なる可能性がある。
PET
MIBGが集積しない腫瘍を有する患者における腫瘍の進展範囲の評価にはフッ素18-フルデオキシグルコースPETが用いられる。[ 27 ]
骨髄穿刺および骨髄生検
骨髄浸潤を除外するため、両側腸骨稜での骨髄穿刺および骨髄生検(コア生検)によって骨髄の評価を行う。 十分な評価とみなすには、コア生検の検体中に骨髄が少なくとも1cm以上(軟骨を除く)含まれていなければならない。多くのCOG研究では2回のコア生検と2回の穿刺が必須とされている。骨髄検査なしで1期と評価される腫瘍には、骨髄検体の採取は必要ない可能性がある。[ 33 ]
病期診断のためのその他の検査および手技
神経芽腫の病期診断に用いられる他の検査および手技としては以下のものがある:
参考文献- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.[PUBMED Abstract]
- Brodeur GM, Seeger RC, Barrett A, et al.: International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6 (12): 1874-81, 1988.[PUBMED Abstract]
- Castleberry RP, Shuster JJ, Smith EI: The Pediatric Oncology Group experience with the international staging system criteria for neuroblastoma. Member Institutions of the Pediatric Oncology Group. J Clin Oncol 12 (11): 2378-81, 1994.[PUBMED Abstract]
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.[PUBMED Abstract]
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.[PUBMED Abstract]
- Brisse HJ, McCarville MB, Granata C, et al.: Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology 261 (1): 243-57, 2011.[PUBMED Abstract]
- Newman EA, Nuchtern JG: Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg 25 (5): 257-264, 2016.[PUBMED Abstract]
- Chen AM, Trout AT, Towbin AJ: A review of neuroblastoma image-defined risk factors on magnetic resonance imaging. Pediatr Radiol 48 (9): 1337-1347, 2018.[PUBMED Abstract]
- Espinoza AF, Bagatell R, McHugh K, et al.: A subset of image-defined risk factors predict completeness of resection in children with high-risk neuroblastoma: An international multicenter study. Pediatr Blood Cancer 71 (10): e31218, 2024.[PUBMED Abstract]
- Brown EG, Adkins ES, Mattei P, et al.: Evaluation of Image-Defined Risk Factor (IDRF) Assessment in Patients With Intermediate-risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Pediatr Surg 60 (1): 161896, 2025.[PUBMED Abstract]
- Avanzini S, Pio L, Erminio G, et al.: Image-defined risk factors in unresectable neuroblastoma: SIOPEN study on incidence, chemotherapy-induced variation, and impact on surgical outcomes. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.[PUBMED Abstract]
- Burchill SA, Beiske K, Shimada H, et al.: Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer 123 (7): 1095-1105, 2017.[PUBMED Abstract]
- Park JR, Bagatell R, Cohn SL, et al.: Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 35 (22): 2580-2587, 2017.[PUBMED Abstract]
- Taggart DR, London WB, Schmidt ML, et al.: Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol 29 (33): 4358-64, 2011.[PUBMED Abstract]
- Monclair T, Mosseri V, Cecchetto G, et al.: Influence of image-defined risk factors on the outcome of patients with localised neuroblastoma. A report from the LNESG1 study of the European International Society of Paediatric Oncology Neuroblastoma Group. Pediatr Blood Cancer 62 (9): 1536-42, 2015.[PUBMED Abstract]
- Cecchetto G, Mosseri V, De Bernardi B, et al.: Surgical risk factors in primary surgery for localized neuroblastoma: the LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J Clin Oncol 23 (33): 8483-9, 2005.[PUBMED Abstract]
- Simon T, Hero B, Benz-Bohm G, et al.: Review of image defined risk factors in localized neuroblastoma patients: Results of the GPOH NB97 trial. Pediatr Blood Cancer 50 (5): 965-9, 2008.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.[PUBMED Abstract]
- Okamatsu C, London WB, Naranjo A, et al.: Clinicopathological characteristics of ganglioneuroma and ganglioneuroblastoma: a report from the CCG and COG. Pediatr Blood Cancer 53 (4): 563-9, 2009.[PUBMED Abstract]
- Morin CE, Hasweh R, Anton C, et al.: Gadolinium-based contrast media does not improve the staging of neuroblastoma image-defined risk factors at diagnosis. Pediatr Blood Cancer 71 (1): e30724, 2024.[PUBMED Abstract]
- Howman-Giles R, Shaw PJ, Uren RF, et al.: Neuroblastoma and other neuroendocrine tumors. Semin Nucl Med 37 (4): 286-302, 2007.[PUBMED Abstract]
- Papathanasiou ND, Gaze MN, Sullivan K, et al.: 18F-FDG PET/CT and 123I-metaiodobenzylguanidine imaging in high-risk neuroblastoma: diagnostic comparison and survival analysis. J Nucl Med 52 (4): 519-25, 2011.[PUBMED Abstract]
- Gauguet JM, Pace-Emerson T, Grant FD, et al.: Evaluation of the utility of (99m) Tc-MDP bone scintigraphy versus MIBG scintigraphy and cross-sectional imaging for staging patients with neuroblastoma. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- Kushner BH, Kramer K, Modak S, et al.: Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. J Clin Oncol 27 (7): 1041-6, 2009.[PUBMED Abstract]
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.[PUBMED Abstract]
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.[PUBMED Abstract]
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.[PUBMED Abstract]
- Lewington V, Lambert B, Poetschger U, et al.: 123I-mIBG scintigraphy in neuroblastoma: development of a SIOPEN semi-quantitative reporting ,method by an international panel. Eur J Nucl Med Mol Imaging 44 (2): 234-241, 2017.[PUBMED Abstract]
- Ladenstein R, Lambert B, Pötschger U, et al.: Validation of the mIBG skeletal SIOPEN scoring method in two independent high-risk neuroblastoma populations: the SIOPEN/HR-NBL1 and COG-A3973 trials. Eur J Nucl Med Mol Imaging 45 (2): 292-305, 2018.[PUBMED Abstract]
- Decarolis B, Schneider C, Hero B, et al.: Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol 31 (7): 944-51, 2013.[PUBMED Abstract]
- Russell HV, Golding LA, Suell MN, et al.: The role of bone marrow evaluation in the staging of patients with otherwise localized, low-risk neuroblastoma. Pediatr Blood Cancer 45 (7): 916-9, 2005.[PUBMED Abstract]
- DuBois SG, Kalika Y, Lukens JN, et al.: Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol 21 (3): 181-9, 1999 May-Jun.[PUBMED Abstract]
- Kramer K, Kushner B, Heller G, et al.: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91 (8): 1510-9, 2001.[PUBMED Abstract]
- 神経芽腫のゲノムおよび生物学的特徴
-
神経芽腫の分子生物学的特徴
神経芽腫の小児は、診断時の臨床的因子と生物学的マーカーに基づいて予測される再発リスクが異なるサブセットに分類できる。
高リスク群神経芽腫の生物学的亜型は、以下に挙げる、互いにほとんど重複のないゲノム変化によって定義することができる:
以下で考察するように、ここに挙げた亜型には特異的な臨床的特徴がある。高リスク群神経芽腫の複数の亜型でみられるALKのバリアントは、約15%の症例で観察され、別のセクションで考察している。
高リスク群神経芽腫症例の大半でみられる高リスク群神経芽腫の重要なゲノム的特徴を以下に考察する。
分節性染色体異常
神経芽腫に高頻度で観察され、SCAの状態を判定する際に用いられるSCAとしては、染色体1p、3p、4p、および11qの欠失や、染色体1q、2p、および17qの増幅などがある。[ 8 ]これらの変化は、蛍光in situハイブリダイゼーション(FISH)法、array comparative genomic hybridization(aCGH)法、次世代シークエンシング法(NGS)など、複数の方法で検出することができる。SCAは高リスク群または4期神経芽腫の大半にみられる。[ 3 ][ 4 ][ 6 ][ 7 ][ 9 ]神経芽腫のすべての患者において、染色体切断点の多さ(すなわち、分節性染色体異常の数の多さ)は、以下の特徴と相関していた:[ 3 ][ 4 ][ 5 ][ 6 ][ 7 ][エビデンスレベルC2]
SCAの有無を判定することは、臨床的に有用である可能性がある。SCAの検出は、臨床的に予後良好な臨床像を呈し治療不成功のリスクが高い患者を特定するのに役立つ可能性がある。以下に例を示す。
MYCN増幅のない切除可能な限局性神経芽腫を対象とした解析では、連続で実施された欧州の2研究と北米のコホート(International Neuroblastoma Staging System[INSS]1期、2A期、2B期を含む)からの症例が、選択されたSCA(すなわち1p、3p、4p、11qの欠失と1q、2p、17qの増幅)について解析された。この研究では、患者の年齢(18カ月未満vs 18カ月以上)および病期(1期vs 2期)によって腫瘍ゲノム情報が予後に及ぼす影響に差がみられることが明らかにされた。 腫瘍残存の有無にかかわらず手術単独での治療が行われた。[ 10 ][エビデンスレベルC1]
遠隔転移のない切除不能な原発性の神経芽腫を有する生後12カ月以上の小児を対象とした研究では、大半の患者でSCAが検出された。年長の小児ほど、SCAを有する割合が高く、腫瘍細胞当たりのSCA数が多い割合が高かった。生後12カ月~18カ月の小児では、SCAの存在がEFSに有意な影響を及ぼしたが、OSに対してはそうではなかった。しかしながら、18カ月以上の小児では、腫瘍の組織型に関係なく、SCAがある小児(67%)とSCAがない小児(100%)の間で、OSに有意差が認められた。[ 7 ]
SCAはまた、MYCN遺伝子増幅のない限局性切除不能または転移性神経芽腫の乳児において再発の予測因子となることも明らかにされた。[ 1 ][ 2 ]MYCN増幅がないINSS 3期の患者133人(生後18カ月以上)を対象とした解析では、SCAにEFS不良との関連が認められ、染色体11qの欠失にはOS不良との独立した関連が認められた。[ 11 ]
染色体11qの欠失は高リスク群神経芽腫症例の約30%にみられるが、MYCN増幅のある腫瘍ではまれにしか観察されない。[ 3 ]染色体11qの欠失は、TERT再構成またはALT経路活性化のいずれかを伴う高リスク群神経芽腫症例において高い頻度で観察される。[ 12 ][ 13 ]また染色体11qの欠失には、以下に記載するように、高リスク群神経芽腫患者におけるEFS不良および導入化学療法に対する反応不良との関連も報告されている:
染色体6q末端の欠失にも予後不良との関連が報告されている。ある国際共同研究では、高リスク群の神経芽腫患者556人が対象とされた。 6q末端の欠失は6%の患者に認められ、それらの患者の10年生存率はわずか3.4%であった。[ 16 ]2つ目の研究では、6q末端の欠失を認める高リスク群神経芽腫患者の予後が非常に不良であることが確認された。両研究のプール解析では、染色体6q末端欠失症例のうちMYCN増幅がみられたのはわずか20%であった。[ 17 ]
高リスク群神経芽腫患者556人を対象として6q末端欠失を有する患者の予後不良を同定した同じ研究では、MYCN座を含まない領域の増幅についても評価された。MYCN以外の増幅領域は18%の患者で検出され、該当する患者の10年生存率は5.8%であった。[ 16 ]
MYCN遺伝子増幅
神経芽腫では16~25%の腫瘍でMYCN増幅が検出される。[ 18 ]高リスク群神経芽腫患者では、40~50%の症例でMYCN増幅がみられる。[ 19 ]
病期を問わず、MYCN遺伝子の増幅は、予後因子のほぼすべての多変量回帰分析において(無増悪期間とOSの両方で)予後不良を強く予測する。[ 1 ][ 2 ]2007年から2017年までに登録された初発患者4,832人を対象としたANBL00B1(NCT00904241)研究では、MYCN増幅がない患者(n = 3,647;81%)における5年EFS率およびOS率は、それぞれ77%および87%であった。一方、腫瘍にMYCN増幅がある患者(n = 827;19%)では、5年EFS率およびOS率はそれぞれ51%および57%であった。[ 9 ]
MYCN増幅のある限局例コホートでは、高二倍体腫瘍の患者は二倍体腫瘍の患者より治療成績が良好である。[ 20 ]しかしながら、MYCN増幅または何らかの分節性染色体異常を伴う高二倍体腫瘍の患者は、MYCN増幅のない高二倍体腫瘍の患者と比較して、相対的に予後不良である。[ 3 ]
最も予後不良の臨床的および病理生物学的特徴には、MYCN増幅とある程度の関連が認められる。International Neuroblastoma Risk Group(INRG)研究の患者7,102人を対象とした多変量ロジスティック回帰分析では、プールされたSCAおよび17q増幅量が予後不良因子となり、MYCN増幅との関連がない場合においてもそうであった。しかしながら、別の予後不良因子となった11qのSCAはMYCN増幅とほぼ完全に相互排他的である。[ 21 ][ 22 ]
INRGデータベースから抽出されたMYCN増幅の有無が既知の患者6,223人で構成されるコホートにおいて、MYCN増幅に関連したOSのハザード比(HR)は6.3(95%信頼区間[CI]、5.7-7.0;P < 0.001)であった。 MYCN増幅のOSに対する予後不良の影響が最も大きくみられたのは、年齢が最も低い集団(生後18カ月未満ではHR、19.6;生後18カ月以上ではHR、3.0)であった。 MYCN増幅の有無が転帰に及ぼす影響が最も大きかった患者集団は、生後18カ月未満や核分裂-核崩壊指数高値、フェリチン低値など、他の特徴では予後良好とされた患者であった。[ 23 ][エビデンスレベルC1]
Intratumoral heterogeneous MYCN amplification(hetMNA)とは、MYCN増幅細胞(細胞集塊と単独で散在する細胞の両方)とMYCN非増幅腫瘍細胞が混在する場合のことである。 HetMNAはまれに報告されている。1つの腫瘍内で空間的にみられる場合もあれば、原発巣と転移巣との間で同時にみられる場合や、経過の中で経時的にみられる場合もある。International Society of Paediatric Oncology Europe Neuroblastoma(SIOPEN)の生物学研究グループは、神経芽腫におけるこの亜型の予後予測上の意義を検討した。MYCN増幅クローンが、それ以外ではMYCN増幅のない神経芽腫に及ぼす予後予測上の意義を解明するべく、1991年から2015年までに診断されてhetMNAが同定された患者99人の腫瘍組織が分析された。 生後18カ月未満の患者では、すべての病期に共通して、年長の患者より成績が良好であった。 ゲノム背景は再発頻度および全生存期間と有意に相関していた。 染色体異常が数的異常のみの症例では再発がみられなかった。 この研究の結果から、hetMNAの腫瘍は年齢や病期などの臨床的パターンも加味した腫瘍のゲノム背景との関連で評価すべきであることが示唆される。限局性かつhetMNAの生後18カ月未満の患者を対象とするさらなる研究が必要である。[ 24 ]
テロメア維持を促進するゲノム変化
染色体の先端であるテロメアの伸長は細胞生存を促進する。テロメアは通常、細胞の複製が起こるたびに短縮していき、最終的に細胞は複製能力を失う。腫瘍にテロメア維持機構がない患者の予後は非常に良好であるが、腫瘍にテロメア維持機構がある患者の予後はかなり不良である。[ 25 ]臨床的/生物学的特徴により定義される低リスク群神経芽腫の腫瘍には、テロメア伸長活性がほとんどみられない。高リスク群神経芽腫腫瘍では、テロメア伸長につながる異常な遺伝学的機序が同定されている。[ 25 ][ 26 ][ 27 ][ 28 ]これまでに以下の3つの機序が報告されており、これらは相互排他的とみられる:
FOXR2の活性化
FOXR2遺伝子の発現は神経芽腫症例の約8%に認められる。FOXR2遺伝子の発現は、正常であれば、男性の生殖組織を除き、出生後にはみられない。[ 34 ]FOXR2の発現は、中枢神経系(CNS)未分化神経外胚葉性腫瘍のサブセット(CNS NB-FOXR2と呼ばれる)でも観察される。[ 35 ]FOXR2の過剰発現は、MYCとMYCNの両方の発現が亢進している神経芽腫腫瘍では実質的に相互排他的であった。FOXR2が活性化された神経芽腫ではMYCN遺伝子の発現量は高くなかったが、FOXR2が発現している症例の遺伝子発現プロファイルは、MYCN増幅がある神経芽腫のそれと酷似していた。FOXR2はMYCNに結合し、MYCN蛋白を安定化させるとみられ、FOXR2が活性化された神経芽腫ではMYCN蛋白が高値となる。この知見は、FOXR2の活性化がある神経芽腫とMYCN増幅がある神経芽腫で遺伝子発現プロファイルが類似していることの説明となる。
FOXR2の活性化がある神経芽腫の観察頻度は、高リスク群症例と非高リスク群症例で同等である。[ 34 ]高リスク群症例のうち、腫瘍にFOXR2の活性化がみられた患者の治療成績は、MYCN増幅がみられた患者の成績と同程度であった。多変量解析では、INSS 4期、生後18カ月以上、およびMYCN増幅とともに、FOXR2の活性化にOS不良との有意な関連が認められた。
神経芽腫におけるエクソンバリアント(ALKバリアントおよび増幅を含む)
成人のがんと比較して、小児に発生する神経芽腫では、蛋白配列に影響を与えるゲノム当たりのバリアント数が少ない(ゲノム当たり10~20個)。[ 39 ]バリアントの頻度が最も高い遺伝子はALKであり、約10%の患者で何らかの変化がみられる(下記参照)。バリアントの頻度が比較的低い他の遺伝子としてATRX、PTPN11、ARID1A、ARID1Bなどがある。[ 26 ][ 27 ][ 30 ][ 40 ][ 41 ][ 42 ][ 43 ]図2に示すように、大半の神経芽腫症例では、反復的な変化がみられる遺伝子にバリアントはみられない。
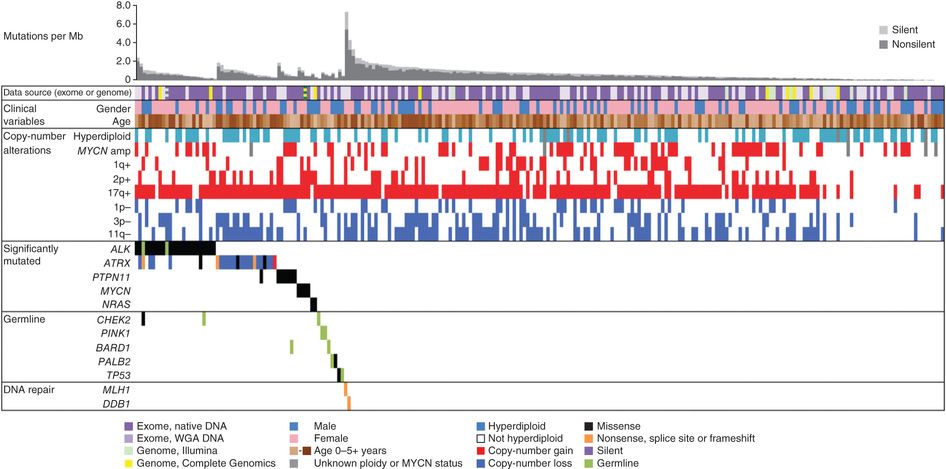
図2. データトラック(行)により神経芽腫症例(列)間の臨床データおよびゲノムデータの比較が容易になる。採用されたデータソースおよびシークエンシング技術は、全ゲノム増幅(WGA)からの全エクソーム配列決定法(WES)(明紫色)、未処理DNAからのWES(濃紫色)、Illumina社のWGS(緑色)、Complete Genomics社のWGS(黄色)であった。縞模様のブロックは2つのアプローチを用いて解析された症例を示している。臨床的変数(clinical variables)としては、性別(男性、青色;女性、ピンク)や年齢(茶色の範囲)が採用された。コピー数変化(copy number alterations)は、フローサイトメトリーで測定した倍数性(高二倍体はDNA指数が1を超えることを意味する)と塩基配列データから得られた臨床的重要なコピー数変化を示している。有意に変異した遺伝子(significantly mutated gene)とは、神経芽腫における背景変異率、遺伝子サイズ、および発現量を踏まえて変異数が統計学的に有意となった遺伝子のことである。生殖細胞系列(germline)は、本コホートでClinVar生殖細胞系列バリアントまたは機能喪失型がん遺伝子バリアントが有意な数認められた遺伝子を示している。DNA修復(DNA repair) は、明らかに変異頻度の高い2つの腫瘍において変異頻度の増加と関連がある可能性のある遺伝子を示している。体細胞変異の予測される影響が凡例に従って色分けされている。Macmillan Publishers Ltdから許諾を得て転載:Nature Genetics (Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma.Nat Genet 45 (3): 279-84, 2013), copyright (2013). ALK遺伝子は、細胞表面受容体型チロシンキナーゼの産生を指示するものであり、発達中の胚および新生児の脳でのみ有意なレベルで発現する。ALKは、神経芽腫でエクソンバリアントが最もよくみられる遺伝子である。ALKにおける病的な生殖細胞系列バリアントは、遺伝性神経芽腫の主要な原因として同定されている。体細胞に生じたALKエクソンの活性化型バリアントも神経芽腫の発がんドライバーであることが明らかになっている。[ 42 ]
