

ご利用について
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of neuroblastoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
CONTENTS
- General Information About Neuroblastoma
-
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[ 1 ][ 2 ] Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ] Between 1975 and 2020, the 5-year survival rate for patients with neuroblastoma increased, from 86% to 93% for children younger than 1 year and from 34% to 83% for children aged 1 to 14 years.[ 2 ][ 3 ]
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Incidence and Epidemiology
Neuroblastoma is the most common extracranial solid tumor in childhood. More than 650 cases are diagnosed each year in the United States.[ 2 ][ 6 ][ 7 ][ 8 ] The prevalence is about 1 case per 7,000 live births. The incidence is 8.3 cases per 1 million per year in children younger than 15 years. The overall incidence of neuroblastoma cases in the United States has remained stable.[ 9 ] About 37% of patients are diagnosed as infants, and 90% are younger than 5 years at diagnosis, with a median age at diagnosis of 17 months.[ 8 ][ 10 ] The data on age at diagnosis show that this is a disease of infancy, with the highest rate of diagnosis in the first month of life.[ 6 ][ 10 ][ 11 ]
Population-based studies (of screening for infants with neuroblastoma) have demonstrated that spontaneous regression of neuroblastoma without clinical detection in the first year of life is at least as prevalent as clinically detected neuroblastoma.[ 12 ][ 13 ][ 14 ]
The United States Cancer Statistics database and the National Program of Cancer Registries survival database were used to describe epidemiological trends in incidence and outcomes in patients with neuroblastoma between 2003 and 2019. Non-Hispanic White patients have a higher risk of developing neuroblastoma than all other race and ethnicity groups. Compared with non-Hispanic White patients, the relative risks were 0.54 for Hispanic patients, 0.64 for non-Hispanic Asian or Pacific Islander patients, 0.69 for non-Hispanic American Indian and Alaska Native patients, and 0.73 for non-Hispanic Black patients.[ 9 ] The 5-year relative survival rates were higher for non-Hispanic White patients (80.7%) and Hispanic patients (80.8%), compared with non-Hispanic Black patients (72.6%).[ 9 ]
Findings from epidemiological studies have not unequivocally linked environmental or other exposures to increased or decreased incidences of neuroblastoma.[ 15 ]
Anatomy
Neuroblastoma originates in the adrenal medulla and paraspinal or periaortic regions where sympathetic nervous system tissue is present (see Figure 1).

Figure 1. Neuroblastoma may be found in the adrenal glands and paraspinal nerve tissue from the neck to the pelvis. Neuroblastoma Screening
Familial neuroblastoma and genetic predisposition
Studies analyzing constitutional DNA in rare cohorts of patients with familial neuroblastoma have provided insight into the complex genetic basis for tumor initiation. About 1% to 2% of patients with neuroblastoma have a family history of the disease. These children are, on average, younger (9 months at diagnosis) than patients without a family history, and about 20% of these patients have multifocal primary neuroblastoma.
Germline variants. Several germline variants have been associated with a genetic predisposition to neuroblastoma, including the following:
Other cancer predisposition syndromes. Children with gene aberrations associated with other cancer predisposition syndromes may be at increased risk of developing neuroblastoma and other malignancies. The following syndromes primarily involve genes in the canonical RAS pathway:
In addition, neuroblastoma has been described in patients with the following syndromes:
With increased availability of sequencing techniques, the spectrum of germline alterations seen in patients with neuroblastoma is expanding. For example, one study identified a series of 11 patients with germline pathogenic variants in SMARCA4.[ 30 ] In another study of 786 patients with neuroblastoma, 13.9% had pathogenic or likely pathogenic germline variants in cancer predisposition genes. BARD1, ERCC2, CHEK2, and MSH3 were the genes in which germline pathogenic variants were most commonly observed. Germline pathogenic variants in BARD1, EZH2, ALK, PTCH1, and MSH3 were specifically enriched in patients with neuroblastoma, compared with controls. Patients with these alterations had inferior survival, compared with patients without these alterations.[ 31 ] Another study replicated the findings that germline alterations in cancer predisposition genes are associated with inferior outcomes. In addition, the researchers showed that the burden of germline functional variants beyond conventional cancer predisposition genes was also prognostic.[ 32 ] For more information about SMARCA4, visit Rhabdoid Tumor Predisposition Syndrome Type 2.
Sporadic neuroblastoma may also have an increased incidence resulting from less potent germline predispositions. Genome-wide association studies have identified several common genomic variants (single nucleotide polymorphisms) with modest effect size that are associated with increased risk of developing neuroblastoma. Most of these genomic risk variants are significantly associated with distinct neuroblastoma phenotypes (i.e., high-risk vs. low-risk disease).[ 33 ]
Neuroblastoma predisposition and surveillance
Screening recommendations from the American Association for Cancer Research (AACR) came from the 2016 Childhood Cancer Predisposition Workshop. The AACR recommends that the following individuals undergo biochemical and radiographic surveillance for early detection of tumors in the first 10 years of life:[ 27 ]
Surveillance consists of the following:[ 27 ]
Surveillance begins at birth or at diagnosis of neuroblastoma predisposition and continues every 3 months until age 6 years, then every 6 months until age 10 years. Patients with Costello syndrome may have elevated urinary catecholamines in the absence of a catecholamine-secreting tumor, so only high or significantly rising levels should prompt investigation beyond ultrasonography and chest x-ray.[ 35 ] Patients with Li-Fraumeni syndrome should not undergo chest x-rays.[ 27 ]
About 5% of children with Beckwith-Wiedemann syndrome have variants that cause decreased activity of CDKN1C. A review of all large studies of genetically subtyped Beckwith-Wiedemann syndrome found 70 children with the CDKN1C variant, 4.6% of whom developed neuroblastoma. There were no cases of Wilms tumor or hepatoblastoma. There is little experience with screening these children for neuroblastoma, so there are no generally accepted guidelines. However, the authors of the study suggest screening with urinary VMA/HVA every 4 to 6 months. Patients with other genetic subtypes of Beckwith-Wiedemann syndrome have a prevalence of neuroblastoma of less than 1%. No neuroblastic tumors were found among 123 children with the genotype gain of methylation at imprinting control region 1.[ 36 ]
General population
Current data do not support neuroblastoma screening in the general public. Screening at the ages of 3 weeks, 6 months, or 1 year did not lead to a reduced incidence of advanced-stage neuroblastoma with unfavorable biological characteristics in older children, nor did it reduce overall mortality from neuroblastoma.[ 13 ][ 14 ] No public health benefits have been shown from screening infants for neuroblastoma at these ages.
Evidence (against neuroblastoma screening):
Clinical Presentation
The most frequent signs and symptoms of neuroblastoma in children are caused by tumor mass and metastases and include the following:
The clinical presentation of neuroblastoma in adolescents is similar to that in children. The only exception is that bone marrow involvement occurs less frequently in adolescents, and there is a greater frequency of metastases in unusual sites such as lung or brain.[ 40 ]
Opsoclonus/myoclonus syndrome
Paraneoplastic neurological findings, including cerebellar ataxia or opsoclonus/myoclonus, occur rarely in children with neuroblastoma.[ 41 ] Of young children presenting with opsoclonus/myoclonus syndrome, about one-half are found to have neuroblastoma.[ 42 ][ 43 ] The incidence in the United Kingdom is estimated at 0.18 cases per 1 million children per year. The average age at diagnosis is 1.5 to 2 years.[ 44 ]
The usual presentation is the onset of progressive neurological dysfunction over a few days before a neuroblastoma is discovered. However, on occasion, neurological symptoms arise long after removal of the primary tumor.[ 42 ][ 45 ][ 46 ] Patients with neuroblastoma who present with opsoclonus/myoclonus syndrome often have neuroblastoma with favorable biological features and have excellent survival rates, although tumor-related deaths have been reported.[ 42 ]
The opsoclonus/myoclonus syndrome appears to be caused by an immunologic mechanism that is not yet fully characterized.[ 42 ] The primary tumor is typically diffusely infiltrated with lymphocytes.[ 47 ] Cerebrospinal fluid shows an increased number of B cells, and oligoclonal immunoglobulin bands are often seen. Steroid-responsive elevations of B-cell–related cytokines are also often seen.[ 48 ]
Genomic copy number profiles were analyzed in 44 cases of neuroblastoma associated with opsoclonus/myoclonus syndrome. Because there were no tumor relapses or disease-related deaths, the overall genomic profile was not prognostically significant.[ 49 ]
Some patients may rapidly respond neurologically to immune interventions or simply to removal of the neuroblastoma, but in many cases, improvement may be slow and partial. While immunological therapy has improved acutely presenting motor deficits and ataxia, its benefit on long-term neuropsychological disability, which primarily consists of cognitive and behavioral deficits, is not clear. The long-term benefits of rapid improvement resulting from treatment, whether of symptoms or of the underlying neuroblastoma, are unclear, but rapid improvement appears to be worthwhile.[ 46 ] [ 50 ]
Treatment with adrenocorticotropic hormones or corticosteroids can be effective for acute symptoms, but some patients do not respond to corticosteroids.[ 45 ][ 51 ] Other therapy with various immunomodulatory drugs, plasmapheresis, intravenous gamma globulin, and rituximab have been reported to be effective in select cases.[ 45 ][ 52 ][ 53 ][ 54 ][ 55 ] Combination immunosuppressive therapy has been explored, with improved short-term results.[ 56 ] The short-term neurological outcomes may be superior in patients treated with chemotherapy, possibly because of its immunosuppressive effects.[ 41 ]
The Children’s Oncology Group (COG) completed the first randomized, open-label, phase III study of patients with opsoclonus/myoclonus ataxia syndrome.[ 57 ] Patients with newly diagnosed neuroblastoma and opsoclonus/myoclonus ataxia syndrome who were younger than 8 years were randomly assigned to receive either intravenous immunoglobulin (IVIG) or no IVIG in addition to prednisone and risk-adapted treatment of the tumor.[ 57 ]
Diagnosis
Diagnostic evaluation of neuroblastoma includes the following:
The diagnosis of neuroblastoma requires the involvement of pathologists who are familiar with childhood tumors. Some neuroblastomas cannot be differentiated morphologically, via conventional light microscopy with hematoxylin and eosin staining alone, from other small round blue cell tumors of childhood, such as lymphomas, Ewing sarcoma, and rhabdomyosarcomas. In such cases, immunohistochemical and cytogenetic analysis may be needed to diagnose a specific small round blue cell tumor.