2つの大規模コホート研究において、ALK異常の臨床像との相関と予後予測上の意義が検討された。COGによる研究では、診断用に採取されたすべてのリスク群を含む1,596の神経芽腫検体について、ALKの状態が調査された。[ 42 ] SIOPENによる別の研究では、高リスク群の神経芽腫患者1,092人が評価された。[ 44 ]
副腎に発生した原発性神経芽腫(n = 646)のゲノムデータと胸部交感神経節に発生した神経芽腫(n = 118)のゲノムデータを比較した研究では、胸部腫瘍の16%にALKバリアントが認められた。[ 45 ]
ロルラチニブ(従来の治療に追加される)などの低分子薬のALKキナーゼ阻害薬が、ALK変異陽性神経芽腫の再発患者(NCT03107988)およびALK活性化を認める高リスク群神経芽腫の初発患者(COG ANBL1531)を対象として検証されている。[ 42 ]詳しい情報については、「神経芽腫の治療」の高リスク群神経芽腫の治療および再発または難治性神経芽腫の治療に関する各セクションを参照のこと。
エクソンバリアントのゲノム進化
神経芽腫の診断から再発までのエクソンバリアントのゲノム進化に関するデータは限られている。 再発と関連のある体細胞遺伝子変化を特定するため、診断時と再発時の神経芽腫腫瘍ペア検体23組に対して全ゲノム配列決定が行われた一方[ 46 ]、2つ目の研究では、診断時と再発時のペア検体16組が評価された。[ 47 ]どちらの研究でも、再発時の検体の方が診断時の検体と比べて、同定されたバリアントの数が多かった。このことは、NGSに供された神経芽腫の腫瘍検体を用いた研究で確認されている。[ 48 ]
高リスク群および再発神経芽腫は広範に転移する性質があることを考慮すると、循環腫瘍DNA(ctDNA)の技術を用いることで、従来の腫瘍生検では検出されない新たなゲノム変化が明らかになる可能性がある。 さらに、これらのアプローチにより、ALK阻害薬による治療を受けた神経芽腫患者において耐性バリアントを検出できることが示されている。[ 50 ][エビデンスレベルC1]ロルラチニブによる治療を受けた患者から採取した一連のctDNA検体を用いた解析では、すべてではないが大半の患者でALK VAFが疾病負荷とともに追跡された。[ 51 ]ロルラチニブの使用中に進行した患者において、ALKの第2の複合バリアントまたは他の遺伝子(RAS経路の遺伝子とTP53を含む)のバリアントが報告されている。[ 51 ][ 52 ]
ディープシークエンシング研究では、276の神経芽腫検体(すべての病期とすべての診断時年齢をカバーしていた)を対象として、2つのALK増幅ホットスポットに限定して超高深度(33,000X)の配列決定が行われた結果、4.8%でクローン性バリアント、さらに5%でサブクローン性バリアントの存在が明らかにされた。この知見は、ALK遺伝子のサブクローン性変異の頻度が高いことを示唆している。[ 53 ]このように、ディープシークエンシングを行うことで、治療中も生存し、増殖して再発巣の一部を構成する可能性がある神経芽腫細胞の微小集団におけるバリアントの存在を明らかにすることができる。
参考文献- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.[PUBMED Abstract]
- Schleiermacher G, Mosseri V, London WB, et al.: Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer 107 (8): 1418-22, 2012.[PUBMED Abstract]
- Janoueix-Lerosey I, Schleiermacher G, Michels E, et al.: Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27 (7): 1026-33, 2009.[PUBMED Abstract]
- Schleiermacher G, Michon J, Ribeiro A, et al.: Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105 (12): 1940-8, 2011.[PUBMED Abstract]
- Carén H, Kryh H, Nethander M, et al.: High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A 107 (9): 4323-8, 2010.[PUBMED Abstract]
- Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al.: Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28 (19): 3122-30, 2010.[PUBMED Abstract]
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.[PUBMED Abstract]
- Bagatell R, Park JR, Acharya S, et al.: Neuroblastoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 22 (6): 413-433, 2024.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Ambros IM, Tonini GP, Pötschger U, et al.: Age Dependency of the Prognostic Impact of Tumor Genomics in Localized Resectable MYCN-Nonamplified Neuroblastomas. Report From the SIOPEN Biology Group on the LNESG Trials and a COG Validation Group. J Clin Oncol 38 (31): 3685-3697, 2020.[PUBMED Abstract]
- Pinto N, Naranjo A, Ding X, et al.: Impact of Genomic and Clinical Factors on Outcome of Children ≥18 Months of Age with Stage 3 Neuroblastoma with Unfavorable Histology and without MYCN Amplification: A Children's Oncology Group (COG) Report. Clin Cancer Res 29 (8): 1546-1556, 2023.[PUBMED Abstract]
- Djos A, Thombare K, Vaid R, et al.: Telomere Maintenance Mechanisms in a Cohort of High-Risk Neuroblastoma Tumors and Its Relation to Genomic Variants in the TERT and ATRX Genes. Cancers (Basel) 15 (24): , 2023.[PUBMED Abstract]
- Yu Y, Zhang M, Yao X, et al.: Translational practice of fluorescence in situ hybridisation to identify neuroblastic tumours with TERT rearrangements. J Pathol Clin Res 9 (6): 475-487, 2023.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.[PUBMED Abstract]
- Depuydt P, Boeva V, Hocking TD, et al.: Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J Natl Cancer Inst 110 (10): 1084-1093, 2018.[PUBMED Abstract]
- Ognibene M, Morini M, Garaventa A, et al.: Identification of a minimal region of loss on chromosome 6q27 associated with poor survival of high-risk neuroblastoma patients. Cancer Biol Ther 21 (5): 391-399, 2020.[PUBMED Abstract]
- Ambros PF, Ambros IM, Brodeur GM, et al.: International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100 (9): 1471-82, 2009.[PUBMED Abstract]
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.[PUBMED Abstract]
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.[PUBMED Abstract]
- Plantaz D, Vandesompele J, Van Roy N, et al.: Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer 91 (5): 680-6, 2001.[PUBMED Abstract]
- Maris JM, Hogarty MD, Bagatell R, et al.: Neuroblastoma. Lancet 369 (9579): 2106-20, 2007.[PUBMED Abstract]
- Campbell K, Shyr D, Bagatell R, et al.: Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 66 (8): e27819, 2019.[PUBMED Abstract]
- Berbegall AP, Bogen D, Pötschger U, et al.: Heterogeneous MYCN amplification in neuroblastoma: a SIOP Europe Neuroblastoma Study. Br J Cancer 118 (11): 1502-1512, 2018.[PUBMED Abstract]
- Ackermann S, Cartolano M, Hero B, et al.: A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362 (6419): 1165-1170, 2018.[PUBMED Abstract]
- Peifer M, Hertwig F, Roels F, et al.: Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526 (7575): 700-4, 2015.[PUBMED Abstract]
- Valentijn LJ, Koster J, Zwijnenburg DA, et al.: TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47 (12): 1411-4, 2015.[PUBMED Abstract]
- Roderwieser A, Sand F, Walter E, et al.: Telomerase is a prognostic marker of poor outcome and a therapeutic target in neuroblastoma. JCO Precis Oncol 3: 1-20, 2019.[PUBMED Abstract]
- Mac SM, D'Cunha CA, Farnham PJ: Direct recruitment of N-myc to target gene promoters. Mol Carcinog 29 (2): 76-86, 2000.[PUBMED Abstract]
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.[PUBMED Abstract]
- Hartlieb SA, Sieverling L, Nadler-Holly M, et al.: Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun 12 (1): 1269, 2021.[PUBMED Abstract]
- Koneru B, Lopez G, Farooqi A, et al.: Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res 80 (12): 2663-2675, 2020.[PUBMED Abstract]
- Meeser A, Bartenhagen C, Werr L, et al.: Reliable assessment of telomere maintenance mechanisms in neuroblastoma. Cell Biosci 12 (1): 160, 2022.[PUBMED Abstract]
- Schmitt-Hoffner F, van Rijn S, Toprak UH, et al.: FOXR2 Stabilizes MYCN Protein and Identifies Non-MYCN-Amplified Neuroblastoma Patients With Unfavorable Outcome. J Clin Oncol 39 (29): 3217-3228, 2021.[PUBMED Abstract]
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.[PUBMED Abstract]
- Amoroso L, Ognibene M, Morini M, et al.: Genomic coamplification of CDK4/MDM2/FRS2 is associated with very poor prognosis and atypical clinical features in neuroblastoma patients. Genes Chromosomes Cancer 59 (5): 277-285, 2020.[PUBMED Abstract]
- Martinez-Monleon A, Kryh Öberg H, Gaarder J, et al.: Amplification of CDK4 and MDM2: a detailed study of a high-risk neuroblastoma subgroup. Sci Rep 12 (1): 12420, 2022.[PUBMED Abstract]
- Gundem G, Levine MF, Roberts SS, et al.: Clonal evolution during metastatic spread in high-risk neuroblastoma. Nat Genet 55 (6): 1022-1033, 2023.[PUBMED Abstract]
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.[PUBMED Abstract]
- Molenaar JJ, Koster J, Zwijnenburg DA, et al.: Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483 (7391): 589-93, 2012.[PUBMED Abstract]
- Sausen M, Leary RJ, Jones S, et al.: Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45 (1): 12-7, 2013.[PUBMED Abstract]
- Bresler SC, Weiser DA, Huwe PJ, et al.: ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26 (5): 682-94, 2014.[PUBMED Abstract]
- Janoueix-Lerosey I, Lequin D, Brugières L, et al.: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455 (7215): 967-70, 2008.[PUBMED Abstract]
- Bellini A, Pötschger U, Bernard V, et al.: Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 39 (30): 3377-3390, 2021.[PUBMED Abstract]
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.[PUBMED Abstract]
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.[PUBMED Abstract]
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.[PUBMED Abstract]
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.[PUBMED Abstract]
- Rosswog C, Fassunke J, Ernst A, et al.: Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer 128 (8): 1559-1571, 2023.[PUBMED Abstract]
- Bosse KR, Giudice AM, Lane MV, et al.: Serial Profiling of Circulating Tumor DNA Identifies Dynamic Evolution of Clinically Actionable Genomic Alterations in High-Risk Neuroblastoma. Cancer Discov 12 (12): 2800-2819, 2022.[PUBMED Abstract]
- Berko ER, Witek GM, Matkar S, et al.: Circulating tumor DNA reveals mechanisms of lorlatinib resistance in patients with relapsed/refractory ALK-driven neuroblastoma. Nat Commun 14 (1): 2601, 2023.[PUBMED Abstract]
- Bobin C, Iddir Y, Butterworth C, et al.: Sequential Analysis of cfDNA Reveals Clonal Evolution in Patients with Neuroblastoma Receiving ALK-Targeted Therapy. Clin Cancer Res 30 (15): 3316-3328, 2024.[PUBMED Abstract]
- Bellini A, Bernard V, Leroy Q, et al.: Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin Cancer Res 21 (21): 4913-21, 2015.[PUBMED Abstract]
- Wang LL, Teshiba R, Ikegaki N, et al.: Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer 113 (1): 57-63, 2015.[PUBMED Abstract]
- がんの小児の治療に関する特別な考慮事項
-
小児および青年期のがんはまれであるが、全体の発生率は1975年以降、緩やかに上昇してきている。[ 1 ] 小児および青年期のがん患者は、小児期および青年期に発生するがんの治療経験を有するがん専門医で構成される集学的チームを擁する医療機関に紹介すべきである。 この集学的チームのアプローチには、至適な生存期間および生活の質を達成するための治療、支持療法、およびリハビリテーションを患児が確実に受けられるようにするべく、以下に該当する小児専門医およびその他の職種の技能が必要とされる:
小児および青年期のがん患者に対する支持療法に関する具体的な情報については、支持療法および緩和ケアに関する要約を参照のこと。
American Academy of Pediatricsによって、小児がん施設向けのガイドラインと小児がん施設ががんの小児および青年期患者の治療において担う役割が概説されている。[ 2 ] それらの施設では、小児および青年期に発生する大半の種類のがんについて臨床試験への参加が可能であり、大半の患者と家族に参加する機会が提示される。 小児および青年期のがんを対象とする臨床試験は一般に、より優れている可能性がある治療法を現行の標準治療と比較するようにデザインされている。 あるがんに対して標準治療が存在しない場合は、他の種類の臨床試験によって新規の治療法が検証される。小児がんに対する根治的治療法の特定において得られた進歩は、その大半が臨床試験を通じて達成されたものである。 現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
参考文献- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.[PUBMED Abstract]
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.[PUBMED Abstract]
- 神経芽腫に対する治療選択肢の概要
-
一般に、神経芽腫の治療は、腫瘍が高リスク群以外(低または中間リスク)と高リスク群のどちらに分類されるかに基づいて決定される。5年生存率は一般に90%以上であるため、高リスク群以外に対する治療の目標は、毒性を最小限に抑えながら根治を得ることである。高リスク群患者の治療成績は、集学的治療法の強度向上に伴い改善が続いてきたものの、依然として至適な水準にはない。
低リスク群、中間リスク群および高リスク群患者に対する治療選択肢を表6に要約する。
表6. 神経芽腫に対する治療選択肢 COGリスク群 治療選択肢 COG = Children's Oncology Group;GM-CSF = 顆粒球マクロファージコロニー刺激因子;HSCT = 造血幹細胞移植 低リスク群神経芽腫 手術とその後の経過観察 場合により生検を行う経過観察 化学療法単独または化学療法と手術の併用(症状がみられるか、手術後に切除不能進行と判定された患者が対象) 放射線療法(緊急治療の場合のみ) 中間リスク群神経芽腫 化学療法単独または化学療法と手術の併用 手術と経過観察(乳児の場合) 放射線療法(進行例に対し必要に応じて) 高リスク群神経芽腫 化学療法、手術、複数回の骨髄破壊的治療とHSCT、放射線療法、ジヌツキシマブとGM-CSF、ならびにイソトレチノインで構成されるレジメン 4S期神経芽腫 支持療法を伴う経過観察(腫瘍の生物学的特徴が良好な無症状の患者が対象) 化学療法(症状のある患者、腫瘍に予後不良の生物学的特徴がみられる患者、および生後3カ月未満の患者が対象) 手術(まれであるが、肝腫大により腎臓またはその他の腹部臓器に障害が起きている患者が対象) 放射線療法(まれであるが、転移による肝腫大に関係した症状がみられる患者が対象) 手術の原則
遠隔転移のない患者では、最初に手術を行うのが標準治療である。この手術の目的は、病期およびリスク群に基づいて以下を達成することである:
COGの報告では、小さな(L1)副腎腫瘤を有する生後6カ月未満の乳児において待機的な経過観察で非常に良好なEFSおよびOSが得られ、大多数の患者で外科的介入を回避することができた。[ 10 ]中間リスク群の神経芽腫を対象とする臨床試験(ANBL0531[NCT00499616])で記載された外科的ガイドラインに従って、4S期神経芽腫の患者では原発巣の切除はルーチンには行われない。ドイツの研究では選択された患者集団に対して生検が行われ、MYCN増幅のないL1期およびL2期の乳児患者を経過観察とし、大半の患者で追加の手術および化学療法が回避されている。[ 11 ]