The minimum criterion for a diagnosis of neuroblastoma, as established by international agreement, is that diagnosis must be based on one of the following:[ 64 ]
- An unequivocal pathological diagnosis made from tumor tissue by light microscopy (with or without immunohistology or electron microscopy).
- The combination of bone marrow aspirate or trephine biopsy containing unequivocal tumor cells (e.g., syncytia or immunocytologically positive clumps of cells) and increased levels of urinary catecholamine metabolites.
Observation and Spontaneous Regression of Fetal/Neonatal Neuroblastoma
The phenomenon of spontaneous regression has been well described in infants with neuroblastoma, especially in infants with the INSS 4S/INRG MS pattern of metastatic spread.[ 65 ] In rare cases, fetal ultrasonography can show suspected neuroblastoma prenatally.[ 66 ] Management recommendations are evolving regarding the need for immediate diagnostic biopsy in infants aged 6 months and younger with suspected neuroblastoma tumors that are likely to spontaneously regress. For more information about INSS 4S/INRG MS disease, see the Evaluation of Primary Tumor and Metastatic Disease section.
Spontaneous regression generally occurs in tumors with the following features:[ 67 ][ 68 ][ 69 ]
Additional features associated with spontaneous regression include the lack of telomerase expression,[ 67 ][ 70 ] the expression of the H-Ras protein,[ 71 ] and the expression of the neurotrophin receptor TrkA, a nerve growth factor receptor.[ 72 ]
Studies have suggested that selected infants who appear to have asymptomatic, small, low-stage adrenal neuroblastoma (detected by screening or during prenatal or incidental ultrasonography) often have tumors that spontaneously regress. These patients may be observed safely without surgical intervention or tissue diagnosis.[ 73 ][ 74 ][ 75 ]
Evidence (observation [spontaneous regression]):
- In a COG study, 83 highly selected infants younger than 6 months with stage 1 small adrenal masses (3.1 cm or less), as defined by imaging studies, were observed without biopsy. Surgical intervention was reserved for those with growth or progression of the mass or increasing concentrations of urinary catecholamine metabolites.[ 76 ]
- A German clinical trial reported on 340 infants with localized neuroblastoma without MYCN amplification. Of these patients, 190 underwent resection, 57 were treated with chemotherapy, and 93 were observed with gross residual tumor.[ 77 ]
- In neuroblastoma screening trials in Quebec, Canada, and Germany, the incidence of neuroblastoma was twice that reported in nonscreened populations, suggesting that many neuroblastomas are never diagnosed clinically and spontaneously regress.[ 12 ][ 13 ][ 14 ]
Prognostic Factors
The prognosis for patients with neuroblastoma is related to the following:
Some of these prognostic factors have been combined to create risk groups to help define treatment. For more information, see the sections on International Neuroblastoma Risk Group Staging System (INRGSS) and Children’s Oncology Group (COG) Neuroblastoma Risk Grouping.
Age at diagnosis
Infants and children
The effect of age at diagnosis on 5-year survival is profound. In the COG ANBL00B1 (NCT00904241) study of 4,832 patients with newly diagnosed neuroblastoma, those younger than 18 months had a 5-year EFS rate of 82% and an OS rate of 91%. In comparison, patients aged 18 months or older had a 5-year EFS rate of 64% and an OS rate of 74%.[ 78 ]
According to the National Childhood Cancer Registry (NCCR), the 5-year relative survival rates from 2014 to 2020 were as follows:[ 2 ]
The effect of patient age on prognosis is strongly influenced by clinical and pathobiological factors, as evidenced by the following:
Adolescents and young adults
Adolescents and adults rarely develop neuroblastoma, accounting for less than 5% of all cases. When neuroblastoma occurs in this age range, it shows a more indolent clinical course than neuroblastoma in younger patients, and it often shows de novo chemotherapy resistance.[ 81 ] Neuroblastoma in adolescents and young adults may also exhibit unusual clinicopathological characteristics such as large tumors, bilateral adrenal disease, and pheochromocytoma-like features.[ 82 ][Level of evidence C1] Neuroblastoma has a worse long-term prognosis in adolescents older than 10 years or in adults, regardless of stage or site.
Although adolescent and young adult patients have infrequent MYCN amplification (9% in patients aged 10–21 years), older children with advanced disease have a poor rate of survival. Tumors from the adolescent and young adult population commonly have segmental chromosomal aberrations, and ALK and ATRX variants are much more frequent.[ 83 ][ 84 ][ 85 ] In adolescents, approximately 40% of the tumors have loss-of-function variants in ATRX, compared with less than 20% in younger children and 0% in infants younger than 1 year.[ 81 ] Complex DNA microarray findings and novel variants have been reported in some patients.[ 82 ][Level of evidence C1]
The 5-year OS rate for adolescent and young adult patients (aged 15–39 years) is 38%.[ 86 ][Level of evidence C1] The 5-year EFS rate is 32% for patients between the ages of 10 years and 21 years, and the OS rate is 46%. For patients with stage 4 disease, the 10-year EFS rate is 3%, and the OS rate is 5%.[ 87 ] Aggressive chemotherapy and surgery have been shown to achieve a minimal disease state in more than 50% of these patients.[ 40 ][ 88 ] Other modalities, such as local radiation therapy, autologous stem cell transplant, and the use of agents with confirmed activity, may improve the poor prognosis for adolescents and adults.[ 87 ][ 88 ]
Adults
The biology of adult-onset neuroblastoma appears to differ from the biology of pediatric or adolescent neuroblastoma based on a single-institution series of 44 patients (aged 18–71 years).[ 89 ]
As noted above, adult-onset neuroblastoma is enriched for activating ALK variants. In a single-institution retrospective study, 13 adults (median age, 34 years; range, 16–71 years) with relapsed, ALK-altered neuroblastoma were treated with lorlatinib. Nine patients (69%) had a complete or partial response, five of whom were previously treated with other ALK inhibitors. Lorlatinib was associated with significant adverse events requiring dose reduction. However, responses were seen using doses below the recommended adult dose.[ 90 ] In another multicenter trial, 15 adults (aged 18 years or older; median age, 24 years) with relapsed or refractory ALK-altered neuroblastoma were treated with lorlatinib. The response rate (complete, partial, and minor response) was 67%.[ 91 ]
Stage of disease
Several image-based and surgery-based systems were used for assigning disease stage of neuroblastoma before the 1990s. In an effort to compare results obtained throughout the world, a surgical pathological staging system, termed the International Neuroblastoma Staging System (INSS), was developed.[ 64 ] The INSS predicted outcome based on stage at diagnosis, although important interactions with biological variables were also found.[ 3 ][ 4 ][ 11 ][ 64 ][ 79 ][ 80 ][ 92 ][ 93 ][ 94 ] However, because surgical approaches differ from one institution to another, INSS stage for patients with locoregional disease may also vary considerably. To define extent of disease at diagnosis in a uniform manner, a presurgical International Neuroblastoma Risk Group staging system (INRGSS) was developed for the International Neuroblastoma Risk Group Classification System.[ 95 ][ 96 ] The INRGSS is currently used in North American and European cooperative group studies. This staging system is not affected by locoregional lymph node involvement.
For the patients with newly diagnosed neuroblastoma enrolled in the ANBL00B1 (NCT00904241) study, the 5-year EFS and OS rates, according to INRGSS stage, were the following:[ 78 ]
For more information, see the following sections:
Tumor histology
Neuroblastoma tumor histology has a significant impact on prognosis and risk group assignment. For more information, see the Histological Classification of Neuroblastic Tumors section and Table 2.