生後18カ月以上の4期患者において化学療法後に原発腫瘍の肉眼的全切除(gross-total resection)(90%超)に利点があるかどうかについては、依然として議論がある。[ 12 ][ 13 ][ 14 ][ 15 ][ 16 ][ 17 ]3期と4期の神経芽腫患者を比較したメタアナリシスから、すべての年齢を併合した場合、肉眼的全切除(90%超)は3期神経芽腫でのみ亜全切除と比べて利点があることが判明した。[ 18 ]ある小規模研究では、術前補助化学療法の終了後、切除の完全性は残っているIDRFの数に影響を受けていたことが示唆された。[ 19 ]経験豊富な外科医が手技を行った場合、4期神経芽腫において原発腫瘍の90%以上の切除によって局所制御率の向上が得られたが、OSには統計学的に有意な影響が示されなかった。[ 20 ]
IDRFに関する詳しい情報については、International Neuroblastoma Risk Group Staging System(INRGSS)のセクションを参照のこと。
放射線療法の役割
現在の治療パラダイムでは、低リスク群または中間リスク群の神経芽腫患者に対する放射線療法は、腫瘍塊により生命または臓器機能を脅かす症状が生じていて、化学療法で十分な効果が迅速に得られなかった腫瘍塊に対してのみ適用される。 これらの患者で放射線療法が用いられる共通の状況としては、以下が挙げられる:
高リスク群患者では放射線療法が標準治療の一部となっており、通常は大量化学療法と造血幹細胞移植に続いて行われる。詳しい情報については、高リスク群神経芽腫の治療のセクションを参照のこと。
神経芽腫の乳児(総じて高リスク群ではない)における放射線療法の利用を制限することは、Childhood Cancer Survivor Studyの長期追跡データによって支持されている。この研究では、放射線療法を受けた乳児において二次悪性腫瘍および有意な慢性疾患の発生率が高いことが示された。[ 23 ][エビデンスレベルC1]
脊髄圧迫の治療
脊髄圧迫は医学的緊急と考えられる。診断および治療に先立つ比較的短期間に症状がみられる場合は、神経学的回復の可能性が比較的高いため、即時の治療が行われる。回復はまた神経学的異常の重症度(脱力 vs 麻痺)にも依存する。脊髄圧迫の治療に化学療法、放射線療法、手術のいずれを用いても神経学的な成績は同程度になるとみられるが、放射線療法の施行頻度は以前より低くなっている。
低リスク群および中間リスク群神経芽腫を対象とする完了済みのCOG臨床試験では、低リスク群または中間リスク群患者の脊髄圧迫には即時の化学療法が推奨された。[ 22 ][ 24 ][ 25 ]この設定で神経学的成績に対するグルココルチコイドの効果を調査した単一の研究において、この治療に初期症状軽減の改善との関連が認められた。 ただし、グルココルチコイドでは晩期に残存する障害を予防できなかった。[ 25 ]
重度の脊髄圧迫が速やかに改善しない小児や症状の悪化がみられる小児には、脳神経外科的介入が有益となる可能性がある。椎弓切除術は後に脊柱後弯を招く可能性があり、化学療法の必要性を必ずしもなくすわけではない。[ 22 ][ 24 ][ 25 ]骨組織を除去しない手技である骨形成的椎弓切除術は、脊椎変形を軽減すると考えられていた。骨形成的椎弓切除術を受けた患者では脊椎固定術を要する進行性の脊柱変形がみられる頻度が低いとの報告があるが、椎弓形成術で機能的な神経学的異常が改善されるとするエビデンスは得られていない。[ 26 ]
髄腔内進展が認められた神経芽腫生存者における長期的な健康問題の負荷は高度である。 髄腔内進展がある患者の治療および転帰に関する28の研究をまとめた系統的レビューでは、診断時の症状の重症度と治療法に長期的な健康問題の存在との関連が最も強く認められた。 特に、神経学的な運動障害の重症度は神経学的予後の予測能が最も高かった。[ 27 ] 診断時の運動障害の重症度には、追跡終了時の脊柱変形および括約筋機能障害との関連がみられる一方、診断時の括約筋機能障害には括約筋の長期的問題との相関が認められた。[ 28 ] このことは、症状が悪化して神経学的機能が完全に喪失する前からの治療開始を支持している。
症状を引き起こしている硬膜外脊髄圧迫がある乳児34人の症例集積研究では、いったん対麻痺が定着してしまうと、手術と化学療法のどちらでも満足できる結果が得られなかった。 グレード3の運動障害および腸機能障害の発生頻度は、症状持続期間の延長に伴って増加した。 症状のある硬膜外脊髄圧迫を有する乳児の大半で後遺症がみられ、約半数の患者では重度であった。[ 29 ]
COG ANBL0531[NCT00499616]研究で治療を受けた中間リスク群患者の解析には、脊髄内病変を有する患者92人が含まれていた。[ 30 ]そのうち42人(46%)に症状が認められた。症状がみられた患者のうち、運動症状および腸管/膀胱症状はそれぞれ73%および88%で完全に消失した。症状がみられた患者42人中22人に椎弓切除術または椎弓形成術が施行されたが、症状改善との有意な関連は認められなかった。
治療中または治療後のサーベイランス
神経芽腫の再発を検出するための画像検査によるサーベイランスの役割は十分に研究されていないが、大半の患者は治療完了後に定期的に画像検査を受けることになる。再発する患者の多くは無症状であり、再発はサーベイランス評価で発見される。 治療後のサーベイランスでは、リスク層別化や病変部位、生体分子マーカー、累積照射線量などの因子を考慮してもよい。[ 31 ][ 32 ][ 33 ]
ある症例集積研究では、完全奏効または極めて良好な部分奏効が得られた後に再発した高リスク群神経芽腫患者154人が検討された。この研究では、113人(73%)が無症状で再発した一方、症状がみられた患者は41人(27%)のみであったことが明らかにされた。メタヨードベンジルグアニジン(MIBG)シンチグラフィは、無症候性再発を検出する上で最も信頼性の高い検査法であった。[ 32 ]
神経芽腫と診断された患者183人を対象とした別の症例集積研究では、50人で再発または進行がみられた。大半の患者で再発が症状/診察、MIBGシンチグラフィ、尿中カテコールアミン、X線または超音波検査やこれらの組合せによって検出された。[ 33 ]
CTによる断層画像の撮影については、放射線被曝量の大きさと、この方法論で発見される再発の割合の小ささから、議論がある。[ 33 ]
治療効果の評価
反応の評価は個々の患者の管理に極めて重要であるが、同時に臨床試験の結果を比較するためにも必要である。 骨および骨髄への転移傾向を伴う疾患としての複雑さを考慮して、国際的なコンセンサス基準が過去数十年で開発され、洗練されてきた。 このInternational Neuroblastoma Response Criteria(INRC)の最新版を以下に示す。
International Neuroblastoma Response Criteria(INRC)
INRCは治療に対する反応を評価するために用いられる。[ 34 ][ 35 ][ 36 ]改定版INRCにおける全体的な効果判定では、原発巣、軟部組織および骨転移、ならびに骨髄の3つの要素における腫瘍の反応を統合して評価する。原発部位と軟部組織の転移部位は、Response Evaluation Criteria in Solid Tumors(RECIST)とヨウ素123(123I)-MIBGシンチグラフィまたはMIBG集積がない腫瘍の場合はフッ素18-フルデオキシグルコース(18F-FDG)PETを用いて評価する。骨髄様転移の評価では、テクネチウムTc 99m(99 mTc)ジホスホン酸塩による骨シンチグラフィに代わって、123I-MIBGシンチグラフィー(またはMIBG非集積例には18F-FDG PET)が用いられるようになった。骨髄は組織学的に評価され、場合により免疫組織化学検査と細胞診または免疫細胞診が併用される。腫瘍浸潤が5%以下の骨髄は微小病変に分類される。尿中カテコールアミン値は反応評価に含まれない。効果判定は完全奏効、部分奏効、最小奏効、安定、または進行のいずれかに分類される。[ 36 ]
INRCの全体的な効果判定基準は以下の通りである:[ 34 ][ 35 ]
初発時に1期または2期とされた乳児でみられた転移病変の発生については、解釈に注意を要する。このような患者における転移パターンが4S期のパターン(皮膚、肝、および/または骨髄への転移、後者の転移は10%未満)と一致している場合、それらの患者は進行性/転移性(典型的にはプロトコルの治療法の除外基準とされる)に分類されない。代わりに、それらの患者は4S期として管理される。
原発腫瘍の反応を3つの腫瘍径すべてで測定する必要があるのか、あるいは、RECISTの腫瘍効果判定のように、単一の最大径のみで等しく有用であるかについては議論がある。[ 37 ]INRCでは後者が採用されている。
参考文献- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.[PUBMED Abstract]
- Park JR, Kreissman SG, London WB, et al.: Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 322 (8): 746-755, 2019.[PUBMED Abstract]
- Newman EA, Nuchtern JG: Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg 25 (5): 257-264, 2016.[PUBMED Abstract]
- Overman RE, Kartal TT, Cunningham AJ, et al.: Optimization of percutaneous biopsy for diagnosis and pretreatment risk assessment of neuroblastoma. Pediatr Blood Cancer 67 (5): e28153, 2020.[PUBMED Abstract]
- Gabra HO, Irtan S, Cross K, et al.: Minimally invasive surgery for neuroblastic tumours: A SIOPEN multicentre study: Proposal for guidelines. Eur J Surg Oncol 48 (1): 283-291, 2022.[PUBMED Abstract]
- Zenitani M, Yoshida M, Matsumoto S, et al.: Feasibility and safety of laparoscopic tumor resection in children with abdominal neuroblastomas. Pediatr Surg Int 39 (1): 91, 2023.[PUBMED Abstract]
- Chang S, Lin Y, Yang S, et al.: Safety and feasibility of laparoscopic resection of abdominal neuroblastoma without image-defined risk factors: a single-center experience. World J Surg Oncol 21 (1): 113, 2023.[PUBMED Abstract]
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.[PUBMED Abstract]
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.[PUBMED Abstract]
- Adkins ES, Sawin R, Gerbing RB, et al.: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39 (6): 931-6, 2004.[PUBMED Abstract]
- Castel V, Tovar JA, Costa E, et al.: The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37 (11): 1574-8, 2002.[PUBMED Abstract]
- La Quaglia MP, Kushner BH, Su W, et al.: The impact of gross total resection on local control and survival in high-risk neuroblastoma. J Pediatr Surg 39 (3): 412-7; discussion 412-7, 2004.[PUBMED Abstract]
- Simon T, Häberle B, Hero B, et al.: Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31 (6): 752-8, 2013.[PUBMED Abstract]
- Englum BR, Rialon KL, Speicher PJ, et al.: Value of surgical resection in children with high-risk neuroblastoma. Pediatr Blood Cancer 62 (9): 1529-35, 2015.[PUBMED Abstract]
- von Allmen D, Davidoff AM, London WB, et al.: Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol 35 (2): 208-216, 2017.[PUBMED Abstract]
- Mullassery D, Farrelly P, Losty PD: Does aggressive surgical resection improve survival in advanced stage 3 and 4 neuroblastoma? A systematic review and meta-analysis. Pediatr Hematol Oncol 31 (8): 703-16, 2014.[PUBMED Abstract]
- Irtan S, Brisse HJ, Minard-Colin V, et al.: Image-defined risk factor assessment of neurogenic tumors after neoadjuvant chemotherapy is useful for predicting intra-operative risk factors and the completeness of resection. Pediatr Blood Cancer 62 (9): 1543-9, 2015.[PUBMED Abstract]
- Wolden SL, Gollamudi SV, Kushner BH, et al.: Local control with multimodality therapy for stage 4 neuroblastoma. Int J Radiat Oncol Biol Phys 46 (4): 969-74, 2000.[PUBMED Abstract]
- Hsu LL, Evans AE, D'Angio GJ: Hepatomegaly in neuroblastoma stage 4s: criteria for treatment of the vulnerable neonate. Med Pediatr Oncol 27 (6): 521-8, 1996.[PUBMED Abstract]
- Katzenstein HM, Kent PM, London WB, et al.: Treatment and outcome of 83 children with intraspinal neuroblastoma: the Pediatric Oncology Group experience. J Clin Oncol 19 (4): 1047-55, 2001.[PUBMED Abstract]
- Friedman DN, Goodman PJ, Leisenring WM, et al.: Long-Term Morbidity and Mortality Among Survivors of Neuroblastoma Diagnosed During Infancy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (8): 1565-1576, 2023.[PUBMED Abstract]
- De Bernardi B, Pianca C, Pistamiglio P, et al.: Neuroblastoma with symptomatic spinal cord compression at diagnosis: treatment and results with 76 cases. J Clin Oncol 19 (1): 183-90, 2001.[PUBMED Abstract]
- Simon T, Niemann CA, Hero B, et al.: Short- and long-term outcome of patients with symptoms of spinal cord compression by neuroblastoma. Dev Med Child Neurol 54 (4): 347-52, 2012.[PUBMED Abstract]
- McGirt MJ, Chaichana KL, Atiba A, et al.: Incidence of spinal deformity after resection of intramedullary spinal cord tumors in children who underwent laminectomy compared with laminoplasty. J Neurosurg Pediatr 1 (1): 57-62, 2008.[PUBMED Abstract]
- Kraal K, Blom T, van Noesel M, et al.: Treatment and outcome of neuroblastoma with intraspinal extension: A systematic review. Pediatr Blood Cancer 64 (8): , 2017.[PUBMED Abstract]
- Angelini P, Plantaz D, De Bernardi B, et al.: Late sequelae of symptomatic epidural compression in children with localized neuroblastoma. Pediatr Blood Cancer 57 (3): 473-80, 2011.[PUBMED Abstract]
- De Bernardi B, Quaglietta L, Haupt R, et al.: Neuroblastoma with symptomatic epidural compression in the infant: the AIEOP experience. Pediatr Blood Cancer 61 (8): 1369-75, 2014.[PUBMED Abstract]
- Voeller J, Katzenstein HM, Naranjo A, et al.: Outcomes of patients with intermediate-risk neuroblastoma presenting with motor deficits relating to intraspinal tumor extension: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 72 (1): e31407, 2025.[PUBMED Abstract]
- Papathanasiou ND, Gaze MN, Sullivan K, et al.: 18F-FDG PET/CT and 123I-metaiodobenzylguanidine imaging in high-risk neuroblastoma: diagnostic comparison and survival analysis. J Nucl Med 52 (4): 519-25, 2011.[PUBMED Abstract]
- Kushner BH, Kramer K, Modak S, et al.: Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. J Clin Oncol 27 (7): 1041-6, 2009.[PUBMED Abstract]
- Owens C, Li BK, Thomas KE, et al.: Surveillance imaging and radiation exposure in the detection of relapsed neuroblastoma. Pediatr Blood Cancer 63 (10): 1786-93, 2016.[PUBMED Abstract]
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.[PUBMED Abstract]
- Brodeur GM, Seeger RC, Barrett A, et al.: International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6 (12): 1874-81, 1988.[PUBMED Abstract]
- Park JR, Bagatell R, Cohn SL, et al.: Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 35 (22): 2580-2587, 2017.[PUBMED Abstract]
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.[PUBMED Abstract]
- 低リスク群神経芽腫の治療
-
神経芽腫の初発患者のうち約半数は高リスク群ではない(すなわち、低リスク群または中間リスク群である)。[ 1 ]それらの患者の生存率は非常に良好であり、5年生存率は低リスク群患者で95%以上、中間リスク群患者で90~95%であることから、これらの患者に対する治療の目標は、毒性を最小限に抑えつつ治癒を達成することである。
神経芽腫の病期分類システム、リスク分類システム、および効果判定基準の定義は、過去20年間で複雑化してきた。 結果として、高リスク群以外の神経芽腫患者を対象とした過去の臨床試験の公表結果では、現行とは異なる病期分類システム(International Neuroblastoma Staging System)や効果判定基準が用いられていたり、プロトコルごとに異なる効果判定基準が用いられていたりするため、試験結果の比較が困難となっている。
低リスク群神経芽腫
過去のChildren's Oncology Group(COG)臨床試験の成功が寄与して、特定の条件を満たす神経芽腫患者では治療強度が減少し続けている。COGのリスク分類によると、低リスク群患者は一般に早期(International Neuroblastoma Risk Group[INRG]のL1期)であり、腫瘍はMYCN増幅を認めず、高二倍体で、予後良好な組織型(FH)を有する。COGリスク分類に関する詳しい情報については、表3を参照のこと。
早期例に関する詳しい情報については、INSS 4S期およびINRG MS期神経芽腫の治療のセクションを参照のこと。
低リスク群神経芽腫に対する治療選択肢
INRG L1期の低リスク群患者では、経験豊富な外科医による手術が選択すべき治療である。ただし、副腎腫瘤のみを有し、充実性なら最大径3.1 cm未満であるか、25%以上の腫瘤が嚢胞性なら最大径5 cm未満である、生後6カ月未満の患者は例外である。これに該当する患者には、生検なしで経過観察とすることが、推奨されるアプローチである。 生物学的特徴が予後良好なものと確定した場合は、手術後の残存病変は再発の危険因子とはみなされず、化学療法の適応とならない。 いくつかの研究により、生物学的特徴が予後良好で残存病変を有する患者では、イベントフリー生存(EFS)率が90%超、全生存(OS)率が99~100%という、非常に良好な治療成績が得られることが示されている。[ 2 ][ 3 ]
INRG MS期の患者で、症状がなく、腫瘍の生物学的特徴が予後良好の場合は、経過観察が望ましいアプローチである。
神経芽腫と推定される患者の一部には、生検なしで経過観察が行われている。COGはANBL1232(NCT02176967)試験(募集は締め切られた)において、この戦略をさらに検討している。[ 4 ][ 5 ]
低リスク群神経芽腫に対する治療選択肢としては以下のものがある:
- 手術とその後の経過観察(INRGでL1期またはMS期)。
- 場合により生検を行う経過観察。
- 化学療法単独または化学療法と手術の併用(症状がみられるか、手術後に切除不能進行と判定された患者が対象)。
- 放射線療法(緊急治療の場合のみ)。
場合により生検を行う経過観察
小さい副腎腫瘍を呈する周産期の神経芽腫の治療に対して生検なしでの経過観察が用いられている。
あるCOG研究により、スクリーニングまたは偶然に行われた超音波検査により生後6カ月未満の乳児に発見され、神経芽腫が疑われた、選択された小さなINSS 1期または2期の副腎腫瘤は、組織学的な確定診断と外科的介入なしでも安全に経過を観察できる可能性があることが確認された。この手法では、新生児患者での潜在的な手術合併症を回避できる。[ 4 ]介入の必要性を示唆する腫瘍の増殖や進展を検出するために、頻回に経過観察を行う。手術なしでの経過観察を許容する基準拡大の検討を含めた追加研究がCOG ANBL1232(NCT02176967)研究として進行中である(募集は締め切られた)。
根拠(生検なしでの経過観察):
- COG-ANBL00P2研究により、3.1cm未満の充実性副腎腫瘍(または5cm未満の嚢胞性副腎腫瘍)でINSS 1期と判定された生後6カ月未満の患者において、待機的な経過観察が安全であることが報告された。[ 4 ]