In the ANBL00B1 (NCT00904241) study of 4,832 patients with newly diagnosed neuroblastoma, 52% of tumors were classified as favorable and 48% as unfavorable, according to the International Neuroblastoma Pathology Classification (INPC). For patients with tumors classified as favorable, the 5-year EFS rate was 88%, and the 5-year OS rate was 96%. For patients with tumors classified as unfavorable, the 5-year EFS rate was 55%, and the 5-year OS rate was 66% (P < .0001).[ 78 ]
Histological characteristics considered prognostically favorable include the following:
High mitosis/karyorrhexis index and undifferentiated tumor cells are considered prognostically unfavorable histological characteristics, but the prognostic value is age dependent.[ 100 ][ 101 ]
A COG study (P9641 [NCT00003119]) investigated the effect of histology, among other factors, on outcome. Of 915 children with stage 1 and stage 2 neuroblastoma without MYCN amplification, 87% were treated with initial surgery and observation. Patients (13%) who had or were at risk of developing symptomatic disease, or who had less than 50% tumor resection at diagnosis, or who had unresectable progressive disease after surgery alone, were treated with chemotherapy and surgery. Those with favorable histological features reported a 5-year EFS rate of 90% to 94% and an OS rate of 99% to 100%. Those with unfavorable histology had an EFS rate of 80% to 86% and an OS rate of 89% to 93%.[ 79 ]
In the COG ANBL0531 (NCT00499616) study for intermediate-risk patients with neuroblastoma, treatment was assigned using a biology-based and response-based algorithm, which included allelic status of 1p36 and 11q23. Patients with MYCN-amplified tumors were excluded.[ 102 ]
A study using data from the INRG Data Commons evaluated the prognostic strength of the underlying INPC histological criteria. The independent prognostic ability of age, histological category, mitosis-karyorrhexis index (MKI), and grade was demonstrated. Four age-related, histological prognostic groups were identified (aged <18 months with low vs. high MKI, and aged ≥18 months with differentiated vs. undifferentiated/poorly differentiated tumors). Compared with survival trees generated with established COG risk criteria, an additional prognostic subgroup was identified and validated when individual histological features were analyzed in lieu of INPC.[ 104 ] The INPC is described in the Histological Classification of Neuroblastic Tumors section.
Biological features
For more information, see the Genomic and Biological Features of Neuroblastoma section.
Site of primary tumor
Clinical and biological features of neuroblastoma differ by primary tumor site. In a study of data on 8,389 patients in clinical trials and compiled by the International Risk Group Project, the following results were observed, confirming the results from much smaller, previous studies with less complete clinical and biological data:[ 105 ]
Using the Therapeutically Applicable Research to Generate Effect Treatments (TARGET) and genome-wide association study data sets, a study compared the genomic and epigenomic data of primary diagnostic neuroblastomas originating in the adrenal gland (n = 646) with that of neuroblastomas originating in the thoracic sympathetic ganglia (n = 118). Neuroblastomas arising in the adrenal gland were more likely to harbor structural DNA aberrations such as MYCN amplification, whereas thoracic tumors showed defects in mitotic checkpoints resulting in hyperdiploidy. Thoracic tumors were more likely to harbor gain-of-function ALK aberrations than were adrenal tumors among all cases (OR, 1.89; P = .04), and among cases without MYCN amplification (OR, 2.86; P = .003). Because 16% of thoracic tumors harbor ALK variants, routine sequencing for these variants in this setting should be considered.[ 106 ]
In the TARGET cohort, 70% of patients with adrenal primary tumors and 51% of patients with thoracic primary tumors had stage 4 disease. In the genome-wide association study without MYCN amplification, 43% of patients with adrenal primary tumors and 17% of patients with thoracic primary tumors had stage 4 disease. By multivariate analysis, adrenal site was an independent predictor of worse outcome in the genome-wide association study cohort but not in the TARGET cohort after adjusting for MYCN amplification status, disease stage, and age of at least 18 months. Adrenal neuroblastoma was not an independent predictor of worse EFS by similar multivariable analysis for either the genome-wide association study or TARGET cohorts.[ 106 ]
It is not clear whether the effect of primary neuroblastoma tumor site on prognosis is entirely dependent on the differences in tumor biology associated with tumor site.
Multifocal neuroblastoma occurs rarely, usually in infants, and generally has a good prognosis.[ 107 ] Familial neuroblastoma and germline ALK gene pathogenic variants should be considered in patients with multiple primary neuroblastomas.
Response to treatment
Response to treatment has been associated with outcome. In patients with intermediate-risk disease who had a poor response to initial therapy in the COG ANBL0531 (NCT00499616) study, 6 of 20 patients subsequently developed progressive or recurrent disease, and one patient died.[ 102 ]
In patients with high-risk disease, the persistence of neuroblastoma cells in bone marrow after induction chemotherapy is associated with a poor prognosis. Sensitive techniques that detect minimal residual disease may be used to assess prognosis.[ 108 ][ 109 ][ 110 ] For example, detection of RNA transcripts expressed by neuroblastoma cells (in the bone marrow) after initial induction chemotherapy in children with high-risk neuroblastoma has been associated with significantly inferior EFS and OS.[ 111 ]
Similarly, the persistence of MIBG-avid tumor, measured as Curie score greater than 2 after completion of induction therapy, predicts a poor prognosis for patients with MYCN-nonamplified high-risk tumors. A Curie score greater than 0 after induction therapy is associated with a worse outcome for high-risk patients with MYCN-amplified disease.[ 112 ][ 113 ] An analysis of North American patients who went on to receive tandem transplants showed that patients with Curie scores greater than 0 at the end of induction therapy had inferior EFS rates.[ 114 ] For more information about Curie scoring, see the Curie and SIOPEN scoring methods section.
In an analysis of patients from four consecutive COG high-risk trials, an end-induction response of partial response (PR) or better, according to the 1993 International Neuroblastoma Response Criteria,[ 64 ] was significantly associated with higher EFS and OS. On multivariable analysis (n = 407), the absence of 11q loss of heterozygosity (LOH) was the only factor that remained significantly associated with PR or better (OR, 1.962 vs. 11q LOH; 95% CI, 1.104–3.487; P = .0216).[ 115 ]
A treatment-associated decrease in mitosis and an increase in histological differentiation of the primary tumor are also prognostic of response.[ 116 ]
The accuracy of prognostication based on decrease in primary tumor size is less clear. In a study conducted by seven large international centers, 229 high-risk patients were treated in a variety of ways. Treatment included chemotherapy, surgical removal of the primary tumor, radiation to the tumor bed, high-dose myeloablative therapy plus stem cell transplant, and, in most cases, isotretinoin and anti-GD2 antibody immunotherapy enhanced by cytokines. Primary tumor response was measured after induction chemotherapy in three ways: as 30% or greater reduction in the longest dimension, 50% or greater reduction in tumor volume, or 65% or greater reduction in tumor volume (calculated from three tumor dimensions, a conventional radiological technique). The measurements were performed at diagnosis and after induction chemotherapy before primary tumor resection. None of the methods of measuring primary tumor response at end of induction chemotherapy predicted survival.[ 117 ]
Levels of LDH and ferritin
Higher serum LDH and ferritin values conferred worse 5-year EFS and OS rates in a large international cohort of patients diagnosed with neuroblastoma (n > 8,575) from 1990 to 2016. Higher serum values for LDH and ferritin also conferred worse 3-year EFS and OS rates in patients with high-risk neuroblastoma after 2009. In a multivariate analysis that adjusted for age at diagnosis, MYCN status, and INSS stage 4 disease, LDH and ferritin maintained independent prognostic ability (P < .0001).[ 118 ][Level of evidence C1]
Although not critically evaluated in the original INRG classification system, subsequent analysis of the INRG Data Commons has clearly demonstrated independent statistical significance of the levels of serum ferritin and LDH on prognosis in all patients and in high-risk patients, including in the time period between 2010 and 2016. Therefore, it was suggested that these two easily obtainable lab values be incorporated into the prognostic classification system of the INRG.[ 118 ]
Treatment era
Between 1975 and 2020, the 5-year survival rate for neuroblastoma in the United States increased from 86% to 93% for children younger than 1 year and from 34% to 83% for children aged 1 to 14 years.[ 2 ][ 3 ] The 5-year relative survival rate for all infants and children with neuroblastoma increased from 46% when diagnosed between 1974 and 1989 to 71% when diagnosed between 1999 and 2005.[ 119 ] More recent estimates from 2014 to 2020 show an even higher 5-year relative survival rate of approximately 85% for infants and children younger than 15 years.[ 2 ] These statistics can be misleading because of the extremely heterogeneous prognosis based on the patient's age, stage, and biology. However, studies demonstrate a significant improvement in survival for high-risk patients diagnosed and treated between 2000 and 2019, compared with patients diagnosed from 1990 to 1999.[ 120 ][ 121 ] For more information, see Table 1. Similarly, the COG ANBL0531 (NCT00499616) study found equivalent outcomes for many subsets of intermediate-risk children who were treated with substantially reduced chemotherapy, compared with the earlier COG-A3961 (NCT00003093) study.[ 102 ]