2B期または3期とみられ、MYCN増幅を認めず、生物学的に予後良好と判定された神経芽腫を有する生後12カ月未満の無症状の乳児において切除を(診断時またはそれ以降に)試みることの必要性については議論がある。ドイツのある臨床試験では、このような患者の一部が生検または部分切除後に化学療法や放射線療法を行わない経過観察とされた。多くの患者が局所において進行せず、初回切除も追加切除も受けなかった。[ 5 ]COG ANBL1232(NCT02176967)試験(募集は締め切られた)では、生物学的特徴が良好なL2腫瘍を有する生後18カ月未満の乳児に対して、腫瘍生検後に経過観察が行われている。
化学療法単独または化学療法と手術の併用
化学療法単独または化学療法と手術の併用は以下の状況で選択される:
根拠(化学療法の中止について):
-
COG-P9641研究は、コンセンサスから導き出された因子に基づくリスク層別化を検証するために実施された初期のCOG研究の1つである。この第III相非ランダム化試験では、INSS 2A期および2B期の乳児および小児患者915人が、診断および生物学的特徴の検討のために組織を採取し、最大限の原発巣切除を安全に行うことを目的とする初回手術を受けた。化学療法は、症状が出現しているか、そうなるリスクがあるか、診断時に切除された腫瘍が50%未満であるか、手術単独の施行後に切除不能進行と判定された患者のみが対象とされた。[
3
]
- 1期:
- 2A期および2B期:
- 初回手術後に経過観察とされた診断時点で無症状患者および術後に化学療法を受けた患者の治療成績:最初の915人のうち、800人が診断時に無症状で、初回手術後に経過観察とされた。この集団内では、11%の患者が再発または進行を経験した。 すぐに化学療法(サイクル数は中央値4、範囲1~8)を受けた患者115人では、81%の患者で非常に良好な部分奏効以上の反応が得られた。 化学療法後には、10%の患者で再発または進行がみられた。
- MYCN増幅:1期患者を対象としてMYCN増幅の影響が解析された。
臨床評価段階にある治療選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーを務める臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録が進行しているものを検索するには、臨床試験アドバンスト・サーチを使用のこと。この検索では、試験の実施場所、治療の種類、薬剤名、その他の基準で絞り込むことができる。臨床試験に関する一般情報も入手することができる。
中間リスク群神経芽腫
2021年のCOGリスク分類によると、限局したINRG L2期の腫瘍(MYCN増幅はなし)が中間リスク群に該当する状況として、以下のものが挙げられる:[ 1 ]
M期(MYCN増幅なし):[ 1 ]
MS期(MYCN増幅なし):[ 1 ]
MS期の腫瘍を有し、状態が不安定なあまり治療開始前に生検を施行できない乳児には、化学療法を開始するとともに、安全になった時点で生検を施行する。
COGリスク分類に関する詳しい情報については、表3を参照のこと。
4S期およびMS期例に関する詳しい情報については、INSS 4S期およびINRG MS期神経芽腫の治療のセクションを参照のこと。
中間リスク群神経芽腫に対する治療選択肢
中間リスク群神経芽腫に対する治療選択肢としては以下のものがある:
- 化学療法単独または化学療法と手術の併用。
- 手術と経過観察(乳児の場合)。
- 放射線療法(進行例に対して必要に応じて)。
化学療法単独または化学療法と手術の併用
中間リスク群に分類された患者では、2、4または8サイクルの術前補助化学療法を伴う外科的な完全切除が成功を収めている。化学療法のレジメンはカルボプラチン、シクロホスファミド、ドキソルビシン、およびエトポシドで構成される。化学療法レジメンによる長期的影響を最小限に抑えるため、各薬剤の累積投与量は低く抑えられる(ANBL0531[NCT00499616])。 原則として、予後不良の生物学的特徴がある患者は8サイクルの化学療法を受け、予後不良の生物学的特徴がない患者は2または4サイクルの化学療法を受けた。 予後良好の生物学的特徴としては、FH、DI > 1、SCAなしなどがある。
治療効果の判定は、Response Evaluation Criteria in Solid Tumors(RECIST)に従って、1次元で測定される。化学療法を(病期、年齢、および生物学的特徴に基づいて)規定されたサイクル数施行した後、部分奏効を超える効果が得られない場合は、手術と追加の化学療法の効果を比較検討するための集学的な議論を行うべきである。 部分奏効以上を達成した患者はサーベイランスに移行する。 化学療法による腫瘍の縮小が50%未満である場合は、外科的切除を考慮すべきである。 外科的切除を受けられず、追加の化学療法を受ける患者については、2サイクルごとに効果判定を繰り返すべきである。 化学療法で十分に縮小しなかった残存腫瘤を評価するために、組織学的分化を検出するための再度の生検が必要になることがある。[ 7 ]
腹部の神経芽腫が腎臓に進展していると考えられる症例では、化学療法の開始前に腎摘出術は行われていない。[ 8 ]腎摘出術は全例で控えるべきである。
ANBL0531(NCT00499616)研究では、中間リスク群の化学療法を8サイクル受けたが目標とする反応が得られなかった患者に対する追加治療として、シクロホスファミドとトポテカンが使用された。[ 9 ][ 10 ]
初回化学療法が限局性神経芽腫を有するすべての中間リスク群患者を適応とするか否かについては、さらなる研究が必要である。
根拠(化学療法単独または化学療法と手術の併用):
- ANBL0531(NCT00499616)研究の目標は、中間リスク群神経芽腫(MYCN増幅なし、年齢および病期が定義に従う)の患者集団に対する治療を低減することであった。治療期間(2、4または8サイクルの中用量の術前補助化学療法)は、臨床的特徴および腫瘍の生物学的特徴(1p36および11q23のアレル状態を含む)と治療に対する反応に基づくアルゴリズムに従って割り付けられた。研究コホート全体(N = 404)での10年EFS率およびOS率は、それぞれ82.0%および94.7%であった。[ 10 ]いくつかの患者集団では、治療期間が短縮し、治療強度が低減された。この研究では、生物学的特徴が良好の生後12~18カ月の4期患者が追加された。[ 9 ]
- ドイツのある臨床試験では、1期、2期、または3期の腫瘍を有し、組織学的に検証がなされ、MYCN増幅が認められない1歳未満の乳児340人が登録された。腫瘍により臓器機能が脅かされている57人の乳児には、診断時に化学療法が施行された。低リスク群手術を受けた190人では、腫瘍が完全またはほぼ完全に切除された。年齢または臓器障害のために高リスク手術ができず切除不能となった乳児計93人は、化学療法なしの経過観察とされた。[ 5 ]
- 中用量の化学療法が効果的であることが、前向き研究であるInfant Neuroblastoma European Study(EURO-INF-NB-STUDY-1999-99.1)で示されている。切除不能かつ非転移性の神経芽腫でMYCN増幅を認めない乳児患者の約半数が安全に外科的切除を受け、長期の有害な影響が回避された。[ 12 ][エビデンスレベルC1]
- 播種性の神経芽腫でMYCN遺伝子の増幅を認めない乳児を対象とした欧州の2つの前向き研究において、INSS 3期または放射線学的骨転移は認めないが骨シンチグラフィ陽性(大半はMIBGシンチグラフィで同定されたが、少数はテクネチウム99m骨シンチグラフィで同定された)の乳児に対して、生命または臓器機能を脅かす症状がみられない限り、化学療法による治療が開始されなかった。実施された場合、化学療法は短期用量および標準用量の化学療法で投与された。[ 13 ]
- COGによる後ろ向き解析では、転移病変があり生物学的特徴が良好な生後12~18カ月の患者が評価された。既存の試験では、これらの患者は高リスク群のレジメンで治療されていた。[ 14 ]
- International Society of Paediatric Oncology Europe Neuroblastoma(SIOPEN)試験では、MYCN増幅がない2期または3期切除不能神経芽腫の乳児患者とINPCで予後良好とされた生後12~18カ月の小児患者に対する治療が前向きに評価された。[ 15 ][エビデンスレベルC2]
手術と経過観察(乳児の場合)
3期または4期神経芽腫を有する無症状の乳児では欧州の一部の研究で手術と経過観察で良好な成績が示されていることから、こうした乳児全例における化学療法の必要性について議論がある。[ 13 ]
根拠(乳児における手術と経過観察):
- フランスのある研究では、原発腫瘍が正中線を越えて浸潤しているか(原発巣がINSS 3期で転移巣が4S期カテゴリーまで)、単純X線撮影および/またはCTで確認される皮質骨の変化を伴わずに骨シンチグラフィで陽性となった乳児患者が4期に分類された。[ 16 ]
- このフランスの研究を基礎とし、MYCN増幅のない播種性神経芽腫の乳児125人(原発巣INSS 3期または骨シンチグラフィ陽性は41人)を対象として、これらの患児で症状がみられない場合に経過観察での管理が可能かどうかを判断するための前向き研究が、SIOPENによって実施された。 ただし、治療担当医は経過観察戦略に常に従ったわけではなかった。[ 13 ]
- ドイツのある前方視的臨床試験では、1期、2期、または3期腫瘍を有し、組織所見が検証され、MYCN増幅が認められない1歳以下の乳児340人が登録された。切除を受けた190人のうち、3期の神経芽腫であったのは8人であった。 年齢または臓器障害のために高リスク手術でなければ切除不能と判断された乳児計93人が化学療法なしの経過観察とされ、そのうち21人が3期患者であった。3期の41人を含む57人には、脅威となる症状をコントロールするために化学療法が行われた。[ 5 ]
臨床評価段階にある治療選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーを務める臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録が進行しているものを検索するには、臨床試験アドバンスト・サーチを使用のこと。この検索では、試験の実施場所、治療の種類、薬剤名、その他の基準で絞り込むことができる。臨床試験に関する一般情報も入手することができる。
参考文献- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Matthay KK, Perez C, Seeger RC, et al.: Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children's Cancer Group study. J Clin Oncol 16 (4): 1256-64, 1998.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.[PUBMED Abstract]
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.[PUBMED Abstract]
- Iehara T, Hamazaki M, Tajiri T, et al.: Successful treatment of infants with localized neuroblastoma based on their MYCN status. Int J Clin Oncol 18 (3): 389-95, 2013.[PUBMED Abstract]
- Bagatell R, Park JR, Acharya S, et al.: Neuroblastoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 22 (6): 413-433, 2024.[PUBMED Abstract]
- Shamberger RC, Smith EI, Joshi VV, et al.: The risk of nephrectomy during local control in abdominal neuroblastoma. J Pediatr Surg 33 (2): 161-4, 1998.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.[PUBMED Abstract]
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.[PUBMED Abstract]
- Rubie H, De Bernardi B, Gerrard M, et al.: Excellent outcome with reduced treatment in infants with nonmetastatic and unresectable neuroblastoma without MYCN amplification: results of the prospective INES 99.1. J Clin Oncol 29 (4): 449-55, 2011.[PUBMED Abstract]
- De Bernardi B, Gerrard M, Boni L, et al.: Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol 27 (7): 1034-40, 2009.[PUBMED Abstract]
- Bender HG, Irwin MS, Hogarty MD, et al.: Survival of Patients With Neuroblastoma After Assignment to Reduced Therapy Because of the 12- to 18-Month Change in Age Cutoff in Children's Oncology Group Risk Stratification. J Clin Oncol 41 (17): 3149-3159, 2023.[PUBMED Abstract]
- Kohler JA, Rubie H, Castel V, et al.: Treatment of children over the age of one year with unresectable localised neuroblastoma without MYCN amplification: results of the SIOPEN study. Eur J Cancer 49 (17): 3671-9, 2013.[PUBMED Abstract]
- Minard V, Hartmann O, Peyroulet MC, et al.: Adverse outcome of infants with metastatic neuroblastoma, MYCN amplification and/or bone lesions: results of the French society of pediatric oncology. Br J Cancer 83 (8): 973-9, 2000.[PUBMED Abstract]
- Kim C, Choi YB, Lee JW, et al.: Excellent treatment outcomes in children younger than 18 months with stage 4 MYCN nonamplified neuroblastoma. Korean J Pediatr 61 (2): 53-58, 2018.[PUBMED Abstract]
- 高リスク群神経芽腫の治療
-
進行および死亡のリスクが最も高い患者集団は、生後18カ月以上の患者、遠隔転移がある患者、MYCN増幅など予後不良の生物学的特徴を示す限局性の患者、および予後不良の組織学的特徴を有する患者である。 Children's Oncology Group(COG)のリスク分類に関する詳しい情報については、表3を参照のこと。
MS期神経芽腫の乳児の約8~10%は、腫瘍にMYCN増幅が認められ、通常は高リスク群対象のプロトコルで治療される。COG ANBL00B1(NCT00904241)試験に登録された患者5,000人のうち、MYCN増幅がみられるMS期の乳児(n = 23)では、5年イベントフリー生存(EFS)率および全生存(OS)率がそれぞれ60%および64%であった。[ 1 ]
現行の治療を受けた高リスク群神経芽腫患者の5年OS率は、2007年から2017年までに診断された患者で約60%であった。[ 1 ]高リスク群神経芽腫の小児は、積極的治療を受けても後に再発する傾向があり、治療完了後5年以上が経過してから再発することもある。[ 2 ][ 3 ]
International Neuroblastoma Risk Group(INRG)のデータベースを用いた研究により、リンパ節に限られる遠隔転移が認められ(4N期とされた)、腫瘍の生物学的特徴が予後良好かつ治療成績が良好(5年OS率、85%)となる傾向がある患者が146人特定された。この知見から、高リスク群の4期患者で構成されるこの非常にまれな特別なサブグループでは、比較的強度の低い治療を考慮できる可能性が示唆される。[ 4 ]51人の患者を対象とした単一施設研究で同様の比較的良好な成績が確認された。[ 5 ]
高リスク群神経芽腫に対する治療選択肢
高リスク群神経芽腫患者の治療成績は、ランダム化試験で最近報告されている生存率の改善にもかかわらず、依然として不良のままである。
高リスク群神経芽腫に対する治療選択肢としては典型的には以下のものがある:
化学療法、手術、骨髄破壊的治療とHSCTを2サイクル行うタンデム移植、放射線療法、ならびにジヌツキシマブ(IL-2/GM-CSFおよびイソトレチノインと併用)
高リスク群神経芽腫の患者に対する治療は一般に以下の3段階に分けられる:
寛解導入期
最も頻用される導入化学療法の基本骨格には、シスプラチンおよびエトポシドとビンクリスチン、シクロホスファミド、およびドキソルビシンの高用量の交互併用が含まれる。[ 6 ]再発患者でみられた抗神経芽腫活性に基づいて、このレジメンにトポテカンとシクロホスファミドが追加された。[ 7 ]4サイクルの化学療法後または導入化学療法終了時の治療に対する反応は、高リスク群治療完了時のEFSと相関する。[ 8 ][ 9 ][ 10 ]
根拠(導入化学療法単独または導入化学療法と追加治療):
- ある研究では、4期の初発患者42人を対象として、導入化学療法のコースごとにGM-CSFおよび低用量インターロイキン2(IL-2)を併用する抗GD2抗体療法が追加され、有望な成績が得られた。[ 11 ]
- 欧州のランダム化比較試験では、高リスク群神経芽腫の初発患者422人を対象として長期の導入化学療法が前向きに検討された。 患者は6コースの化学療法による標準の導入化学療法を受ける群と、トポテカン、シクロホスファミド、およびエトポシドの追加2コースで治療開始後に標準の導入化学療法を行う実験的な導入化学療法を受ける群のいずれかにランダムに割り付けられた。[ 12 ][エビデンスレベルB3]
- 欧州の研究グループにより、高リスク群神経芽腫患者に対する導入化学療法レジメンを検討する別のランダム化試験が完了した。 計630人の患者がシスプラチン、ビンクリスチン、カルボプラチン、エトポシド、およびシクロホスファミドで構成されるレジメン(rCOJECレジメン;n = 313)またはMemorial Sloan Kettering Cancer Center N5導入化学レジメン(MSKCC-N5;n = 317)のいずれかを受ける群にランダムに割り付けられた。[ 13 ][エビデンスレベルB1]
化学療法で反応がみられた後は、大半の治療プロトコルで原発腫瘍の切除が推奨されている。肉眼的全切除(gross-total resection)が有益かどうかについては議論がある。[ 14 ]
根拠(原発腫瘍の切除の達成度):
- COG A3973(NCT00004188)研究では、導入化学療法後に肉眼的全切除が試みられた患者220人の手術が中央審査された。外科医の推定によって切除の達成度が90%以上または90%未満と判定されたが、画像に基づく中央判定との一致度はわずか63%であったことが明らかにされた。[ 15 ][エビデンスレベルC1]
- 高リスク群神経芽腫の小児87人を対象とした単一施設後ろ向き研究により、near-total resection(90%超)との比較で肉眼的全切除(gross-total resection)に有意な利益が示されなかった。[ 16 ][エビデンスレベルC2]
高リスク群の転移例において診断時または化学療法施行後に腫瘍を完全切除する積極的な外科的アプローチの潜在的有益性については、明確には示されていない。いくつかの研究から、診断時点での原発巣の完全切除による生存率の改善が報告されている。しかしながら、このような患者の治療成績は、外科的切除の達成度よりも腫瘍の生物学的特徴により強く依存する可能性があり、腫瘍の生物学的特徴のみで切除可能性が規定される可能性もある。[ 17 ][ 18 ][ 19 ]
生後18カ月以上の4期患者において化学療法後の原発腫瘍に対する肉眼的全切除に利点があるかどうかについては議論がある。[ 15 ][ 18 ][ 19 ][ 20 ]一部の研究では、不完全切除を受けた患者の経過が完全切除を受けた患者よりも不良であった。[ 21 ]これらの成績については、完全切除不能であった腫瘍の予後不良な生物学的特徴によるものであった可能性もあれば、腫瘍体積の縮小によるものであった可能性もある。[ 22 ][エビデンスレベルB1]腎摘出術を必要とする状況での完全切除は、標準化学療法には腎毒性があり、また完全切除が予後に及ぼす効果が証明されていないことから、推奨されない。
大半のグループによる研究では、原発腫瘍の外科的切除が寛解導入期に行われている。しかしながら、JN-H-11試験では、全身療法の優先順位を決定することを目標として、造血幹細胞移植を伴う大量化学療法の後に遅延切除を行う戦略の実行可能性が評価された。[ 23 ]完全切除または90%を超える切除の達成割合は、導入化学療法中に手術が施行された過去の試験でみられた割合と同程度であった。過去試験での値と比べて、腎摘出術の施行率は数値上低く(5.8% vs 17%)、3年累積局所再発率は数値上高かった(17.3% vs 11.6%)。
高リスク例では、導入化学療法の終了時に典型的にはフルセットの病勢評価が行われる。従来の導入化学療法の終了時に残存病変が認められる患者の管理は標準化されていない。ある後ろ向き研究では、導入化学療法終了時の効果判定が部分奏効以下であった高リスク群患者201例が解析された。患者は大量化学療法(コホート1)、ブリッジング療法(通常は化学免疫療法またはヨウ素131メタヨードベンジルグアニジン[MIBG])からの大量化学療法(コホート2)、または大量化学療法ではない追加治療(コホート3)のいずれかを直ちに受けるように選択された。[ 24 ]
これらの後ろ向きデータは、従来の導入化学療法に対して不完全奏効であった患者におけるブリッジング療法の役割を示唆している。
地固め療法
高リスク群患者を対象とする治療の地固め療法の段階では、骨髄破壊的化学療法とHSCTが行われるが、そこでは(導入化学療法中に収集した)自家造血幹細胞を移植して骨髄に定着させなければ致死的となる骨髄破壊的化学療法によって、微小残存腫瘍(MRD)の根絶を図る。いくつかの大規模ランダム化比較研究により、3年EFS率は従来の化学療法(22~31%)と比べてHSCT(31~47%)で改善することが示された。[ 25 ][ 26 ][ 27 ] 以前は、HSCTの前処置の一部として全身照射が用いられていた。北米の最新のプロトコルでは、シクロホスファミド/チオテパおよびカルボプラチン/エトポシド/メルファランによる化学療法とHSCTを2サイクル行うタンデム移植が用いられている。[ 28 ][エビデンスレベルC1]欧州では、ブスルファン/メルファランとHSCTも臨床試験で評価されている。
根拠(骨髄破壊的化学療法および造血幹細胞移植):
- 地固め療法に関する欧州の大規模多施設試験において、多剤併用レジメンによる導入療法(シスプラチン、カルボプラチン、シクロホスファミド、ビンクリスチン、およびエトポシドと場合によりトポテカン、ビンクリスチン、およびドキソルビシン)を完了して十分な反応が得られた患者が、ブスルファン/メルファランまたはカルボプラチン/エトポシド/メルファランのいずれかを受ける群にランダムに割り付けられた。[ 29 ][エビデンスレベルA1]
- あるランダム化臨床試験(COG-ANBL0532)では、骨髄破壊的化学療法と造血幹細胞移植を併用する治療の有効性が2サイクルと1サイクルの2条件で検証された。[ 30 ][エビデンスレベルA1] 6サイクルの導入化学療法を受けた生後18カ月以上の4期神経芽腫患者が、カルボプラチン/エトポシド/メルファランの投与と自家HSCTを1回ずつ行うシングル移植と、シクロホスファミド/チオテパに続いて低用量でカルボプラチン/エトポシド/メルファランを投与する治療と自家HSCTを複数回ずつ行うタンデム移植にランダムに割り付けられた。 2つ目の試験では、腫瘍床に対する放射線照射後に、大半の患者がイソトレチノイン単独またはイソトレチノインとジヌツキシマブおよび免疫増強の併用を受ける群にランダムに割り付けられた。
- 更新されたCochraneレビューでは、骨髄移植を標準化学療法と比較した3つのランダム化臨床試験が評価された。[ 25 ][ 26 ][ 27 ][ 31 ][ 32 ]
- Center for International Blood and Marrow Transplant Researchに提出された同種移植症例147例のレビューにより、同種移植レシピエントが以前に自家移植を受けていた場合も含めて、自家移植に対する同種移植の優位性は示されなかった。[ 33 ]
別のランダム化試験では、移植前に採取された自家造血幹細胞から神経芽腫細胞をパージしても優位性は示されなかった。[ 34 ]
移植に関する詳しい情報については、小児における自家造血幹細胞移植および小児がんに対する造血幹細胞移植および細胞療法を参照のこと。