参考文献- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.[PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.[PUBMED Abstract]
- Gurney JG, Ross JA, Wall DA, et al.: Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992. J Pediatr Hematol Oncol 19 (5): 428-32, 1997 Sep-Oct.[PUBMED Abstract]
- United States Census Bureau: Age and Sex Composition in the United States: 2018. U.S. Census Bureau, 2018. Available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Mahapatra S, Challagundla KB: Neuroblastoma. Treasure Island, FL: StatPearls Publishing LLC, 2022. Available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Campbell K, Siegel DA, Umaretiya PJ, et al.: A comprehensive analysis of neuroblastoma incidence, survival, and racial and ethnic disparities from 2001 to 2019. Pediatr Blood Cancer 71 (1): e30732, 2024.[PUBMED Abstract]
- London WB, Castleberry RP, Matthay KK, et al.: Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol 23 (27): 6459-65, 2005.[PUBMED Abstract]
- Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012. Also available online. Last accessed May 22, 2024.[PUBMED Abstract]
- Takeuchi LA, Hachitanda Y, Woods WG, et al.: Screening for neuroblastoma in North America. Preliminary results of a pathology review from the Quebec Project. Cancer 76 (11): 2363-71, 1995.[PUBMED Abstract]
- Woods WG, Gao RN, Shuster JJ, et al.: Screening of infants and mortality due to neuroblastoma. N Engl J Med 346 (14): 1041-6, 2002.[PUBMED Abstract]
- Schilling FH, Spix C, Berthold F, et al.: Neuroblastoma screening at one year of age. N Engl J Med 346 (14): 1047-53, 2002.[PUBMED Abstract]
- Heck JE, Ritz B, Hung RJ, et al.: The epidemiology of neuroblastoma: a review. Paediatr Perinat Epidemiol 23 (2): 125-43, 2009.[PUBMED Abstract]
- Mossé YP, Laudenslager M, Longo L, et al.: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455 (7215): 930-5, 2008.[PUBMED Abstract]
- Mosse YP, Laudenslager M, Khazi D, et al.: Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 75 (4): 727-30, 2004.[PUBMED Abstract]
- Raabe EH, Laudenslager M, Winter C, et al.: Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 27 (4): 469-76, 2008.[PUBMED Abstract]
- van Limpt V, Schramm A, van Lakeman A, et al.: The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 23 (57): 9280-8, 2004.[PUBMED Abstract]
- Serra A, Häberle B, König IR, et al.: Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol 30 (10): 728-32, 2008.[PUBMED Abstract]
- Satgé D, Moore SW, Stiller CA, et al.: Abnormal constitutional karyotypes in patients with neuroblastoma: a report of four new cases and review of 47 others in the literature. Cancer Genet Cytogenet 147 (2): 89-98, 2003.[PUBMED Abstract]
- Mosse Y, Greshock J, King A, et al.: Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol 4 (12): 769-71, 2003.[PUBMED Abstract]
- Gillani R, Collins RL, Crowdis J, et al.: Rare germline structural variants increase risk for pediatric solid tumors. Science 387 (6729): eadq0071, 2025.[PUBMED Abstract]
- Moroni I, Bedeschi F, Luksch R, et al.: Costello syndrome: a cancer predisposing syndrome? Clin Dysmorphol 9 (4): 265-8, 2000.[PUBMED Abstract]
- Cotton JL, Williams RG: Noonan syndrome and neuroblastoma. Arch Pediatr Adolesc Med 149 (11): 1280-1, 1995.[PUBMED Abstract]
- Gutmann DH, Ferner RE, Listernick RH, et al.: Neurofibromatosis type 1. Nat Rev Dis Primers 3: 17004, 2017.[PUBMED Abstract]
- Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.[PUBMED Abstract]
- Bougnères P, Pantalone L, Linglart A, et al.: Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab 93 (10): 3971-80, 2008.[PUBMED Abstract]
- Maas SM, Vansenne F, Kadouch DJ, et al.: Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 170 (9): 2248-60, 2016.[PUBMED Abstract]
- Witkowski L, Nichols KE, Jongmans M, et al.: Germline pathogenic SMARCA4 variants in neuroblastoma. J Med Genet 60 (10): 987-992, 2023.[PUBMED Abstract]
- Kim J, Vaksman Z, Egolf LE, et al.: Germline pathogenic variants in neuroblastoma patients are enriched in BARD1 and predict worse survival. J Natl Cancer Inst 116 (1): 149-159, 2024.[PUBMED Abstract]
- Seo ES, Lee JW, Lim J, et al.: Germline functional variants contribute to somatic mutation and outcomes in neuroblastoma. Nat Commun 15 (1): 8360, 2024.[PUBMED Abstract]
- Tolbert VP, Coggins GE, Maris JM: Genetic susceptibility to neuroblastoma. Curr Opin Genet Dev 42: 81-90, 2017.[PUBMED Abstract]
- Matser YAH, Verly IRN, van der Ham M, et al.: Optimising urinary catecholamine metabolite diagnostics for neuroblastoma. Pediatr Blood Cancer 70 (6): e30289, 2023.[PUBMED Abstract]
- Kratz CP, Rapisuwon S, Reed H, et al.: Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 157 (2): 83-9, 2011.[PUBMED Abstract]
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.[PUBMED Abstract]
- Citak C, Karadeniz C, Dalgic B, et al.: Intestinal lymphangiectasia as a first manifestation of neuroblastoma. Pediatr Blood Cancer 46 (1): 105-7, 2006.[PUBMED Abstract]
- Bourdeaut F, de Carli E, Timsit S, et al.: VIP hypersecretion as primary or secondary syndrome in neuroblastoma: A retrospective study by the Société Française des Cancers de l'Enfant (SFCE). Pediatr Blood Cancer 52 (5): 585-90, 2009.[PUBMED Abstract]
- Mahoney NR, Liu GT, Menacker SJ, et al.: Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol 142 (4): 651-9, 2006.[PUBMED Abstract]
- Conte M, Parodi S, De Bernardi B, et al.: Neuroblastoma in adolescents: the Italian experience. Cancer 106 (6): 1409-17, 2006.[PUBMED Abstract]
- Matthay KK, Blaes F, Hero B, et al.: Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer Lett 228 (1-2): 275-82, 2005.[PUBMED Abstract]
- Rudnick E, Khakoo Y, Antunes NL, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children's Cancer Group Study. Med Pediatr Oncol 36 (6): 612-22, 2001.[PUBMED Abstract]
- Antunes NL, Khakoo Y, Matthay KK, et al.: Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol 22 (4): 315-20, 2000 Jul-Aug.[PUBMED Abstract]
- Pang KK, de Sousa C, Lang B, et al.: A prospective study of the presentation and management of dancing eye syndrome/opsoclonus-myoclonus syndrome in the United Kingdom. Eur J Paediatr Neurol 14 (2): 156-61, 2010.[PUBMED Abstract]
- Pranzatelli MR: The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol 15 (3): 186-228, 1992.[PUBMED Abstract]
- Mitchell WG, Davalos-Gonzalez Y, Brumm VL, et al.: Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and neurologic sequelae. Pediatrics 109 (1): 86-98, 2002.[PUBMED Abstract]
- Cooper R, Khakoo Y, Matthay KK, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: histopathologic features-a report from the Children's Cancer Group. Med Pediatr Oncol 36 (6): 623-9, 2001.[PUBMED Abstract]
- Pranzatelli MR, Tate ED, McGee NR: Demographic, Clinical, and Immunologic Features of 389 Children with Opsoclonus-Myoclonus Syndrome: A Cross-sectional Study. Front Neurol 8: 468, 2017.[PUBMED Abstract]
- Hero B, Clement N, Øra I, et al.: Genomic Profiles of Neuroblastoma Associated With Opsoclonus Myoclonus Syndrome. J Pediatr Hematol Oncol 40 (2): 93-98, 2018.[PUBMED Abstract]
- Catsman-Berrevoets CE, Aarsen FK, van Hemsbergen ML, et al.: Improvement of neurological status and quality of life in children with opsoclonus myoclonus syndrome at long-term follow-up. Pediatr Blood Cancer 53 (6): 1048-53, 2009.[PUBMED Abstract]
- Connolly AM, Pestronk A, Mehta S, et al.: Serum autoantibodies in childhood opsoclonus-myoclonus syndrome: an analysis of antigenic targets in neural tissues. J Pediatr 130 (6): 878-84, 1997.[PUBMED Abstract]
- Bell J, Moran C, Blatt J: Response to rituximab in a child with neuroblastoma and opsoclonus-myoclonus. Pediatr Blood Cancer 50 (2): 370-1, 2008.[PUBMED Abstract]
- Corapcioglu F, Mutlu H, Kara B, et al.: Response to rituximab and prednisolone for opsoclonus-myoclonus-ataxia syndrome in a child with ganglioneuroblastoma. Pediatr Hematol Oncol 25 (8): 756-61, 2008.[PUBMED Abstract]
- Pranzatelli MR, Tate ED, Travelstead AL, et al.: Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol 28 (9): 585-93, 2006.[PUBMED Abstract]
- Ertle F, Behnisch W, Al Mulla NA, et al.: Treatment of neuroblastoma-related opsoclonus-myoclonus-ataxia syndrome with high-dose dexamethasone pulses. Pediatr Blood Cancer 50 (3): 683-7, 2008.[PUBMED Abstract]
- Pranzatelli MR, Tate ED: Dexamethasone, Intravenous Immunoglobulin, and Rituximab Combination Immunotherapy for Pediatric Opsoclonus-Myoclonus Syndrome. Pediatr Neurol 73: 48-56, 2017.[PUBMED Abstract]
- de Alarcon PA, Matthay KK, London WB, et al.: Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Health 2 (1): 25-34, 2018.[PUBMED Abstract]
- Kumar P, Willard VW, Embry L, et al.: Late cognitive and adaptive outcomes of patients with neuroblastoma-associated opsoclonus-myoclonus-ataxia-syndrome: A report from the Children's Oncology Group. Pediatr Blood Cancer 71 (7): e31039, 2024.[PUBMED Abstract]
- Vik TA, Pfluger T, Kadota R, et al.: (123)I-mIBG scintigraphy in patients with known or suspected neuroblastoma: Results from a prospective multicenter trial. Pediatr Blood Cancer 52 (7): 784-90, 2009.[PUBMED Abstract]
- Yang J, Codreanu I, Servaes S, et al.: I-131 MIBG post-therapy scan is more sensitive than I-123 MIBG pretherapy scan in the evaluation of metastatic neuroblastoma. Nucl Med Commun 33 (11): 1134-7, 2012.[PUBMED Abstract]
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.[PUBMED Abstract]
- Pio L, Brisse HJ, Alaggio R, et al.: Image-guided core-needle or surgical biopsy for neuroblastoma diagnosis in children: A systematic review and meta-analysis from the International Society of Pediatric Surgical Oncology (IPSO). Pediatr Blood Cancer 71 (2): e30789, 2024.[PUBMED Abstract]
- Schoeman S, Bagatell R, Cahill AM, et al.: Percutaneous biopsy for the diagnosis, risk stratification, and molecular profiling of neuroblastoma: A single-center retrospective study. Pediatr Blood Cancer 71 (4): e30887, 2024.[PUBMED Abstract]