骨髄破壊的療法後には(完全切除が得られたかどうかにかかわらず)原発部位に対する放射線照射が適応となる。[ 35 ][ 36 ]; [ 37 ][エビデンスレベルC1]肉眼的残存病変に対する追加照射を前向きに評価したANBL0532(NCT00567567)試験では、局所制御の改善が示されなかった。[ 38 ][エビデンスレベルC1]放射線療法の至適線量は特定されていない。[ 39 ]
根拠(不完全切除に対する追加照射を伴う放射線療法 vs 追加照射を伴わない放射線療法):
- 不完全切除後の局所再発率が高いことを受けて、COG ANBL0532(NCT00567567)試験では、肉眼的残存腫瘍がある患者に対する追加照射の潜在的有益性が前向きに評価され、その結果が追加照射を行わなかった高リスク群神経芽腫を対象とする先行のCOG臨床試験(A3973[NCT00004188])で得られた結果と比較された。ANBL0532試験の患者は全員、導入化学療法後に術前の原発部位に対して21.6Gyの放射線照射を受けた。[ 38 ][エビデンスレベルC1]
HSCTに先行する外科的切除の達成度を問わずに行う広範なリンパ節照射は、局所進行率とOSに関して有益な効果をもたらさなかった。[ 40 ][エビデンスレベルC1]
局所領域再発に関する詳細な後ろ向き多施設レビューでは、48.4%が照射野内での再発で、別の19.4%が辺縁部での再発であったことが示された。[ 41 ]これらの知見は、放射線療法アプローチのさらなる最適化が依然として必要であることを示唆している。
病勢制御を最大化するために、原発腫瘍床に対する放射線照射と同時に行う骨転移巣の治療も考慮される。 転移部位に対する放射線療法は、症例ごとの状況に応じて、または研究に登録された患者に対するプロトコルガイドラインに従って決定される。多くの小児が広範な骨転移がある状態で診断される。最初の骨転移部位すべてに対する照射は現実的でないため、HSCT前のMIBGによる評価で反応がみられない部位を治療対象とするのが一般的である。[ 42 ][ 43 ][ 44 ]診断時に同定された転移部位のうちフロントラインの放射線療法で照射対象とされなかった部位では、照射対象とされた転移部位と比べて、初回再発時に病変が存在するリスクが高いようであった。[ 42 ]ある単一施設研究では、転移部位に対する放射線療法を行わないフロントライン治療の終了時に骨組織へのMIBG集積の残存がみられた患者24人中17人で再発がみられた。17人のうち、13人(76.5%)で過去の骨病変部位で再発がみられた。[ 45 ]
高リスク群のM期神経芽腫の小児159人を対象とした症例集積研究では、化学療法に対する反応にかかわらず、転移巣の数が多すぎない限り、すべての転移部位に対して局所照射が行われた。[ 46 ]
これらの観察結果は、高リスク群患者で導入化学療法後もMIBG集積により持続が認められる転移巣に対する現行の照射パラダイムの裏付けとなる。 骨髄の50%を超える照射は勧められない。[ 46 ]
導入化学療法後にびまん性の骨転移が残存する症例では、大量化学療法を施行した後、地固め放射線療法の施行を決定する前に再び効果判定を行う。
高リスク群神経芽腫の患者に対する陽子線治療の予備的成績が公表され、有効性と毒性が許容可能な水準であることが示されている。[ 47 ]
地固め後療法期
地固め後療法は、HSCT後に残存している可能性があるMRDに対する治療としてデザインされる。[ 31 ]HSCT後の寛解期にある高リスク群患者について、ジヌツキシマブをGM-CSFおよびイソトレチノインと併用する治療でEFSが改善することが示された。[ 48 ][ 49 ]
根拠(すべての治療):
- あるランダム化試験では、大量化学療法で腫瘍細胞を除去した上での自家骨髄移植が3サイクルの集中的な地固め化学療法と比較された。患者はさらに、化学療法または自家骨髄移植の完了後に、治療を止める群とイソトレチノインを6カ月間投与する群にランダムに割り付けられた。以下に示すEFSおよびOSの結果は各ランダム化時点からの成績を反映している。[ 25 ];[ 31 ][エビデンスレベルA1]
- 単一施設の後ろ向き研究において、自家HSCTまたは従来の化学療法のいずれかに続いてGM-CSFの投与と3F8による抗GD2抗体療法を受けた患者が比較された。[ 50 ]初回治療で紹介された患者と追加治療で紹介された患者が混在していて、さらに難治例と再発例も含まれており、その一部は紹介元の医療機関で自家HSCTを受けていた。自家HSCT群では、最初の化学療法または自家HSCTから、GM-CSFの投与と3F8による抗GD2抗体療法を開始するまでの期間が有意に長かった。また自家HSCT群では、超高リスク群患者の割合も有意に高かった。
- あるCOG第III相試験(ANBL0032[NCT00026312])では、HSCTの既往がある患者が、イソトレチノインに加えてGM-CSFおよびIL-2とともにジヌツキシマブを投与する治療とイソトレチノイン単剤による治療のいずれかにランダムに割り付けられた。[ 48 ]
- 欧州のある研究において、大量化学療法と自家HSCTの併用に続く維持療法として、ジヌツキシマブβ(マウス細胞ではなくハムスター細胞で製造されるジヌツキシマブ)単独とジヌツキシマブβ + IL-2の皮下投与が比較された。すべての被験者が追加でイソトレチノインの投与を受けた。[ 56 ]
- 2つ目のSIOPEN試験では、以下のことが報告された:[ 56 ]
- ランダム化第II相試験として実施された3つ目のSIOPEN試験では、IL-2を併用する治療とIL-2を併用しない治療が比較された。[ 57 ]
COGはSIOPENのデータに基づき、標準の地固め後免疫療法からIL-2を除外した。
再発神経芽腫の治療に放射性MIBG療法が用いられており、ある程度の成功を収めている。この治療法については、安全であり、高リスク群神経芽腫の初発患者に対する治療レジメンに組み込むことが可能であることが示されている。[ 58 ]初発の高リスク群神経芽腫に対する複合的治療に放射性MIBG療法を組み込んだランダム化試験(ANBL1531[NCT03126916])が登録完了となっている。
高リスク群神経芽腫の小児を対象とした多施設第II相臨床試験において、経口のオルニチン脱炭酸酵素阻害薬であるエフロルニチン(かつてはジフルオロメチルオルニチン[DFMO]として知られていた)を使用する2年間の継続療法が評価された。[ 59 ]その研究では、ANBL0032(NCT00026312)試験で治療を受けた患者の一部と比較して生存期間が改善されたと結論されたが、過去データとの比較であることと患者選択での潜在的バイアスから、この知見の妥当性は限定的である。最新の報告として、ANBL0032試験でエフロルニチンによる治療を受けた患者とエフロルニチンによる治療を受けなかった患者を比較した傾向スコアマッチング解析の結果が報告されている。[ 60 ]入手可能であった患者特性の差は傾向スコアマッチングによって概ね均衡化された。マッチングを行った解析では、エフロルニチン投与コホートの患者はエフロルニチン非投与コホートの患者と比較してEFS率およびOS率が統計学的に有意に高かった(4年EFS率84%vs 73%、4年OS率96%vs 84%)。著者らは、この非ランダム化比較では交絡因子が制御されていなかった可能性があることを指摘した。これらの結果に基づき、FDAは2023年12月、継続療法としてのエフロルニチンの使用を承認した。
GD2/GD3ガングリオシドワクチンは、標準治療完了後の初回寛解の状態にある患者を対象として研究されている。初回寛解の患者を主な対象としたランダム化試験では、β-グルカンをGD2/GD3ワクチンとともに早期に導入することで、毒性を増強することなく、GD2/GD3抗体価の上昇がみられた。無増悪生存(PFS)率は両治療群で同程度であった。しかしながら、治療群にかかわらず、抗体価が高い患者ではPFS率が良好であった。[ 61 ][エビデンスレベルB1]
臨床評価段階にある治療選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーを務める臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている国および/または医療機関が主導する臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録が進行しているものを検索するには、臨床試験アドバンスト・サーチを使用のこと。この検索では、試験の実施場所、治療の種類、薬剤名、その他の基準で絞り込むことができる。臨床試験に関する一般情報も入手することができる。
参考文献- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Cotterill SJ, Pearson AD, Pritchard J, et al.: Late relapse and prognosis for neuroblastoma patients surviving 5 years or more: a report from the European Neuroblastoma Study Group "Survey". Med Pediatr Oncol 36 (1): 235-8, 2001.[PUBMED Abstract]
- Mertens AC, Yasui Y, Neglia JP, et al.: Late mortality experience in five-year survivors of childhood and adolescent cancer: the Childhood Cancer Survivor Study. J Clin Oncol 19 (13): 3163-72, 2001.[PUBMED Abstract]
- Morgenstern DA, London WB, Stephens D, et al.: Metastatic neuroblastoma confined to distant lymph nodes (stage 4N) predicts outcome in patients with stage 4 disease: A study from the International Neuroblastoma Risk Group Database. J Clin Oncol 32 (12): 1228-35, 2014.[PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Cardenas FI, et al.: Stage 4N neuroblastoma before and during the era of anti-GD2 immunotherapy. Int J Cancer 153 (12): 2019-2031, 2023.[PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Bonilla MA, et al.: Highly effective induction therapy for stage 4 neuroblastoma in children over 1 year of age. J Clin Oncol 12 (12): 2607-13, 1994.[PUBMED Abstract]
- Park JR, Scott JR, Stewart CF, et al.: Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high-risk neuroblastoma: a Children's Oncology Group study. J Clin Oncol 29 (33): 4351-7, 2011.[PUBMED Abstract]
- Decarolis B, Schneider C, Hero B, et al.: Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol 31 (7): 944-51, 2013.[PUBMED Abstract]
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.[PUBMED Abstract]
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.[PUBMED Abstract]
- Furman WL, McCarville B, Shulkin BL, et al.: Improved Outcome in Children With Newly Diagnosed High-Risk Neuroblastoma Treated With Chemoimmunotherapy: Updated Results of a Phase II Study Using hu14.18K322A. J Clin Oncol 40 (4): 335-344, 2022.[PUBMED Abstract]
- Berthold F, Faldum A, Ernst A, et al.: Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: results of the randomized open-label GPOH trial NB2004-HR. Ann Oncol 31 (3): 422-429, 2020.[PUBMED Abstract]
- Garaventa A, Poetschger U, Valteau-Couanet D, et al.: Randomized Trial of Two Induction Therapy Regimens for High-Risk Neuroblastoma: HR-NBL1.5 International Society of Pediatric Oncology European Neuroblastoma Group Study. J Clin Oncol 39 (23): 2552-2563, 2021.[PUBMED Abstract]
- De Ioris MA, Crocoli A, Contoli B, et al.: Local control in metastatic neuroblastoma in children over 1 year of age. BMC Cancer 15: 79, 2015.[PUBMED Abstract]
- von Allmen D, Davidoff AM, London WB, et al.: Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol 35 (2): 208-216, 2017.[PUBMED Abstract]
- Englum BR, Rialon KL, Speicher PJ, et al.: Value of surgical resection in children with high-risk neuroblastoma. Pediatr Blood Cancer 62 (9): 1529-35, 2015.[PUBMED Abstract]
- DeCou JM, Bowman LC, Rao BN, et al.: Infants with metastatic neuroblastoma have improved survival with resection of the primary tumor. J Pediatr Surg 30 (7): 937-40; discussion 940-1, 1995.[PUBMED Abstract]
- Castel V, Tovar JA, Costa E, et al.: The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37 (11): 1574-8, 2002.[PUBMED Abstract]
- Simon T, Häberle B, Hero B, et al.: Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31 (6): 752-8, 2013.[PUBMED Abstract]
- Adkins ES, Sawin R, Gerbing RB, et al.: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39 (6): 931-6, 2004.[PUBMED Abstract]
- Seemann NM, Erker C, Irwin MS, et al.: Survival effect of complete surgical resection of the primary tumor in patients with metastatic, high-risk neuroblastoma in a large Canadian cohort. Pediatr Blood Cancer 70 (6): e30286, 2023.[PUBMED Abstract]
- Holmes K, Pötschger U, Pearson ADJ, et al.: Influence of Surgical Excision on the Survival of Patients With Stage 4 High-Risk Neuroblastoma: A Report From the HR-NBL1/SIOPEN Study. J Clin Oncol 38 (25): 2902-2915, 2020.[PUBMED Abstract]
- Yoneda A, Shichino H, Hishiki T, et al.: A nationwide phase II study of delayed local treatment for children with high-risk neuroblastoma: The Japan Children's Cancer Group Neuroblastoma Committee Trial JN-H-11. Pediatr Blood Cancer 71 (6): e30976, 2024.[PUBMED Abstract]
- Desai AV, Applebaum MA, Karrison TG, et al.: Efficacy of post-induction therapy for high-risk neuroblastoma patients with end-induction residual disease. Cancer 128 (15): 2967-2977, 2022.[PUBMED Abstract]
- Matthay KK, Villablanca JG, Seeger RC, et al.: Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med 341 (16): 1165-73, 1999.[PUBMED Abstract]
- Berthold F, Boos J, Burdach S, et al.: Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol 6 (9): 649-58, 2005.[PUBMED Abstract]
- Pritchard J, Cotterill SJ, Germond SM, et al.: High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatr Blood Cancer 44 (4): 348-57, 2005.[PUBMED Abstract]
- Elborai Y, Hafez H, Moussa EA, et al.: Comparison of toxicity following different conditioning regimens (busulfan/melphalan and carboplatin/etoposide/melphalan) for advanced stage neuroblastoma: Experience of two transplant centers. Pediatr Transplant 20 (2): 284-9, 2016.[PUBMED Abstract]
- Ladenstein R, Pötschger U, Pearson ADJ, et al.: Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol 18 (4): 500-514, 2017.[PUBMED Abstract]
- Park JR, Kreissman SG, London WB, et al.: Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 322 (8): 746-755, 2019.[PUBMED Abstract]
- Matthay KK, Reynolds CP, Seeger RC, et al.: Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J Clin Oncol 27 (7): 1007-13, 2009.[PUBMED Abstract]
- Yalçin B, Kremer LC, Caron HN, et al.: High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst Rev 8: CD006301, 2013.[PUBMED Abstract]
- Hale GA, Arora M, Ahn KW, et al.: Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience. Bone Marrow Transplant 48 (8): 1056-64, 2013.[PUBMED Abstract]
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.[PUBMED Abstract]
- Haas-Kogan DA, Swift PS, Selch M, et al.: Impact of radiotherapy for high-risk neuroblastoma: a Children's Cancer Group study. Int J Radiat Oncol Biol Phys 56 (1): 28-39, 2003.[PUBMED Abstract]
- Casey DL, Kushner BH, Cheung NK, et al.: Local Control With 21-Gy Radiation Therapy for High-Risk Neuroblastoma. Int J Radiat Oncol Biol Phys 96 (2): 393-400, 2016.[PUBMED Abstract]
- Gatcombe HG, Marcus RB, Katzenstein HM, et al.: Excellent local control from radiation therapy for high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 74 (5): 1549-54, 2009.[PUBMED Abstract]
- Liu KX, Naranjo A, Zhang FF, et al.: Prospective Evaluation of Radiation Dose Escalation in Patients With High-Risk Neuroblastoma and Gross Residual Disease After Surgery: A Report From the Children's Oncology Group ANBL0532 Study. J Clin Oncol 38 (24): 2741-2752, 2020.[PUBMED Abstract]
- Casey DL, Kushner BH, Cheung NV, et al.: Dose-escalation is needed for gross disease in high-risk neuroblastoma. Pediatr Blood Cancer 65 (7): e27009, 2018.[PUBMED Abstract]
- Braunstein SE, London WB, Kreissman SG, et al.: Role of the extent of prophylactic regional lymph node radiotherapy on survival in high-risk neuroblastoma: A report from the COG A3973 study. Pediatr Blood Cancer 66 (7): e27736, 2019.[PUBMED Abstract]
- Liu KX, Shaaban SG, Chen JJ, et al.: Patterns of recurrence after radiotherapy for high-risk neuroblastoma: Implications for radiation dose and field. Radiother Oncol 198: 110384, 2024.[PUBMED Abstract]
- Polishchuk AL, Li R, Hill-Kayser C, et al.: Likelihood of bone recurrence in prior sites of metastasis in patients with high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 89 (4): 839-45, 2014.[PUBMED Abstract]