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.[PUBMED Abstract]
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.[PUBMED Abstract]
- Jennings RW, LaQuaglia MP, Leong K, et al.: Fetal neuroblastoma: prenatal diagnosis and natural history. J Pediatr Surg 28 (9): 1168-74, 1993.[PUBMED Abstract]
- Brodeur GM: Spontaneous regression of neuroblastoma. Cell Tissue Res 372 (2): 277-286, 2018.[PUBMED Abstract]
- Guan J, Hallberg B, Palmer RH: Chromosome Imbalances in Neuroblastoma-Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers (Basel) 13 (23): , 2021.[PUBMED Abstract]
- Schneiderman J, London WB, Brodeur GM, et al.: Clinical significance of MYCN amplification and ploidy in favorable-stage neuroblastoma: a report from the Children's Oncology Group. J Clin Oncol 26 (6): 913-8, 2008.[PUBMED Abstract]
- Hiyama E, Hiyama K, Yokoyama T, et al.: Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1 (3): 249-55, 1995.[PUBMED Abstract]
- Kitanaka C, Kato K, Ijiri R, et al.: Increased Ras expression and caspase-independent neuroblastoma cell death: possible mechanism of spontaneous neuroblastoma regression. J Natl Cancer Inst 94 (5): 358-68, 2002.[PUBMED Abstract]
- Brodeur GM, Minturn JE, Ho R, et al.: Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res 15 (10): 3244-50, 2009.[PUBMED Abstract]
- Yamamoto K, Ohta S, Ito E, et al.: Marginal decrease in mortality and marked increase in incidence as a result of neuroblastoma screening at 6 months of age: cohort study in seven prefectures in Japan. J Clin Oncol 20 (5): 1209-14, 2002.[PUBMED Abstract]
- Okazaki T, Kohno S, Mimaya J, et al.: Neuroblastoma detected by mass screening: the Tumor Board's role in its treatment. Pediatr Surg Int 20 (1): 27-32, 2004.[PUBMED Abstract]
- Fritsch P, Kerbl R, Lackner H, et al.: "Wait and see" strategy in localized neuroblastoma in infants: an option not only for cases detected by mass screening. Pediatr Blood Cancer 43 (6): 679-82, 2004.[PUBMED Abstract]
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.[PUBMED Abstract]
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.[PUBMED Abstract]
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.[PUBMED Abstract]
- McCarthy LC, Chastain K, Flatt TG, et al.: Neuroblastoma in Adolescents and Children Older than 10 Years: Unusual Clinicopathologic and Biologic Features. J Pediatr Hematol Oncol 41 (8): 586-595, 2019.[PUBMED Abstract]
- Mazzocco K, Defferrari R, Sementa AR, et al.: Genetic abnormalities in adolescents and young adults with neuroblastoma: A report from the Italian Neuroblastoma group. Pediatr Blood Cancer 62 (10): 1725-32, 2015.[PUBMED Abstract]
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.[PUBMED Abstract]
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.[PUBMED Abstract]
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.[PUBMED Abstract]
- Mossé YP, Deyell RJ, Berthold F, et al.: Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer 61 (4): 627-35, 2014.[PUBMED Abstract]
- Kushner BH, Kramer K, LaQuaglia MP, et al.: Neuroblastoma in adolescents and adults: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 41 (6): 508-15, 2003.[PUBMED Abstract]
- Suzuki M, Kushner BH, Kramer K, et al.: Treatment and outcome of adult-onset neuroblastoma. Int J Cancer 143 (5): 1249-1258, 2018.[PUBMED Abstract]
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.[PUBMED Abstract]
- Goldsmith KC, Park JR, Kayser K, et al.: Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med 29 (5): 1092-1102, 2023.[PUBMED Abstract]
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.[PUBMED Abstract]
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.[PUBMED Abstract]
- Campbell K, Gastier-Foster JM, Mann M, et al.: Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children's Oncology Group. Cancer 123 (21): 4224-4235, 2017.[PUBMED Abstract]
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.[PUBMED Abstract]
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.[PUBMED Abstract]
- Kubota M, Suita S, Tajiri T, et al.: Analysis of the prognostic factors relating to better clinical outcome in ganglioneuroblastoma. J Pediatr Surg 35 (1): 92-5, 2000.[PUBMED Abstract]
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.[PUBMED Abstract]
- Isaacs H: Fetal and neonatal neuroblastoma: retrospective review of 271 cases. Fetal Pediatr Pathol 26 (4): 177-84, 2007 Jul-Aug.[PUBMED Abstract]
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.[PUBMED Abstract]
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.[PUBMED Abstract]
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.[PUBMED Abstract]
- Vo KT, Matthay KK, Neuhaus J, et al.: Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol 32 (28): 3169-76, 2014.[PUBMED Abstract]
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.[PUBMED Abstract]
- Hiyama E, Yokoyama T, Hiyama K, et al.: Multifocal neuroblastoma: biologic behavior and surgical aspects. Cancer 88 (8): 1955-63, 2000.[PUBMED Abstract]
- Burchill SA, Lewis IJ, Abrams KR, et al.: Circulating neuroblastoma cells detected by reverse transcriptase polymerase chain reaction for tyrosine hydroxylase mRNA are an independent poor prognostic indicator in stage 4 neuroblastoma in children over 1 year. J Clin Oncol 19 (6): 1795-801, 2001.[PUBMED Abstract]
- Seeger RC, Reynolds CP, Gallego R, et al.: Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 18 (24): 4067-76, 2000.[PUBMED Abstract]
- Bochennek K, Esser R, Lehrnbecher T, et al.: Impact of minimal residual disease detection prior to autologous stem cell transplantation for post-transplant outcome in high risk neuroblastoma. Klin Padiatr 224 (3): 139-42, 2012.[PUBMED Abstract]
- van Zogchel LMJ, Decarolis B, van Wezel EM, et al.: Sensitive liquid biopsy monitoring correlates with outcome in the prospective international GPOH-DCOG high-risk neuroblastoma RT-qPCR validation study. J Exp Clin Cancer Res 43 (1): 331, 2024.[PUBMED Abstract]
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.[PUBMED Abstract]
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.[PUBMED Abstract]
- Streby KA, Parisi MT, Shulkin BL, et al.: Impact of diagnostic and end-of-induction Curie scores with tandem high-dose chemotherapy and autologous transplants for metastatic high-risk neuroblastoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 70 (8): e30418, 2023.[PUBMED Abstract]
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.[PUBMED Abstract]
- George RE, Perez-Atayde AR, Yao X, et al.: Tumor histology during induction therapy in patients with high-risk neuroblastoma. Pediatr Blood Cancer 59 (3): 506-10, 2012.[PUBMED Abstract]
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.[PUBMED Abstract]
- Moroz V, Machin D, Hero B, et al.: The prognostic strength of serum LDH and serum ferritin in children with neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 67 (8): e28359, 2020.[PUBMED Abstract]
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.[PUBMED Abstract]
- Bagatell R, DuBois SG, Naranjo A, et al.: Children's Oncology Group's 2023 blueprint for research: Neuroblastoma. Pediatr Blood Cancer 70 Suppl 6 (Suppl 6): e30572, 2023.[PUBMED Abstract]
- Histological Classification of Neuroblastic Tumors
-
Neuroblastomas are classified as one of the small round blue cell tumors of childhood. They are a heterogenous group of tumors composed of cellular aggregates with varying degrees of differentiation, from mature ganglioneuromas to less-mature ganglioneuroblastomas to immature neuroblastomas. These differences reflect the varying malignant potential of these tumors.[ 1 ]
International Neuroblastoma Pathology Classification (INPC) System
The INPC system was derived from the experience with the original Shimada classification, and the two systems are compared in Table 1. The INPC involves histological evaluation of tumor specimens obtained before therapy for the following morphological features:[ 2 ][ 3 ][ 4 ][ 5 ][ 6 ]
Favorable and unfavorable prognoses are defined based on these histological parameters and patient age. The prognostic significance of this classification system, and of related systems using similar criteria, has been confirmed in several studies (see Table 1).[ 2 ][ 3 ][ 4 ][ 6 ]
Table 1. Prognostic Evaluation of Neuroblastic Tumors According to the International Neuroblastoma Pathology Classification (Shimada System)a International Neuroblastoma Pathology Classification Original Shimada Classification Prognostic Group MKI = mitosis-karyorrhexis index. aReprinted with permission. Copyright © 1999 American Cancer Society. All rights reserved.[ 2 ] Hiroyuki Shimada, Inge M. Ambros, Louis P. Dehner, Jun-ichi Hata, Vijay V. Joshi, Borghild Roald, Daniel O. Stram, Robert B. Gerbing, John N. Lukens, Katherine K. Matthay, Robert P. Castleberry, The International Neuroblastoma Pathology Classification (the Shimada System), Cancer, volume 86, issue 2, pages 364–72. bSubtypes of neuroblastoma are described in detail elsewhere.[ 7 ] cRare subtype, especially diagnosed in this age group. Further investigation and analysis required. dPrognostic grouping for these tumor categories is not related to patient age. Neuroblastoma: (Schwannian stroma-poor)b Stroma-poor Favorable: Favorable Favorable <1.5 y Poorly differentiated or differentiating & low or intermediate MKI tumor 1.5–5 y Differentiating & low MKI tumor Unfavorable: Unfavorable Unfavorable <1.5 y a) undifferentiated tumorc b) high MKI tumor 1.5–5 y a) undifferentiated or poorly differentiated tumor b) intermediate or high MKI tumor ≥5 y All tumors Ganglioneuroblastoma, intermixed (Schwannian stroma-rich) Stroma-rich intermixed (favorable) Favorabled Ganglioneuroma: (Schwannian stroma-dominant) Maturing Well differentiated (favorable) Favorabled Mature Ganglioneuroma Ganglioneuroblastoma, nodular (composite Schwannian stroma-rich/stroma-dominate and stroma-poor) Stroma-rich nodular (unfavorable) Unfavorabled Most neuroblastomas with MYCN amplification have unfavorable INPC histology, but about 7% of tumors have favorable histology. The tumors generally do not express MYCN, even with the gene being amplified, and these patients have a more favorable prognosis than patients whose tumors are MYCN amplified and overexpress MYCN.[ 8 ]
The individual components of INPC data from the INRG Data Commons (18,865 patients) were analyzed, and the analysis validated the independent prognostic ability of age at diagnosis, histological category, MKI, and grade of differentiation. Four histological prognostic groups of patients were identified (aged <18 months with low vs. high MKI; aged >18 months with differentiating vs. undifferentiating/poorly differentiating tumors). Also, by using a risk schema devoid of the confounding of age and INPC, this analysis identified a novel and unfavorable subgroup of patients older than 547 days with stage 1 or 2, MYCN-nonamplified, intermediate or high MKI diploid tumors who had a very poor event-free survival (EFS) rate of 46%.[ 9 ][Level of evidence C1]
In some cases, biopsy may not be fully representative of the type of neuroblastic tumor present. For example, in one study of 125 patients with a biopsy diagnosis of ganglioneuroma or ganglioneuroblastoma, intermixed went on to undergo surgical resections. The pathological diagnosis changed in 39% of the cases, including 14 cases (12%) in which pathology changed to neuroblastoma or ganglioneuroblastoma, nodular.[ 10 ]
参考文献- Joshi VV, Silverman JF: Pathology of neuroblastic tumors. Semin Diagn Pathol 11 (2): 107-17, 1994.[PUBMED Abstract]
- Shimada H, Ambros IM, Dehner LP, et al.: The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86 (2): 364-72, 1999.[PUBMED Abstract]
- Shimada H, Umehara S, Monobe Y, et al.: International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (9): 2451-61, 2001.[PUBMED Abstract]
- Goto S, Umehara S, Gerbing RB, et al.: Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (10): 2699-708, 2001.[PUBMED Abstract]
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.[PUBMED Abstract]
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.[PUBMED Abstract]
- Shimada H, Ambros IM, Dehner LP, et al.: Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer 86 (2): 349-63, 1999.[PUBMED Abstract]
- Suganuma R, Wang LL, Sano H, et al.: Peripheral neuroblastic tumors with genotype-phenotype discordance: a report from the Children's Oncology Group and the International Neuroblastoma Pathology Committee. Pediatr Blood Cancer 60 (3): 363-70, 2013.[PUBMED Abstract]
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.[PUBMED Abstract]
- Burnand KM, Neville J, Budzanowski A, et al.: Management of Ganglioneuroma and Ganglioneuroblastoma Intermixed: A United Kingdom Children's Cancer and Leukaemia Group (UK CCLG) Nationwide Study Report. Pediatr Blood Cancer 72 (2): e31445, 2025.[PUBMED Abstract]
- Neuroblastoma Staging and Risk Classification Systems
-
International Neuroblastoma Staging System (INSS)
The INSS was developed and adopted by the Children's Oncology Group (COG) in 1986 and by cooperative groups in Europe and Japan in 1993. The INSS is a postsurgical staging system that uses tumor location with respect to midline structures, lymph node status, and, importantly, extent of upfront surgical resection to determine whether a locoregional tumor is INSS stage 1, 2A, 2B, or 3.[ 1 ][ 2 ] This system represented the first step in harmonizing disease staging and risk stratification worldwide. As a result of further advances in the understanding of neuroblastoma biology and genetics, a risk classification system was developed that incorporates clinical and biological factors in addition to INSS stage to facilitate risk group and treatment assignment for COG studies.[ 1 ][ 2 ][ 3 ][ 4 ] The final use of the INSS by the COG was for the intermediate-risk ANBL0531 (NCT00499616) study, which was closed in 2014.
International Neuroblastoma Risk Group Staging System (INRGSS)
To create a staging system independent of surgical resection extent, the INRGSS was developed in 2005 using image-defined risk factors (IDRFs) to categorize locoregional tumors as L1 (IDRFs absent), L2 (IDRFs present), M (metastatic), or MS (the equivalent of 4S in the INSS). For example, in the case of spinal cord compression, an IDRF is present when more than one-third of the spinal canal in the axial plane is invaded, when the leptomeningeal spaces are not visible, or when the spinal cord magnetic resonance signal intensity is abnormal. For more information about the INRGSS, see Table 2 and the lists of IDRFs (original IDRFs and COG IDRFs).
Presence of IDRFs has been associated with an increase in intraoperative complications, incomplete tumor resection, and worse survival in numerous studies.[ 5 ][ 6 ][ 7 ] Since 2014, COG and International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) clinical trials have used the INRGSS, a preoperative staging system that was developed specifically for the International Neuroblastoma Risk Group (INRG) classification system (see Table 2), in place of the INSS.
Table 2. International Neuroblastoma Risk Group Staging Systema Stage Description IDRFs = image-defined risk factors; INSS = International Neuroblastoma Staging System. aAdapted from Monclair et al.[ 5 ]; [ 6 ] L1 Localized tumor not involving vital structures as defined by the list of IDRFsa and confined to one body compartment. L2 Locoregional tumor with presence of one or more IDRFs.a M Distant metastatic disease (except stage MS). MS Metastatic disease in children younger than 18 months with metastases confined to skin, liver, and/or bone marrow. The primary tumor can be INSS stage 1, 2, or 3. IDRFs, as defined in the original literature, include the following:[ 5 ][ 7 ]
COG IDRFs, using an anatomical localization approach, include the following:[ 6 ][ 8 ]; [ 7 ][Level of evidence C1]
Assessment of surgical resectability must include IDRFs. The more IDRFs present, the higher the morbidity of the operation and the lower the chance of complete resection. The presence of two or more IDRFs should prompt a discussion regarding up-front chemotherapy rather than surgical resection at diagnosis. To decrease morbidity, it is critical to avoid up-front surgical resection with invasive tumors. An international analysis demonstrated that specific IDRFs present at diagnosis or before surgery may be associated with a lower likelihood of achieving a greater than 90% resection of the primary tumor.[ 9 ] Concordance in assessing IDRFs between local investigators and central reviewers was assessed in the context of a COG intermediate-risk trial and showed agreement in only 51.9% of cases.[ 10 ]
Neoadjuvant chemotherapy is not always effective in eliminating IDRFs. A retrospective study in the European Unresectable Neuroblastoma trial from 2001 to 2006 examined data from 143 patients with INSS stage 3 neuroblastoma who were older than 1 year without MYCN amplification. All patients had surgical risk factors that deemed the tumors unresectable. In a centrally reviewed subset, unfavorable histology by International Neuroblastoma Pathology Classification was found in 53% of patients. At diagnosis, 228 IDRFs were identified.[ 8 ]; [ 11 ][Level of evidence C1]
The INRGSS staging system is one of the prognostic markers included in the INRG Classification System.[ 12 ] For more information, see Table 4.
The INRGSS includes four disease stages: L1, L2, M, or MS. Localized tumors are classified as stage L1 or L2 disease based on whether one or more of the 20 IDRFs are present.[ 5 ]
The INRG Task Force has also reported consensus techniques for detecting and quantifying neuroblastoma in bone marrow, both at diagnosis and after treatment. Quantification of bone marrow metastatic disease may result in more accurate assessment of response to treatment,[ 13 ] and it is now incorporated into the International Neuroblastoma Response Criteria, which assess response to therapy.[ 14 ]
The decision by the INRG Task Force to define MS disease differently from 4S disease was based on reports in which small numbers of infants with L2 primary tumors and 4S metastatic patterns, including those aged 12 to 18 months, had favorable outcomes.[ 5 ][ 15 ] A subsequent study analyzing INRG data demonstrated that a number of biological characteristics predicted poor outcome for patients with MS disease who were aged 12 to 18 months at diagnosis. However, long-term outcomes of toddlers, aged 12 to 18 months, with favorable-biology MS disease were similar to those of infants younger than 12 months with INSS stage 4S neuroblastoma.[ 15 ]
By combining the INRGSS, age, and biological factors, each patient is assigned an INRG risk group that is prognostic of outcome and guides the appropriate risk-based treatment approach. The validity of the INRGSS was explored in the following retrospective studies of localized neuroblastoma with previously defined INSS stage without MYCN amplification:
Most international protocols have begun to incorporate the collection and use of IDRFs to define INRG stage, which is used in risk stratification and assignment of therapy.[ 17 ][ 18 ] The COG has been collecting and evaluating INRGSS data since 2006. A COG trial that opened in 2014 uses the INRGSS along with input from the surgeon to determine therapy for subsets of patients not at high risk, including those with L1, L2, and MS disease (ANBL1232 [NCT02176967], closed to accrual). Note that the INSS allows patients up to age 12 months to be classified as stage 4S, while the INRGSS allows patients up to age 18 months to be staged as MS. The primary tumor in INSS stage 4S must be INSS stage 1 or 2, while the primary tumor in MS can be L1 or L2, which includes INSS stages 1, 2, or 3. The INRGSS is used in ongoing COG studies and does not depend on a resection variable, but rather on pretreatment imaging combined with age and biological variables. It is anticipated that the use of standardized nomenclature will contribute substantially to more uniform staging and facilitate comparisons of clinical trials conducted in different parts of the world.