- Li R, Polishchuk A, DuBois S, et al.: Patterns of Relapse in High-Risk Neuroblastoma Patients Treated With and Without Total Body Irradiation. Int J Radiat Oncol Biol Phys 97 (2): 270-277, 2017.[PUBMED Abstract]
- Mazloom A, Louis CU, Nuchtern J, et al.: Radiation therapy to the primary and postinduction chemotherapy MIBG-avid sites in high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 90 (4): 858-62, 2014.[PUBMED Abstract]
- Rossillon L, Edeline V, Agrigoroaie L, et al.: Rationale for irradiation of persisting oligo-skeletal metastases to improve survival of metastatic neuroblastoma patients with a poor response to chemotherapy: A retrospective study. Pediatr Blood Cancer 72 (1): e31350, 2025.[PUBMED Abstract]
- Casey DL, Pitter KL, Kushner BH, et al.: Radiation Therapy to Sites of Metastatic Disease as Part of Consolidation in High-Risk Neuroblastoma: Can Long-term Control Be Achieved? Int J Radiat Oncol Biol Phys 100 (5): 1204-1209, 2018.[PUBMED Abstract]
- Hattangadi JA, Rombi B, Yock TI, et al.: Proton radiotherapy for high-risk pediatric neuroblastoma: early outcomes and dose comparison. Int J Radiat Oncol Biol Phys 83 (3): 1015-22, 2012.[PUBMED Abstract]
- Yu AL, Gilman AL, Ozkaynak MF, et al.: Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363 (14): 1324-34, 2010.[PUBMED Abstract]
- Cheung NK, Cheung IY, Kushner BH, et al.: Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 30 (26): 3264-70, 2012.[PUBMED Abstract]
- Kushner BH, Ostrovnaya I, Cheung IY, et al.: Lack of survival advantage with autologous stem-cell transplantation in high-risk neuroblastoma consolidated by anti-GD2 immunotherapy and isotretinoin. Oncotarget 7 (4): 4155-66, 2016.[PUBMED Abstract]
- Yu AL, Gilman AL, Ozkaynak MF, et al.: Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin Cancer Res 27 (8): 2179-2189, 2021.[PUBMED Abstract]
- Desai AV, Gilman AL, Ozkaynak MF, et al.: Outcomes Following GD2-Directed Postconsolidation Therapy for Neuroblastoma After Cessation of Random Assignment on ANBL0032: A Report From the Children's Oncology Group. J Clin Oncol 40 (35): 4107-4118, 2022.[PUBMED Abstract]
- Forlenza CJ, Boudreau JE, Zheng J, et al.: KIR3DL1 Allelic Polymorphism and HLA-B Epitopes Modulate Response to Anti-GD2 Monoclonal Antibody in Patients With Neuroblastoma. J Clin Oncol 34 (21): 2443-51, 2016.[PUBMED Abstract]
- Venstrom JM, Zheng J, Noor N, et al.: KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin Cancer Res 15 (23): 7330-4, 2009.[PUBMED Abstract]
- Erbe AK, Wang W, Carmichael L, et al.: Neuroblastoma Patients' KIR and KIR-Ligand Genotypes Influence Clinical Outcome for Dinutuximab-based Immunotherapy: A Report from the Children's Oncology Group. Clin Cancer Res 24 (1): 189-196, 2018.[PUBMED Abstract]
- Ladenstein R, Pötschger U, Valteau-Couanet D, et al.: Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 19 (12): 1617-1629, 2018.[PUBMED Abstract]
- Ladenstein RL, Poetschger U, Valteau-Couanet D, et al.: Randomization of dose-reduced subcutaneous interleukin-2 (scIL2) in maintenance immunotherapy (IT) with anti-GD2 antibody dinutuximab beta (DB) long-term infusion (LTI) in front–line high-risk neuroblastoma patients: Early results from the HR-NBL1/SIOPEN trial. [Abstract]J Clin Oncol 37 (suppl 15): A-10013, 2019. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Weiss BD, Yanik G, Naranjo A, et al.: A safety and feasibility trial of 131 I-MIBG in newly diagnosed high-risk neuroblastoma: A Children's Oncology Group study. Pediatr Blood Cancer 68 (10): e29117, 2021.[PUBMED Abstract]
- Sholler GLS, Ferguson W, Bergendahl G, et al.: Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci Rep 8 (1): 14445, 2018.[PUBMED Abstract]
- Oesterheld J, Ferguson W, Kraveka JM, et al.: Eflornithine as Postimmunotherapy Maintenance in High-Risk Neuroblastoma: Externally Controlled, Propensity Score-Matched Survival Outcome Comparisons. J Clin Oncol 42 (1): 90-102, 2024.[PUBMED Abstract]
- Cheung IY, Mauguen A, Modak S, et al.: Effect of Oral β-Glucan on Antibody Response to Ganglioside Vaccine in Patients With High-Risk Neuroblastoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 9 (2): 242-250, 2023.[PUBMED Abstract]
- INSS 4S期およびINRG MS期神経芽腫の治療
-
International Neuroblastoma Staging System(INSS)4S期の患者は、生後12カ月未満であり、原発巣の進展度はINSS 1期または2期である。International Neuroblastoma Risk Group(INRG)の病期分類では、MS期の患者は生後18カ月未満であり、原発巣の進展度には規定がない。どちらの病期分類システムも、限定的な遠隔転移パターンの定義は同じである。
INRG Task Forceによる4S期のカテゴリーを新たなMS期の定義に置き換える決定は、L2期の原発巣と4S期パターンの転移巣を有する少数の乳児(生後12~18カ月の患児を含む)は治療成績が良好であったという報告に基づくものであった。[ 1 ][ 2 ]その後、実際のINRGデータの研究により、生後12~18カ月のMS期患者ではいくつかの生物学的特徴が成績不良の予測因子となったことと、従来の4S期として診断された患児と長期成績が同程度になったのは予後良好の生物学的特徴を有する患児のみであったことが明らかにされた。[ 2 ]
INRG MS期の乳児は、生物学的特徴が予後良好であり、積極的治療をあまり受けないにもかかわらず、成績が比較的良好である。5年無イベント生存率(EFS)は86%、全生存(OS)率は95%であった。MYCN増幅のある患者では、5年EFS率が60%、OS率が65%であった。[ 3 ]
4S/MS期神経芽腫患者の多くは治療を必要としない。しかしながら、予後不良の生物学的特徴を有する腫瘍または進行する肝腫大および臓器障害により症状がみられる患者は死亡リスクが高く、低用量から中用量の化学療法で治療される。 それらの患者の8~10%はMYCN増幅がみられるため、高リスク群患者用のレジメンで治療される。[ 4 ]
4S/MS期神経芽腫に対するChildren's Oncology Group(COG)分類スキーマに関する詳しい情報については、表3を参照のこと。
4S期/MS期神経芽腫に対する治療選択肢
4S/MS期神経芽腫の治療に標準のアプローチは存在しない。
4S/MS期神経芽腫に対する治療選択肢としては以下のものがある:
- 支持療法を伴う経過観察(腫瘍の生物学的特徴が良好な無症状の患者が対象)
- 化学療法(症状のある患者と腫瘍に予後不良な生物学的特徴がある患者が対象)
- 手術(まれであるが、肝腫大により腎臓またはその他の腹部臓器に障害が起きている患者が対象)
- 放射線療法(まれであるが、転移による肝腫大に関係した症状がみられる患者が対象)
原発巣の切除には治療成績改善との関連が認められない。[ 5 ][ 6 ][ 7 ]肝臓に巨大な4S/MS期神経芽腫を有する乳児は、まれに病勢制御を目的とする化学療法および/または放射線療法により肝硬変を発症し、同所性肝移植が有益となる可能性がある。[ 8 ]
支持療法を伴う経過観察
支持療法を伴う経過観察は、腫瘍の生物学的特徴が予後良好である無症状の患者の治療に用いられる。
4S/MS期の小児の治療は臨床像に依存する。[ 5 ][ 6 ] 巨大腫瘤が臓器障害や死亡リスクの原因になっている場合を除き、大半の患者は治療を必要としない。
化学療法
化学療法は、症状のある患者または腫瘍に予後不良な生物学的特徴がある患者に用いられる。生後数週間で急速な腫瘍増大の所見を認めた患者には、不可逆的となる可能性のある腹部コンパートメント症候群と肝および腎不全を回避するため、化学療法による即時介入を必要とする。[ 9 ]
INSS 4S/MS期の神経芽腫と診断された乳児は、特に肝腫大を認める場合または生後2カ月未満で高リスク群の特徴を認める場合、急速な臨床的悪化をみる可能性があり、早期の治療開始が有益となる可能性がある。[ 9 ]4S期で化学療法が有益となる患児を同定することは困難とされてきた。
そこで、この4S期の患者集団に対する評価を改善するべく、増悪や状態悪化の徴候および症状を測定するためのスコアリングシステムが開発された。[ 10 ] このスコアリングシステムが後ろ向きに評価され、臨床経過の予測能が示されたことを受けて、INSS 4S期の神経芽腫患者の管理指針として前向きに適用されている。[ 10 ][ 11 ]このスコアリングシステムは、4S/MS期の乳児に対する化学療法による介入の指針となるべく、上で考察した最も年齢層の低い乳児を対象としたANBL0531(NCT00499616)研究の結果に基づいて修正されている。[ 9 ]
症状のある患者の治療には、さまざまな化学療法レジメン(シクロホスファミド単剤、カルボプラチン/エトポシド、シクロホスファミド/ドキソルビシン/ビンクリスチン)が用いられている。アプローチとしては、生存率の低下に寄与する毒性作用を回避するため、症状が持続している場合に限定して化学療法を施行する。 また非常に幼若または低体重の乳児に対しては、化学療法の各サイクル後の顆粒球コロニー刺激因子の投与とともに比較的低用量で化学療法を施行する治療が、しばしば推奨される。
根拠(4S/MS期例に対する化学療法):
- COG ANBL0531(NCT00499616)試験では、臓器障害がみられるか臓器機能不全が切迫しているか、予後不良な生物学的特徴(予後不良な組織像または二倍体のDNA index[DI])を有する、MYCN増幅のない4S期患者が前向きの評価対象とされた。 49人が登録され、そのうち41人に症状がみられ、28人に予後不良の生物学的特徴が認められた。 患者は腫瘍の生物学的特徴、患者の年齢、および症状に基づいて2、4、または8サイクルの化学療法に割り付けられた。[ 9 ][エビデンスレベルC1]
- COG-P9641(NCT00003119)試験には80人の4S期患者が登録された。無症状の4S期神経芽腫患者41人が手術または生検単独で治療され、39人が手術および化学療法で治療された。[ 12 ]
- またCOG-P9641試験では、生物学的に予後良好な(MYCN増幅を認めない)INSS 4S期の神経芽腫を有する無症状の乳児には、疾患または臨床症状の出現まで化学療法が行われなかった。[ 12 ]
- COG-ANBL0531試験では、症状、年齢、および腫瘍の生物学的特徴に基づいて治療が割り付けられた。[ 9 ]
-
MYCN増幅がない4S期の腫瘍またはINSS 3期の原発腫瘍および/または皮質骨の変化[単純X線撮影および/またはCTで確認]がない骨シンチグラフィ陽性例を有する乳児125人を対象として、前向き研究が実施された。[
11
] 最初の治療の決定には治療前症状スコアが用いられた。症状スコアが低い患児(n = 86)には経過観察が推奨され、症状スコアが高い患児(n = 37)には化学療法が推奨された。
症状スコアが高い患者に対する化学療法としては、カルボプラチンとエトポシドを3日間コースで2~4コース投与するレジメンがあった。症状が持続するか進行がみられた場合は、シクロホスファミド、ドキソルビシン、およびビンクリスチンの5日間投与が最大4コース行われた。 半数の患者が原発腫瘍の完全または部分切除を受けた。
手術
ときに、肝臓が大きくなりすぎて腎臓やその他の腹部臓器に障害が生じている場合には、開腹による減圧が必要になることがあるが[ 13 ][ 14 ]、この状況は典型的には化学療法の適応にもなる。同様に、緊急の外科的腹部減圧によって、呼吸の悪化を回避し、換気を改善することもできる。[ 13 ][ 14 ]
放射線療法
化学療法に反応しなかった症状のあるMS期(4S期)神経芽腫の乳児に著明な肝腫大が認められるまれな症例では、非常に低線量の放射線療法が用いられてきた。症状のあるMS期神経芽腫の乳児41人を対象とした症例集積研究では、5人に放射線療法が行われ、そのうち3人が死亡した。[ 9 ]
臨床評価段階にある治療選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーを務める臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録が進行しているものを検索するには、臨床試験アドバンスト・サーチを使用のこと。この検索では、試験の実施場所、治療の種類、薬剤名、その他の基準で絞り込むことができる。臨床試験に関する一般情報も入手することができる。
参考文献- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.[PUBMED Abstract]
- Taggart DR, London WB, Schmidt ML, et al.: Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol 29 (33): 4358-64, 2011.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Canete A, Gerrard M, Rubie H, et al.: Poor survival for infants with MYCN-amplified metastatic neuroblastoma despite intensified treatment: the International Society of Paediatric Oncology European Neuroblastoma Experience. J Clin Oncol 27 (7): 1014-9, 2009.[PUBMED Abstract]
- Guglielmi M, De Bernardi B, Rizzo A, et al.: Resection of primary tumor at diagnosis in stage IV-S neuroblastoma: does it affect the clinical course? J Clin Oncol 14 (5): 1537-44, 1996.[PUBMED Abstract]
- Katzenstein HM, Bowman LC, Brodeur GM, et al.: Prognostic significance of age, MYCN oncogene amplification, tumor cell ploidy, and histology in 110 infants with stage D(S) neuroblastoma: the pediatric oncology group experience--a pediatric oncology group study. J Clin Oncol 16 (6): 2007-17, 1998.[PUBMED Abstract]
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.[PUBMED Abstract]
- Steele M, Jones NL, Ng V, et al.: Successful liver transplantation in an infant with stage 4S(M) neuroblastoma. Pediatr Blood Cancer 60 (3): 515-7, 2013.[PUBMED Abstract]
- Twist CJ, Naranjo A, Schmidt ML, et al.: Defining Risk Factors for Chemotherapeutic Intervention in Infants With Stage 4S Neuroblastoma: A Report From Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (2): 115-124, 2019.[PUBMED Abstract]
- Hsu LL, Evans AE, D'Angio GJ: Hepatomegaly in neuroblastoma stage 4s: criteria for treatment of the vulnerable neonate. Med Pediatr Oncol 27 (6): 521-8, 1996.[PUBMED Abstract]
- De Bernardi B, Gerrard M, Boni L, et al.: Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol 27 (7): 1034-40, 2009.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Keene DJ, Minford J, Craigie RJ, et al.: Laparostomy closure in stage 4S neuroblastoma. J Pediatr Surg 46 (1): e1-4, 2011.[PUBMED Abstract]
- Harper L, Perel Y, Lavrand F, et al.: Surgical management of neuroblastoma-related hepatomegaly: do material and method really count? Pediatr Hematol Oncol 25 (4): 313-7, 2008.[PUBMED Abstract]
- 再発または難治性神経芽腫の治療
-
成熟による腫瘍の増大は、生検の施行と組織学的検討によって腫瘍の進行と鑑別すべきである。メタヨードベンジルグアニジン(MIBG)の集積を示す成熟中の病変が持続して認められることがあるが、これは治療成績にあまり影響を及ぼさず、低リスクおよび中間リスク群患者では特にその傾向が強い。[ 1 ]ヨウ素131-MIBG(131I-MIBG)による治療を受けた高リスク群の難治性または再発神経芽腫患者14人を対象とした23組のMIBGおよびPET所見の解析から、MIBGシンチグラフィは転移性骨病変の検出においてフッ素18-フルデオキシグルコース(18F-FDG)PETより感度が高いことが明らかにされたが、軟部組織病変に対しては18F-FDG PETの方が感度が高い傾向が認められた。[ 2 ]
サブクローン性のALKバリアントやその他のMAPK経路異常が診断時から存在していて、その後の再発時にクローン性増殖を認めることがある。そのため、進行を続ける腫瘍の検体採取を経時的に行うことで、治療対象となりうるバリアントを同定できる可能性がある。[ 3 ][ 4 ]同じ神経芽腫患者の原発腫瘍と再発腫瘍を比較した包括的な分子生物学的解析により、広範なクローン濃縮といくつかの新規のバリアントが明らかになり、多くの腫瘍でRAS-MAPK経路に新しいバリアントやクローン濃縮されたバリアントが認められた。これは診断時に高リスク群と判定された患者と低リスク群と判定された患者の両方に該当した。[ 5 ][ 6 ]詳しい情報については、神経芽腫のゲノムおよび生物学的特徴のセクションを参照のこと。
NCI-COG Pediatric MATCH試験に登録された小児(n = 59)および若年成人(n = 1)患者から採取された再発および難治性神経芽腫の腫瘍検体に対する塩基配列決定により、60検体中27検体(45%)においてMATCH研究で治療対象と判断されたゲノム変化が検出された。[ 7 ]ALKのホットスポット変異が最も頻度が高く、腫瘍60検体中19検体(31.7%)で報告された。MAPK経路のバリアント(NF1、NRAS)は60検体中4検体(6.7%)で検出され、FGFR1のバリアントは60検体中3検体(5%)で検出された。
初発時に高リスク群と診断された小児において神経芽腫が再発した場合、さらに強力な治療を行っても、通常は予後不良である。[ 8 ][ 9 ][ 10 ][ 11 ]しかしながら、こうした患者で別の化学療法レジメンにより何カ月もの延命が得られる場合もしばしばある。[ 12 ][ 13 ]この状況の患者には臨床試験への参加が適切であり、参加を勧めてもよい。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
再発神経芽腫の患者における予後因子
1989年から2017年までに診断/登録された患者を対象としたInternational Neuroblastoma Risk Group(INRG)のデータベースを用いて、再発パターンの包括的解析が行われた。[ 14 ][エビデンスレベルC1]
INRGデータベースを用いて、INRG Staging System(INRGSS)病期分類のMSパターンを呈した神経芽腫の再発または進行後の生存予後を予測する臨床的および生物学的特徴が検討された。1984年から2021年までに診断されて適格基準を満たした患者1,511人のうち、209人でイベントの発生が確認された。適格基準としては、INRGSSでMS期またはInternational Neuroblastoma Staging System(INSS)で4S期の初回診断時点で生後365日未満である患者と、INSS病期4期で転移が肝臓、皮膚、骨髄に限局している生後365~546日の患者とされた。[ 15 ][エビデンスレベルC1]
International Neuroblastoma Risk Group Projectでは、世界中から厳選された臨床試験群に含まれる大規模臨床試験に登録された神経芽腫患者計2,266人を対象として、再発後の生存期間と関連を示す(診断時に定義される)臨床的および生物学的特徴について、survival-tree analysisが実施された。[ 8 ]このsurvival-tree analysisにより、以下のことが明らかになった:
診断時に規定される予後因子のうち再発後の生存期間を予測する有意な因子としては、以下のものがある:[ 8 ]
Children's Oncology Group(COG)が報告した低リスク群および中間リスク群神経芽腫患者における再発の経験では、大半の患者が救済可能であることが示された。COGの報告では、中間リスク群患者における3年イベントフリー生存(EFS)率は88%、OS率は96%で、低リスク群患者における5年EFS率は89%、OS率は97%であった。[ 16 ][ 17 ]さらに、初発時に低リスク群または中間リスク群と診断された患者の大半では、局所再発または4S期パターンの再発は、骨髄破壊的治療と造血幹細胞移植を併用することなく、経過観察のみ、手術単独、または中用量の化学療法を行う治療戦略が成功する可能性がある。
初発時に高リスク群と判定された神経芽腫の小児における再発後のOSは、一般に極めて不良である。しかしながら、完全寛解または微小残存(MRD)後の初回再発で再発巣が単一部位の軟部組織腫瘤であった患者では(数例では再発時に骨髄または骨病変もみられた)、5年OS率が35%であった。すべての患者が軟部組織病変の外科的切除を受けた。 MYCN増幅および多発性の軟部組織病変には、進行後の生存期間短縮との関連が認められた。[ 18 ]診断時のInternational Neuroblastoma Pathology Classificationが予後不良であるか、MYCN遺伝子の増幅が認められる、局所再発を来した比較的年長の小児は予後不良であり、手術または積極的な多剤併用化学療法で治療されることもあれば、臨床試験への登録を勧めてもよい。
INSSに基づくリスク群別に見た再発神経芽腫に対する治療選択肢を表7に要約する。
表7. 再発神経芽腫に対する治療選択肢 COGリスク群 治療選択肢 COG = Children's Oncology Group;131I-MIBG = ヨウ素131-メタヨードベンジルグアニジン 初発時に低リスク群に分類された患者における局所領域再発 手術とその後の経過観察または化学療法 化学療法と場合によりその後の手術 初発時に低リスク群に分類された患者における転移性再発 経過観察(乳児において転移巣が4S期のパターンで認められる場合) 化学療法 手術とその後の化学療法 初発時に中間リスク群に分類された患者における局所領域再発 手術(完全切除) 手術(不完全切除)とその後の化学療法 放射線療法(化学療法およびセカンドルック手術の後に進行がみられる患者に対してのみ) 初発時に中間リスク群に分類された患者における転移性再発 高リスク群治療 初発時に高リスク群に分類された患者における再発 化学療法と免疫療法の併用 131I-MIBGの単独、他の治療との併用、その後に造血幹細胞移植 ALKバリアントを有する患者に対するALK阻害薬など、新規の治療法 化学療法 免疫療法 中枢神経系での再発 手術および放射線療法 化学療法と手術および放射線療法の併用 新規の治療アプローチ 初発時に低リスク群に分類された患者の再発神経芽腫
局所領域再発
初発時に低リスク群に分類されて局所領域再発を来した神経芽腫に対する治療選択肢としては以下のものがある:
- 手術とその後の経過観察または化学療法
- 化学療法と場合によりその後の手術
局所または領域再発病変は可能であれば切除する。
腫瘍の生物学的特徴が良好で、計画した治療の完了後3カ月以上経過後に領域再発を来した患児は、再発巣を完全ないしほぼ完全(90%以上)に切除されていれば、経過観察とする。腫瘍の生物学的特徴が良好でも、切除の達成度がほぼ全切除(near total resection)に満たない患児は、化学療法で治療する。[ 16 ][ 17 ][ 19 ]
局所領域再発時点で1歳未満の乳児で腫瘍に予後不良の生物学的特徴が認められる場合は、腫瘍が完全またはほぼ完全に切除されていれば、経過観察とする。同様の乳児で切除がほぼ完全(near total)に満たない場合には、化学療法で治療する。 化学療法は中用量のカルボプラチン、シクロホスファミド、ドキソルビシン、およびエトポシドの併用か、シクロホスファミドおよびトポテカンの併用で構成される。 過去のCOG試験(COG-P9641およびCOG-A3961)で採用されていたように、長期的な影響を最小限にとどめるために各薬剤の累積投与量を低く抑える。[ 16 ][ 17 ][ 19 ]
根拠(手術とその後の経過観察または化学療法):
- 1期、2A期、2B期、および4S期神経芽腫の低リスク群患者を対象としたCOG研究では、915人が登録され、そのうち800人が無症状で、手術単独での治療後に経過観察とされた。残りの患者は化学療法を単独または手術との併用で受けた。[ 17 ]
転移再発または標準治療で難治性
初発時に低リスク群に分類されて転移性再発を来した神経芽腫に対する治療選択肢としては以下のものがある:
- 経過観察
- 化学療法(患者の年齢、腫瘍の生物学的特徴、および前治療に基づく;初回診断時と同様の中間リスク群または高リスク群治療を含めてもよい)
- 手術とその後の化学療法
初発時に低リスク群に分類され、再発時の年齢が1歳未満である患者の転移性再発または進行性神経芽腫は、以前のCOG試験(COG-P9641およびCOG-A3961)で定義されていたように、腫瘍の生物学的特徴に応じて以下のように治療される:
- 生物学的特徴がすべて良好で、4S期パターンの転移巣であり、診断後3カ月以内に再発または進行がみられた場合には、症状を指標とした経過観察を行う。
- 遠隔転移を伴う進行または再発が診断後3カ月以上経過してからみられたか、4S期のパターンに該当しない場合には、可能であれば原発腫瘍を切除して、化学療法を施行する。
化学療法は中間的用量のカルボプラチン、シクロホスファミド、ドキソルビシン、およびエトポシドで構成される。 過去のCOG試験(COG-P9641およびCOG-A3961)で採用されていたように、長期的な影響を最小限にとどめるために各薬剤の累積投与量を低く抑える。
初発時には低リスク群に分類され、生後18カ月以上で転移性再発または進行を来した、4S期以外の再発パターンを呈する患者は、通常は予後不良であり、以下のような治療を受ける:
- 高リスク群治療
転移性再発を来した神経芽腫患者は、高リスク群神経芽腫の初発患者と同様に治療される。詳しい情報については、高リスク群神経芽腫に対する治療選択肢のセクションを参照のこと。
初発時に中間リスク群に分類された患者の再発神経芽腫
COG ANBL0531(NCT00499616)研究では、中間リスク群神経芽腫の初発患者に対してカルボプラチン、エトポシド、シクロホスファミド、およびドキソルビシンで構成される化学療法が行われた。 研究登録後3年以内に転移のない進行性疾患を発症した患者に対しては、救済療法(retrieval therapy)がプロトコルに盛り込まれた。 シクロホスファミドおよびトポテカンが最大6サイクルまで投与可能とされた。シクロホスファミドおよびトポテカンの投与を受けた患者29人中、18人がイベントフリー生存を維持し、9人が再発し、2人が死亡した。8サイクルの化学療法に対して当初十分な反応がみられなかった20人の患者には、シクロホスファミドおよびトポテカンによる治療が行われた。 この20人中、9人が非常に良好な部分奏効以上を達成したが、6人は進行または再発となり、1人が死亡した。この結果から、8サイクルの化学療法後に一定の治療エンドポイントを達成できない患者には、より積極的な治療が必要であることが示唆される。[ 19 ]
中間リスク群神経芽腫を対象としたCOG研究(COG-A3961)には、479人の患者が登録され、うち42人で進行がみられた。再発率は生物学的特徴が予後良好な患者で10%、予後不良の生物学的特徴がある患者で17%であった。30人で局所領域再発、11人で転移性再発、1人で両形態の再発がみられた。42人中6人が原病死した一方、36人が治療に反応した。このように、中間リスク群神経芽腫で進行がみられた患者の大半が救済可能である。[ 16 ]これらの結果を他のCOG中間リスク群研究(ANBL0531)の結果と比較することは、2つの研究で適格患者の分類が異なることから、不可能である。[ 19 ]
局所領域再発
初発時に中間リスク群に分類され局所領域再発を来した神経芽腫に対する治療選択肢としては以下のものがある:
- 手術(完全切除)
- 手術(不完全切除)とその後の化学療法
- 放射線療法。放射線療法は、化学療法およびセカンドルック手術の後に進行がみられた患者に限定して考慮される。[ 16 ]
生物学的特徴が予後良好な神経芽腫の局所領域再発のうち、化学療法終了からの経過期間が3カ月以上であるものは、外科的に治療できる可能性がある。 切除がほぼ完全に満たない場合には、追加の化学療法を行うこともできる。 化学療法は、患者が過去に受けた化学療法の内容に基づいて選択すべきである。[ 16 ]
転移性再発
初発時に中間リスク群に分類され転移性再発を来した神経芽腫に対する治療選択肢としては以下のものがある:
- 高リスク群治療
転移性再発を来した神経芽腫患者は、高リスク群神経芽腫の初発患者と同様に治療される。詳しい情報については、高リスク群神経芽腫に対する治療選択肢のセクションを参照のこと。
初発時に高リスク群に分類された患者の再発または難治性神経芽腫
初発時に高リスク群に分類された患者の再発は非常に予後不良である。[ 8 ]臨床試験への参加を考慮してもよい。患者の治療計画の一部として緩和ケアも考慮すべきである。
いくつかの試験を対象とする解析として、最近の初期相のCOG試験で再発または進行がみられた神経芽腫患者383人が組み入れられた。1年無増悪生存(PFS)率は21%、4年PFS率は6%であった。OS率は1年時点で57%、4年時点で20%であった。その後も再発や進行がみられなかった患者は10%に満たない。MYCN増幅はPFSおよびOS率低下の予測因子であった。[ 20 ]初発時から高リスク群神経芽腫の小児における再発後のOSは一般にきわめて不良であるが、ある単一施設研究では、完全寛解またはMRD達成後に初回再発を来し、再発巣が単一部位の軟部組織腫瘤(2~3人では再発時に骨髄または骨病変がみられた)であった高リスク群神経芽腫患者において、35%の5年OS率が得られた。[ 18 ]
初発時に高リスク群に分類された患者の再発または難治性神経芽腫に対する治療選択肢としては以下のものがある:
- 化学療法と免疫療法の併用
- 131I-MIBG。131I-MIBGの単独、他の治療との併用、その後に造血幹細胞移植。
- 新規の治療法
- 化学療法(第I/II相試験)
- 免疫療法。新規の抗GD2薬が再発または難治性神経芽腫患者を対象として評価されている。抗GD2抗体Hu14.18をIL-2と化学的に結合させた薬剤が顆粒球マクロファージコロニー刺激因子(GM-CSF)と併用されており、このレジメンの第II相試験において少数例の持続的反応が報告された。[ 29 ]
化学療法と免疫療法の併用により、進行がみられた高リスク群患者で最良の奏効率および奏効期間が得られる。
根拠(化学療法と免疫療法の併用):
- ANBL1221(NCT01767194)試験は、再発または難治性神経芽腫患者のコホートにおいて抗GD2療法と化学療法の併用を評価した最初の多施設共同試験であった。初回再発または進行の患者がイリノテカン/テモゾロミド/ジヌツキシマブ/GM-CSF(I/T/DIN/GM-CSF)またはテモゾロミド/イリノテカン/テムシロリムスのいずれかにランダムに割り付けられた。[ 21 ]; [ 30 ][エビデンスレベルC3]
- 初回再発時にI/T/DIN/GM-CSFによる化学免疫療法を受けた高リスク群神経芽腫患者146人を対象とした後ろ向きコホート研究では、以下の結果が報告された:[ 31 ][エビデンスレベルC2]
- 化学免疫療法の基本骨格としてのイリノテカン/テモゾロミド以外による化学療法については、利用可能なデータが限られている。あるレジストリー研究では、再発または進行性の高リスク群神経芽腫でトポテカン/シクロホスファミドおよびジヌツキシマブによる治療を受けた患者24人が対象とされた。[ 32 ]
根拠(131I-MIBG単独または他の治療法との併用):
- 再発または難治性神経芽腫の小児に対しては、131I-MIBGが有効な症状緩和薬となっており、臨床研究では単独または化学療法および造血幹細胞移植との併用で考慮されることがある。[ 33 ][ 34 ][ 35 ][ 36 ][ 37 ][ 38 ]; [ 39 ][ 40 ][エビデンスレベルC1]
- 131I-MIBG療法による治療を受けた200人以上の小児患者を対象とした北米の後ろ向き研究では、再発または進行を来した患児が診断以降安定または不変で推移していた患児と比較された。[ 41 ]
- 131I-MIBG、ビンクリスチン、およびイリノテカンの投与と自家造血幹細胞移植(HSCT)に続いてブスルファン/メルファランと自家HSCTを併用するタンデム移植による地固め療法が、患者8人を対象とする後ろ向き研究で報告された。[ 40 ]
- 1回の自家HSCTと131I-MIBGおよびカルボプラチン/エトポシド/メルファランを併用する治療が追加の患者で評価された。治療は導入化学療法の完了後に行われた。[ 42 ]
- ランダム化第II相試験に評価可能な患者105人が組み入れられ、131I-MIBG単独、131I-MIBGとイリノテカンおよびビンクリスチンの併用、または131I-MIBGとボリノスタットの併用のいずれかによる治療が行われた。[ 43 ]
- ある単群第II相試験では、再発または難治性の神経芽腫患者30人が131I-MIBGとトポテカンによる治療を受けた。[ 44 ]
- 131I-MIBGには二次性悪性腫瘍のリスクとの関連が認められる。神経芽腫の5年生存者563例を対象とした報告では、15.5%が過去に131I-MIBG療法を受けていた。[ 45 ]
根拠(化学療法):
- イリノテカンとテモゾロミドの併用
- ある後ろ向き研究では、イホスファミド、カルボプラチン、およびエトポシドの投与を92サイクル受けた患者74人の成績が報告された。この多剤併用療法に対する反応後に末梢造血幹細胞移植を受けた患者37人が含まれていた。[ 47 ]
- トポテカンとシクロホスファミドまたはエトポシドを併用する治療法が、初回治療でトポテカンの投与を受けなかった再発例に対して用いられている。[ 28 ][エビデンスレベルA2]
- BEACON試験では、テモゾロミドを投与する群とトポテカンおよびテモゾロミドを投与する群(両群とも一部はベバシズマブを併用)に患者がランダムに割り付けられた。[ 26 ]
- 高用量でカルボプラチン、イリノテカン、および/またはテモゾロミドを投与する治療が、難治性の患者または治療(トポテカンを含む治療)終了後に新たに再発を来した患者に対して用いられている(客観的奏効率68%)。しかしながら、このレジメンは治療中に進行した患者の治療には用いられない。[ 48 ]
再発した神経芽腫患者には、他にも様々なアプローチの免疫療法が用いられている。抗GD2モノクローナル抗体の単剤療法には、この状況で活性が示されている。例えば、ある第II相試験では、再発または難治性の高リスク群神経芽腫の小児40人を対象として、10日間にわたるジヌツキシマブの長期投与が評価された。この研究では客観的奏効率が26%と報告された。このアプローチは忍容性が高く、グレード4またはグレード5の事象は発生しなかった。[ 49 ]
再発または進行性神経芽腫における同種移植は歴史的に成功率が低い。ある後ろ向きレジストリー研究では、自家HSCT後の同種HSCTは有益とならないようであった。再発が依然として治療不成功の最も頻度の高い原因となっている。[ 50 ]51人の患者(うち44人は再発または難治性の高リスク群神経芽腫患者)を対象として強度減弱前処置による同種HSCTを評価した多施設共同第II相試験においても、同様の結論に達した。5年無病生存(DFS)率は11.8%であった。[ 51 ]
HLA半合致移植後にGD2標的療法を用いることが、より有望な戦略となる可能性がある。 再発した神経芽腫患者68人を対象とした試験では、HLA半合致移植後にジヌツキシマブとインターロイキン2の皮下投与を併用することが可能とされ、移植片対宿主病の発生率は低かった。5年EFS率は43%であった。ジヌツキシマブによる治療開始時点で完全または部分奏効が得られていた患者では、成績がより良好であった。 移植後も腫瘍がある患者では、抗GD2免疫療法での完全奏効率が35%であった。[ 52 ]
静脈内投与されたモノクローナル抗体の抗腫瘍活性を再現する可能性がある宿主抗ガングリオシド抗体を誘導するようにデザインされたワクチンの臨床試験が複数進行中である。 被験者には広範な免疫賦活性作用を有し、抗GD2/GD3モノクローナル抗体と相乗効果を示すβグルカン治療も行われる。高リスク群神経芽腫の小児15人を対象とした第I相研究では、この治療法は用量制限毒性を示すことなく認容性を示した。[ 53 ]2回目以降の完全寛解または非常に良好な部分寛解の達成後に抗GD2免疫療法およびイソトレチノインによる地固め療法と場合により維持療法を受けた患者において、長期の無増悪生存例が報告されている。 それらの患者には、過去に抗GD2免疫療法およびイソトレチノインによる治療を受けていた患者が含まれている。[ 54 ]
ある第I/II相試験では、GD2を標的とする自己キメラ抗原受容体発現(CAR)T細胞の使用が、再発または難治性の高リスク群神経芽腫の治療において実行可能かつ安全であった。この治療で63%の奏効率が得られた。[ 55 ]これらの知見は、この同じ集団においてGD2を標的とする他のCAR-T細胞アプローチが小さな活性しか示さなかった過去の報告と対照的である。
中枢神経系における再発神経芽腫
中枢神経系進展は、初発時にはまれであるが、再発神経芽腫では3~10%の患者にみられる。 高リスク群神経芽腫患者を対象とした試験で治療を受けた4期患者1,977人にみられた1161回の初回再発では、中枢神経系再発が全転移再発の6%を占めた。[ 56 ]初発患者に対する最初の治療では中枢神経系が十分に治療できないため、中枢神経系が再発につながる聖域部位(sanctuary site)として浮上している。[ 56 ][ 57 ][ 58 ]
International Society of Paediatric Oncology Europe Neuroblastoma(SIOPEN)試験で同定された中枢神経系再発の有意な危険因子は、診断時点の患者因子および疾患因子であった。具体的な因子としては、女性であること(HR、2.0;P = 0.016)、MYCN増幅(HR、2.4;P = 0.0008)、肝病変(HR、2.5;P = 0.01)、複数の器官系/身体区画への転移(HR、7.1;P = 0.047)などがあった。 大量化学療法と免疫療法のいずれにも、再発リスク上昇との関連はみられなかった。研究者は経時的に報告された中枢神経系再発の発生率が安定していたことに注目した。[ 56 ]
中枢神経系再発はほぼ全例で死に至り、死亡までの期間中央値は6カ月である。SIOPEN試験では、再発後1年および3年OS率は、それぞれ25%および7%であった。[ 56 ]中枢神経系のみに再発を認める患者では、長期生存が得られる可能性がある。[ 56 ]
中枢神経系の再発神経芽腫に対する治療選択肢としては以下のものがある:
- 手術および放射線療法
- 化学療法(テモゾロミドを含むレジメンなど)と手術および放射線療法の併用
- 新規の治療アプローチ
現行の治療アプローチには一般に、中枢神経系における肉眼的および顕微鏡的な残存病変とさらなる再発の予兆となりうる全身的な微小残存病変を根絶することが含まれる。脳神経外科的介入は、浮腫の軽減、出血のコントロール、および治療開始前のbulky腫瘍の切除を目的として行われる。
再発した転移性の中枢神経系神経芽腫の患者を対象として、放射性ヨウ素標識抗B7H3モノクローナル抗体の髄腔内投与を18Gyまたは21Gyの頭蓋脊髄照射および中枢神経系の肉眼的病変に対する追加照射と併用する脳室内コンパートメント放射免疫療法(intraventricular compartmental radioimmunotherapy)の単一施設試験において、ある程度の成功が得られている。[ 13 ]5年中枢神経系DFS率は約69%、5年OS率は約45%であった。[ 59 ][エビデンスレベルC2]
最初の中枢神経系再発後に長期生存が得られた患者では、頭蓋脊髄照射(CSI)後に2回目の再発を来すことがある。 2回目の中枢神経系再発を経験した患者に関する公表データは限られている。2回目の中枢神経系再発は予後不良を示唆する。[ 57 ]
最初の中枢神経系再発に対してCSIによる治療を受けた患者128人を対象とした単一施設研究では、40人において2回目の中枢神経系再発がみられ、最初のCSIから2回目の再発までの期間の中央値は6.3カ月であった。2回目の中枢神経系再発がみられた患者の治療成績は不良であるが、2回目の中枢神経系再発時の放射線療法による治療にOS延長との関連が報告されている。[ 60 ][エビデンスレベルC1]
再発または難治性神経芽腫を対象として臨床評価段階にある治療選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーを務める臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている国および/または医療機関が主導する臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録が進行しているものを検索するには、臨床試験アドバンスト・サーチを使用のこと。この検索では、試験の実施場所、治療の種類、薬剤名、その他の基準で絞り込むことができる。臨床試験に関する一般情報も入手することができる。
参考文献- Marachelian A, Shimada H, Sano H, et al.: The significance of serial histopathology in a residual mass for outcome of intermediate risk stage 3 neuroblastoma. Pediatr Blood Cancer 58 (5): 675-81, 2012.[PUBMED Abstract]
- Taggart DR, Han MM, Quach A, et al.: Comparison of iodine-123 metaiodobenzylguanidine (MIBG) scan and [18F]fluorodeoxyglucose positron emission tomography to evaluate response after iodine-131 MIBG therapy for relapsed neuroblastoma. J Clin Oncol 27 (32): 5343-9, 2009.[PUBMED Abstract]
- Schleiermacher G, Javanmardi N, Bernard V, et al.: Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 32 (25): 2727-34, 2014.[PUBMED Abstract]
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.[PUBMED Abstract]
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.[PUBMED Abstract]
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.[PUBMED Abstract]
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.[PUBMED Abstract]
- London WB, Castel V, Monclair T, et al.: Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol 29 (24): 3286-92, 2011.[PUBMED Abstract]
- Pole JG, Casper J, Elfenbein G, et al.: High-dose chemoradiotherapy supported by marrow infusions for advanced neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 9 (1): 152-8, 1991.[PUBMED Abstract]
- Castel V, Cañete A, Melero C, et al.: Results of the cooperative protocol (N-III-95) for metastatic relapses and refractory neuroblastoma. Med Pediatr Oncol 35 (6): 724-6, 2000.[PUBMED Abstract]
- Lau L, Tai D, Weitzman S, et al.: Factors influencing survival in children with recurrent neuroblastoma. J Pediatr Hematol Oncol 26 (4): 227-32, 2004.[PUBMED Abstract]
- Saylors RL, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001.[PUBMED Abstract]