Children’s Oncology Group (COG) Neuroblastoma Risk Grouping
The COG ANBL00B1 (NCT00904241) biology study served as the infrastructure for rapid and reliable acquisition of the clinical and biological prognostic markers used for risk classification and clinical trial eligibility between 2000 and 2023. The APEC14B1 trial is currently used to facilitate risk group. For more information about the COG risk categories, see Table 3.
Based on data from 4,832 patients who were enrolled from 2007 to 2017 in the ANBL00B1 study, the COG has updated the risk classification.[ 19 ] Patients are defined as having low-, intermediate-, or high-risk disease based on clinical and biological factors (see Table 3).
Table 3. Risk Groups Used by the Children’s Oncology Group Committee High-Risk Disease 1. Stage M, aged ≥18 months, regardless of other features 2. Stage M, aged <18 months with MYCN-amplified disease 3. Stage MS or L2 with MYCN-amplified disease 4. Stage L2 with unfavorable histology, aged ≥18 months 5. Stage M or MS 12–18 months with at least one unfavorable feature: —Unfavorable histology — Segmental chromosomal aberrations —Diploid tumor 6. Stage L1 incompletely resected tumor with MYCN amplification Low-Risk Disease 1. Stage L1 with MYCN-nonamplified disease regardless of other features 2. Stage L1 completely resected with MYCN-amplified disease 3. Stage MS, aged <12 months with all favorable features: —Asymptomatic —Favorable histology —No segmental chromosomal aberrations —Hyperdiploid tumor Intermediate-Risk Disease All other groups not meeting the definition of high-risk or low-risk disease.a aFor complete classification, see Irwin MS et al.[ 19 ] International Neuroblastoma Risk Group (INRG)
Combinations of prognostic factors (clinical and biological features) have been used for decades to risk-stratify patients and inform treatment assignment.[ 12 ] Schema differ across international cooperative groups. The INRG Task Force has led efforts to develop uniform approaches for staging and pretreatment risk classification, as outlined below.[ 20 ] The algorithms that use these factors to determine risk are complex and change slightly based on new knowledge. The INRG Classification System was designed based on survival-tree analyses of 35 prognostic factors in more than 8,800 patients with neuroblastoma from a variety of clinical trials. The underlying histological features in the INPC (Shimada system) were included in the analysis:[ 20 ][ 21 ]
The INRG classification schema assigns neuroblastoma patients to one of 16 pretreatment risk groups based on INRG stage, age, histological category, grade of tumor differentiation, MYCN amplification, 11q aberration (the only segmental chromosomal aberration studied), and ploidy. Four levels of risk were defined according to outcomes among 8,800 patients with high-quality data, as they had been entered in clinical trials (see Table 4).
In the overall risk grouping, histological category is an important risk determinant for all stage L1 and L2 tumors, and grade of differentiation is prognostic in neuroblastomas and nodular ganglioneuroblastomas in patients older than 18 months. The goals of the INRG are to increase international collaboration and classify patients uniformly so that the results of clinical trials conducted around the world can be compared.[ 20 ]
Table 4. International Neuroblastoma Risk Group (INRG) Pretreatment Classification Schemaa INRG Stage Histological Category Grade of Tumor Differentiation 11q Aberration Ploidy Pretreatment Risk Group GN = ganglioneuroma; GNB = ganglioneuroblastoma; NA = not amplified. aReprinted with permission. © (2015) American Society of Clinical Oncology. All rights reserved. Pinto N et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma, J Clin Oncol 33 (27), 2015: 3008–3017.[ 12 ] L1/L2 GN maturing, GNB intermixed A (very low) L1 Any, except GN maturing or GNB intermixed NA B (very low) Amplified K (high) L2 Aged <18 mo Any, except GN maturing or GNB intermixed NA No D (low) Yes G (intermediate) Aged ≥18 mo GNB nodular neuroblastoma Differentiating NA No E (low) Yes H (intermediate) Poorly differentiated or undifferentiated NA H (intermediate) Amplified N (high) M Aged <18 mo NA Hyperdiploid F (low) Aged <12 mo NA Diploid I (intermediate) Aged 12 to <18 mo NA Diploid J (intermediate) Aged <18 mo Amplified O (high) Aged ≥18 mo P (high) MS Aged <18 mo NA No C (very low) Yes Q (high) Amplified R (high) Because patient age is used in all risk stratification systems, a cellular classification system that did not employ patient age was desirable, and underlying histological criteria, rather than INPC or Shimada Classification, was used in the survival-tree analyses to select prognostic criteria for the INRG Classification System. Histological findings discriminated prognostic groups most clearly in two subsets of patients, as shown in Table 5.
Table 5. Histological Discrimination of International Neuroblastoma Risk Group Subsets of Neuroblastoma Patientsa INSS Stage/Histological Subtype Number of Cases EFS (%) OS (%) EFS = event-free survival; GN = ganglioneuroma; GNB = ganglioneuroblastoma; INSS = International Neuroblastoma Staging System; NB = neuroblastoma; OS = overall survival. aAdapted from Cohn et al.[ 20 ] INSS stage 1, 2, 3, 4S 5,131 83 ± 1 91 ± 1 GN, maturing 162 97 ± 2 98 ± 2 GNB, intermixed NB 4,970 83 ± 1 90 ± 1 GNB, nodular INSS stage 2, 3; age >547 d 260 69 ± 3 81 ± 2 11q normal and differentiating 16 80 ± 16 100 11q aberration or undifferentiated 49 61 ± 11 73 ± 11 The INRG histological subsets are incorporated into the INRG Risk Classification Schema.
Evaluation of Primary Tumor and Metastatic Disease
Approximately 70% of patients with neuroblastoma have metastatic disease at diagnosis. A thorough evaluation for metastatic disease is performed before therapy initiation. The studies described below are typically performed.[ 1 ]
Computed tomography (CT) and magnetic resonance imaging (MRI) scan
Metaiodobenzylguanidine (MIBG) scan
The extent of metastatic disease is assessed by MIBG scan, which is applicable to all sites of disease, including soft tissue, bone marrow, and cortical bone. Approximately 90% of neuroblastomas will be MIBG avid. The MIBG scan has a sensitivity and specificity of 90% to 99%, and MIBG avidity is equally distributed between primary and metastatic sites.[ 23 ] Although iodine I 123 (123I) has a shorter half-life, it is preferred over 131I because of its lower radiation dose, better quality images, reduced thyroid toxicity, and lower cost.
Imaging with 123I-MIBG is optimal for identifying soft tissue and bony metastases. It was shown to be superior to positron emission tomography–computed tomography (PET-CT) in one prospective comparison.[ 24 ] In a retrospective review of 132 children with neuroblastoma, technetium Tc 99m-methylene diphosphonate (99mTc-MDP) bone scintigraphy failed to identify unique sites of metastatic disease that would change the disease stage or clinical management determined using 123I-MIBG or PET scanning. Bone scans are not used as part of standard staging for neuroblastoma.[ 25 ]
Baseline MIBG scans performed at diagnosis are excellent for monitoring disease response and performing posttherapy surveillance.[ 26 ] A retrospective analysis of paired 123I-MIBG and PET scans in 60 patients with newly diagnosed neuroblastoma demonstrated that for patients with INSS stage 1 and stage disease, PET was superior at determining the extent of primary disease and more sensitive in detecting residual masses. In contrast, for stage 4 disease, 123I-MIBG imaging was superior in detecting bone marrow and bony metastases.[ 27 ]
Curie and SIOPEN scoring methods
Multiple groups have investigated a semiquantitative scoring method to evaluate disease extent and prognostic value. The most common scoring methods in use for evaluation of disease extent and response are the Curie and the SIOPEN methods.
The German Pediatric Oncology Group compared the prognostic value of the Curie and SIOPEN scoring methods in a retrospective study of 58 patients with stage 4 neuroblastoma who were older than 1 year. The study found concordance in prognostic value (of these two methods) at diagnosis and after induction chemotherapy. At diagnosis, a Curie score of 2 or lower and a SIOPEN score of 4 or lower (best cutoff) correlated with significantly better EFS and overall survival (OS) rates, compared with higher scores. After four cycles of induction chemotherapy, patients with a complete response by SIOPEN and Curie scoring had a better outcome than patients with residual uptake in metastases. However, subsequent resolution of MIBG-positive metastases occurring between the fourth and sixth cycles of chemotherapy did not affect prognosis.[ 32 ]
The cited clinical trials did not include postinduction-phase assessments of Curie or SIOPEN scores after transplant and immunotherapy. Cutoffs and outcomes associated with those assessments may differ from the preinduction and postinduction scores.