- Kramer K, Kushner BH, Modak S, et al.: Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol 97 (3): 409-18, 2010.[PUBMED Abstract]
- Vo KT, DuBois SG, Neuhaus J, et al.: Pattern and predictors of sites of relapse in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 69 (9): e29616, 2022.[PUBMED Abstract]
- Campbell K, Kao PC, Naranjo A, et al.: Clinical and biological features prognostic of survival after relapse or progression of INRGSS stage MS pattern neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 70 (2): e30054, 2023.[PUBMED Abstract]
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Murphy JM, Lim II, Farber BA, et al.: Salvage rates after progression of high-risk neuroblastoma with a soft tissue mass. J Pediatr Surg 51 (2): 285-8, 2016.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- London WB, Bagatell R, Weigel BJ, et al.: Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children's Oncology Group early-phase trials. Cancer 123 (24): 4914-4923, 2017.[PUBMED Abstract]
- Mody R, Naranjo A, Van Ryn C, et al.: Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol 18 (7): 946-957, 2017.[PUBMED Abstract]
- Foster JH, Voss SD, Hall DC, et al.: Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children's Oncology Group Study (ADVL0912). Clin Cancer Res 27 (13): 3543-3548, 2021.[PUBMED Abstract]
- Goldsmith KC, Park JR, Kayser K, et al.: Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med 29 (5): 1092-1102, 2023.[PUBMED Abstract]
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.[PUBMED Abstract]
- Cole KA, Ijaz H, Surrey LF, et al.: Pediatric phase 2 trial of a WEE1 inhibitor, adavosertib (AZD1775), and irinotecan for relapsed neuroblastoma, medulloblastoma, and rhabdomyosarcoma. Cancer 129 (14): 2245-2255, 2023.[PUBMED Abstract]
- Moreno L, Weston R, Owens C, et al.: Bevacizumab, Irinotecan, or Topotecan Added to Temozolomide for Children With Relapsed and Refractory Neuroblastoma: Results of the ITCC-SIOPEN BEACON-Neuroblastoma Trial. J Clin Oncol 42 (10): 1135-1145, 2024.[PUBMED Abstract]
- Corbacioglu S, Lode H, Ellinger S, et al.: Irinotecan and temozolomide in combination with dasatinib and rapamycin versus irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma (RIST-rNB-2011): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol 25 (7): 922-932, 2024.[PUBMED Abstract]
- London WB, Frantz CN, Campbell LA, et al.: Phase II randomized comparison of topotecan plus cyclophosphamide versus topotecan alone in children with recurrent or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol 28 (24): 3808-15, 2010.[PUBMED Abstract]
- Shusterman S, Naranjo A, Van Ryn C, et al.: Antitumor Activity and Tolerability of hu14.18-IL2 with GMCSF and Isotretinoin in Recurrent or Refractory Neuroblastoma: A Children's Oncology Group Phase II Study. Clin Cancer Res 25 (20): 6044-6051, 2019.[PUBMED Abstract]
- Mody R, Yu AL, Naranjo A, et al.: Irinotecan, Temozolomide, and Dinutuximab With GM-CSF in Children With Refractory or Relapsed Neuroblastoma: A Report From the Children's Oncology Group. J Clin Oncol 38 (19): 2160-2169, 2020.[PUBMED Abstract]
- Lerman BJ, Li Y, Carlowicz C, et al.: Progression-Free Survival and Patterns of Response in Patients With Relapsed High-Risk Neuroblastoma Treated With Irinotecan/Temozolomide/Dinutuximab/Granulocyte-Macrophage Colony-Stimulating Factor. J Clin Oncol 41 (3): 508-516, 2023.[PUBMED Abstract]
- Raiser P, Schleiermacher G, Gambart M, et al.: Chemo-immunotherapy with dinutuximab beta in patients with relapsed/progressive high-risk neuroblastoma: does chemotherapy backbone matter? Eur J Cancer 202: 114001, 2024.[PUBMED Abstract]
- DuBois SG, Groshen S, Park JR, et al.: Phase I Study of Vorinostat as a Radiation Sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for Patients with Relapsed or Refractory Neuroblastoma. Clin Cancer Res 21 (12): 2715-21, 2015.[PUBMED Abstract]
- Polishchuk AL, Dubois SG, Haas-Kogan D, et al.: Response, survival, and toxicity after iodine-131-metaiodobenzylguanidine therapy for neuroblastoma in preadolescents, adolescents, and adults. Cancer 117 (18): 4286-93, 2011.[PUBMED Abstract]
- Matthay KK, Yanik G, Messina J, et al.: Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol 25 (9): 1054-60, 2007.[PUBMED Abstract]
- Matthay KK, Tan JC, Villablanca JG, et al.: Phase I dose escalation of iodine-131-metaiodobenzylguanidine with myeloablative chemotherapy and autologous stem-cell transplantation in refractory neuroblastoma: a new approaches to Neuroblastoma Therapy Consortium Study. J Clin Oncol 24 (3): 500-6, 2006.[PUBMED Abstract]
- Matthay KK, Quach A, Huberty J, et al.: Iodine-131--metaiodobenzylguanidine double infusion with autologous stem-cell rescue for neuroblastoma: a new approaches to neuroblastoma therapy phase I study. J Clin Oncol 27 (7): 1020-5, 2009.[PUBMED Abstract]
- DuBois SG, Chesler L, Groshen S, et al.: Phase I study of vincristine, irinotecan, and ¹³¹I-metaiodobenzylguanidine for patients with relapsed or refractory neuroblastoma: a new approaches to neuroblastoma therapy trial. Clin Cancer Res 18 (9): 2679-86, 2012.[PUBMED Abstract]
- Johnson K, McGlynn B, Saggio J, et al.: Safety and efficacy of tandem 131I-metaiodobenzylguanidine infusions in relapsed/refractory neuroblastoma. Pediatr Blood Cancer 57 (7): 1124-9, 2011.[PUBMED Abstract]
- French S, DuBois SG, Horn B, et al.: 131I-MIBG followed by consolidation with busulfan, melphalan and autologous stem cell transplantation for refractory neuroblastoma. Pediatr Blood Cancer 60 (5): 879-84, 2013.[PUBMED Abstract]
- Zhou MJ, Doral MY, DuBois SG, et al.: Different outcomes for relapsed versus refractory neuroblastoma after therapy with (131)I-metaiodobenzylguanidine ((131)I-MIBG). Eur J Cancer 51 (16): 2465-72, 2015.[PUBMED Abstract]
- Yanik GA, Villablanca JG, Maris JM, et al.: 131I-metaiodobenzylguanidine with intensive chemotherapy and autologous stem cell transplantation for high-risk neuroblastoma. A new approaches to neuroblastoma therapy (NANT) phase II study. Biol Blood Marrow Transplant 21 (4): 673-81, 2015.[PUBMED Abstract]
- DuBois SG, Granger MM, Groshen S, et al.: Randomized Phase II Trial of MIBG Versus MIBG, Vincristine, and Irinotecan Versus MIBG and Vorinostat for Patients With Relapsed or Refractory Neuroblastoma: A Report From NANT Consortium. J Clin Oncol 39 (31): 3506-3514, 2021.[PUBMED Abstract]
- Sevrin F, Kolesnikov-Gauthier H, Cougnenc O, et al.: Phase II study of 131 I-metaiodobenzylguanidine with 5 days of topotecan for refractory or relapsed neuroblastoma: Results of the French study MIITOP. Pediatr Blood Cancer 70 (11): e30615, 2023.[PUBMED Abstract]
- Westerveld ASR, Tytgat GAM, van Santen HM, et al.: Long-Term Risk of Subsequent Neoplasms in 5-Year Survivors of Childhood Neuroblastoma: A Dutch Childhood Cancer Survivor Study-LATER 3 Study. J Clin Oncol 43 (2): 154-166, 2025.[PUBMED Abstract]
- Bagatell R, London WB, Wagner LM, et al.: Phase II study of irinotecan and temozolomide in children with relapsed or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol 29 (2): 208-13, 2011.[PUBMED Abstract]
- Kushner BH, Modak S, Kramer K, et al.: Ifosfamide, carboplatin, and etoposide for neuroblastoma: a high-dose salvage regimen and review of the literature. Cancer 119 (3): 665-71, 2013.[PUBMED Abstract]
- Kushner BH, Kramer K, Modak S, et al.: Differential impact of high-dose cyclophosphamide, topotecan, and vincristine in clinical subsets of patients with chemoresistant neuroblastoma. Cancer 116 (12): 3054-60, 2010.[PUBMED Abstract]
- Lode HN, Ehlert K, Huber S, et al.: Long-term, continuous infusion of single-agent dinutuximab beta for relapsed/refractory neuroblastoma: an open-label, single-arm, Phase 2 study. Br J Cancer 129 (11): 1780-1786, 2023.[PUBMED Abstract]
- Hale GA, Arora M, Ahn KW, et al.: Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience. Bone Marrow Transplant 48 (8): 1056-64, 2013.[PUBMED Abstract]
- Prete A, Lanino E, Saglio F, et al.: Phase II Study of Allogeneic Hematopoietic Stem Cell Transplantation for Children with High-Risk Neuroblastoma Using a Reduced-Intensity Conditioning Regimen: Results from the AIEOP Trial. Transplant Cell Ther 30 (5): 530.e1-530.e8, 2024.[PUBMED Abstract]
- Flaadt T, Ladenstein RL, Ebinger M, et al.: Anti-GD2 Antibody Dinutuximab Beta and Low-Dose Interleukin 2 After Haploidentical Stem-Cell Transplantation in Patients With Relapsed Neuroblastoma: A Multicenter, Phase I/II Trial. J Clin Oncol 41 (17): 3135-3148, 2023.[PUBMED Abstract]
- Kushner BH, Cheung IY, Modak S, et al.: Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res 20 (5): 1375-82, 2014.[PUBMED Abstract]
- Kushner BH, Ostrovnaya I, Cheung IY, et al.: Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-GD2 immunotherapy and isotretinoin: a prospective Phase II study. Oncoimmunology 4 (7): e1016704, 2015.[PUBMED Abstract]
- Del Bufalo F, De Angelis B, Caruana I, et al.: GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N Engl J Med 388 (14): 1284-1295, 2023.[PUBMED Abstract]
- Berlanga P, Pasqualini C, Pötschger U, et al.: Central nervous system relapse in high-risk stage 4 neuroblastoma: The HR-NBL1/SIOPEN trial experience. Eur J Cancer 144: 1-8, 2021.[PUBMED Abstract]
- Kramer K, Kushner B, Heller G, et al.: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91 (8): 1510-9, 2001.[PUBMED Abstract]
- Matthay KK, Brisse H, Couanet D, et al.: Central nervous system metastases in neuroblastoma: radiologic, clinical, and biologic features in 23 patients. Cancer 98 (1): 155-65, 2003.[PUBMED Abstract]
- Luo LY, Kramer K, Cheung NV, et al.: Reduced-dose craniospinal irradiation for central nervous system relapsed neuroblastoma. Pediatr Blood Cancer 67 (9): e28364, 2020.[PUBMED Abstract]
- Tringale KR, Wolden SL, Casey DL, et al.: Clinical outcomes of pediatric patients receiving multimodality treatment of second central nervous system relapse of neuroblastoma. Pediatr Blood Cancer 70 (2): e30075, 2023.[PUBMED Abstract]
- 本要約の最新更新(2025/04/28)
-
PDQがん情報要約は、定期的にレビューされ、新たな情報が得られ次第更新される。 このセクションでは、上記の日付で本要約に加えられた最新の変更内容を記載する。
本要約は包括的にレビューされ、大幅に改訂された。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。 本要約は独自の文献レビューの結果を反映するものであり、NCIまたはNIHの方針声明を示すものではない。 PDQ要約の更新におけるPDQ Editorial Boardsの役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® Cancer Information for Health Professionalsを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、神経芽腫の治療について、包括的な、専門家の査読を経た、そして根拠に基づいた情報を提供する。本要約は、患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。 医療上の意思決定のための正式なガイドラインや推奨事項を提供するものではない。
査読者および更新情報
本要約は、編集に関して米国国立がん研究所(NCI)から独立しているPDQ Pediatric Treatment Editorial Boardにより定期的にレビューされ、随時更新される。 本要約は独自の文献レビューの結果を反映するものであり、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、直近で発表された記事をレビューし、個々の記事について以下を行うべきかどうかを判断する:
要約の変更は、発表された記事の根拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容についてコメントまたは質問がある場合は、NCIウェブサイトのEmail UsからCancer.govに連絡されたい。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
エビデンスレベル
本要約で引用されている参考文献の一部にはエビデンスレベルの指定が明記されている。それらの指定は、読者が特定の介入またはアプローチの利用を支持している根拠の強さを評価する上で参考になることを意図したものである。PDQ Pediatric Treatment Editorial Boardは、エビデンスレベルの指定を策定するにあたり公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。ただし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋】.”のような一文を記載することができる。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Neuroblastoma Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq. Accessed <MM/DD/YYYY>.[PMID: 26389190]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な根拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。