PET scan
Fluorine F 18-fludeoxyglucose PET scans are used to evaluate extent of disease in patients with tumors that are not MIBG avid.[ 27 ]
Bone marrow aspiration and biopsy
Bone marrow is assessed by bilateral iliac crest marrow aspirates and trephine (core) bone marrow biopsies to exclude bone marrow involvement. To be considered adequate, core biopsy specimens must contain at least 1 cm of marrow, excluding cartilage. Many COG studies require two core biopsies and two aspirates. Bone marrow sampling may not be necessary for tumors that are otherwise stage 1.[ 33 ]
Other staging tests and procedures
Other tests and procedures used to stage neuroblastoma include the following:
参考文献- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.[PUBMED Abstract]
- Brodeur GM, Seeger RC, Barrett A, et al.: International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6 (12): 1874-81, 1988.[PUBMED Abstract]
- Castleberry RP, Shuster JJ, Smith EI: The Pediatric Oncology Group experience with the international staging system criteria for neuroblastoma. Member Institutions of the Pediatric Oncology Group. J Clin Oncol 12 (11): 2378-81, 1994.[PUBMED Abstract]
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.[PUBMED Abstract]
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.[PUBMED Abstract]
- Brisse HJ, McCarville MB, Granata C, et al.: Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology 261 (1): 243-57, 2011.[PUBMED Abstract]
- Newman EA, Nuchtern JG: Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg 25 (5): 257-264, 2016.[PUBMED Abstract]
- Chen AM, Trout AT, Towbin AJ: A review of neuroblastoma image-defined risk factors on magnetic resonance imaging. Pediatr Radiol 48 (9): 1337-1347, 2018.[PUBMED Abstract]
- Espinoza AF, Bagatell R, McHugh K, et al.: A subset of image-defined risk factors predict completeness of resection in children with high-risk neuroblastoma: An international multicenter study. Pediatr Blood Cancer 71 (10): e31218, 2024.[PUBMED Abstract]
- Brown EG, Adkins ES, Mattei P, et al.: Evaluation of Image-Defined Risk Factor (IDRF) Assessment in Patients With Intermediate-risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Pediatr Surg 60 (1): 161896, 2025.[PUBMED Abstract]
- Avanzini S, Pio L, Erminio G, et al.: Image-defined risk factors in unresectable neuroblastoma: SIOPEN study on incidence, chemotherapy-induced variation, and impact on surgical outcomes. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.[PUBMED Abstract]
- Burchill SA, Beiske K, Shimada H, et al.: Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer 123 (7): 1095-1105, 2017.[PUBMED Abstract]
- Park JR, Bagatell R, Cohn SL, et al.: Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 35 (22): 2580-2587, 2017.[PUBMED Abstract]
- Taggart DR, London WB, Schmidt ML, et al.: Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol 29 (33): 4358-64, 2011.[PUBMED Abstract]
- Monclair T, Mosseri V, Cecchetto G, et al.: Influence of image-defined risk factors on the outcome of patients with localised neuroblastoma. A report from the LNESG1 study of the European International Society of Paediatric Oncology Neuroblastoma Group. Pediatr Blood Cancer 62 (9): 1536-42, 2015.[PUBMED Abstract]
- Cecchetto G, Mosseri V, De Bernardi B, et al.: Surgical risk factors in primary surgery for localized neuroblastoma: the LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J Clin Oncol 23 (33): 8483-9, 2005.[PUBMED Abstract]
- Simon T, Hero B, Benz-Bohm G, et al.: Review of image defined risk factors in localized neuroblastoma patients: Results of the GPOH NB97 trial. Pediatr Blood Cancer 50 (5): 965-9, 2008.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.[PUBMED Abstract]
- Okamatsu C, London WB, Naranjo A, et al.: Clinicopathological characteristics of ganglioneuroma and ganglioneuroblastoma: a report from the CCG and COG. Pediatr Blood Cancer 53 (4): 563-9, 2009.[PUBMED Abstract]
- Morin CE, Hasweh R, Anton C, et al.: Gadolinium-based contrast media does not improve the staging of neuroblastoma image-defined risk factors at diagnosis. Pediatr Blood Cancer 71 (1): e30724, 2024.[PUBMED Abstract]
- Howman-Giles R, Shaw PJ, Uren RF, et al.: Neuroblastoma and other neuroendocrine tumors. Semin Nucl Med 37 (4): 286-302, 2007.[PUBMED Abstract]
- Papathanasiou ND, Gaze MN, Sullivan K, et al.: 18F-FDG PET/CT and 123I-metaiodobenzylguanidine imaging in high-risk neuroblastoma: diagnostic comparison and survival analysis. J Nucl Med 52 (4): 519-25, 2011.[PUBMED Abstract]
- Gauguet JM, Pace-Emerson T, Grant FD, et al.: Evaluation of the utility of (99m) Tc-MDP bone scintigraphy versus MIBG scintigraphy and cross-sectional imaging for staging patients with neuroblastoma. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- Kushner BH, Kramer K, Modak S, et al.: Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. J Clin Oncol 27 (7): 1041-6, 2009.[PUBMED Abstract]
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.[PUBMED Abstract]
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.[PUBMED Abstract]
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.[PUBMED Abstract]
- Lewington V, Lambert B, Poetschger U, et al.: 123I-mIBG scintigraphy in neuroblastoma: development of a SIOPEN semi-quantitative reporting ,method by an international panel. Eur J Nucl Med Mol Imaging 44 (2): 234-241, 2017.[PUBMED Abstract]
- Ladenstein R, Lambert B, Pötschger U, et al.: Validation of the mIBG skeletal SIOPEN scoring method in two independent high-risk neuroblastoma populations: the SIOPEN/HR-NBL1 and COG-A3973 trials. Eur J Nucl Med Mol Imaging 45 (2): 292-305, 2018.[PUBMED Abstract]
- Decarolis B, Schneider C, Hero B, et al.: Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol 31 (7): 944-51, 2013.[PUBMED Abstract]
- Russell HV, Golding LA, Suell MN, et al.: The role of bone marrow evaluation in the staging of patients with otherwise localized, low-risk neuroblastoma. Pediatr Blood Cancer 45 (7): 916-9, 2005.[PUBMED Abstract]
- DuBois SG, Kalika Y, Lukens JN, et al.: Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol 21 (3): 181-9, 1999 May-Jun.[PUBMED Abstract]
- Kramer K, Kushner B, Heller G, et al.: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91 (8): 1510-9, 2001.[PUBMED Abstract]
- Genomic and Biological Features of Neuroblastoma
-
Molecular features of neuroblastoma
Children with neuroblastoma can be divided into subsets with different predicted risks of relapse based on clinical factors and biological markers at the time of diagnosis.
Biological subtypes of high-risk neuroblastoma can be defined by the mostly nonoverlapping genomic alterations listed below:
The subtypes listed have specific clinical characteristics, as discussed below. Variants in ALK, which occur across the different subtypes of high-risk neuroblastoma, are observed in approximately 15% of cases and are discussed separately.
Key genomic characteristics of high-risk neuroblastoma that are present in most cases of high-risk neuroblastoma are discussed below.
Segmental chromosomal aberrations (SCAs)
The SCAs frequently observed in neuroblastoma and used when assigning SCA status include losses of or at chromosome arms 1p, 3p, 4p, and 11q and gains of or at chromosome arms 1q, 2p, and 17q.[ 8 ] These alterations can be detected by multiple methods, including fluorescence in situ hybridization (FISH), array comparative genomic hybridization (aCGH), and next-generation sequencing (NGS) assays. SCAs are present in most high-risk and/or stage 4 neuroblastoma tumors.[ 3 ][ 4 ][ 6 ][ 7 ][ 9 ] Among all patients with neuroblastoma, a higher number of chromosome breakpoints (i.e., a higher number of SCAs) correlated with the following:[ 3 ][ 4 ][ 5 ][ 6 ][ 7 ][Level of evidence C2]
Determining the presence of SCAs is potentially clinically useful. Detecting SCAs can help distinguish patients with clinically favorable presentation who are at higher risk of treatment failure. Examples are provided below.
In an analysis of localized, resectable, non-MYCN amplified neuroblastoma, cases from two consecutive European studies and a North American cohort (including International Neuroblastoma Staging System [INSS] stages 1, 2A, and 2B) were analyzed for selected SCAs (namely loss of 1p, 3p, 4p, and 11q and gain of 1q, 2p, and 17q). The study revealed a different prognostic impact of tumor genomics depending on patient age (<18 months vs. >18 months) and stage (1 vs. 2). Patients were treated with surgery alone regardless of a tumor residuum.[ 10 ][Level of evidence C1]
In a study of children older than 12 months who had unresectable primary neuroblastomas without metastases, SCAs were found in most patients. Older children were more likely to have them and to have more SCAs per tumor cell. In children aged 12 to 18 months, the presence of SCAs had a significant effect on EFS but not on OS. However, in children older than 18 months, there was a significant difference in OS between children with SCAs (67%) and children without SCAs (100%), regardless of tumor histology.[ 7 ]
SCAs were also found to be predictive of recurrence in infants with localized unresectable or metastatic neuroblastoma without MYCN gene amplification.[ 1 ][ 2 ] An analysis of 133 patients (aged ≥18 months) with INSS stage 3 tumors without MYCN amplification demonstrated that SCAs were associated with inferior EFS, and chromosome 11q loss was independently associated with worse OS.[ 11 ]
Chromosome 11q loss occurs in approximately 30% of high-risk neuroblastoma cases, but it is uncommonly observed in tumors with MYCN amplification.[ 3 ] Chromosome 11q loss is frequently observed in high-risk neuroblastoma cases with either TERT rearrangements or with ALT pathway activation.[ 12 ][ 13 ] Chromosome 11q loss has also been associated with inferior EFS and poor response to induction therapy in patients with high-risk neuroblastoma, as described below:
Distal chromosome 6q losses have also been associated with poor outcome. An international collaboration studied 556 patients with high-risk neuroblastoma. Distal 6q losses were found in 6% of patients and were associated with a 10-year survival rate of only 3.4%.[ 16 ] A second study confirmed the very poor prognosis of patients with high-risk neuroblastoma who have distal 6q loss. Pooling across both studies, MYCN amplification occurred in only 20% of cases with distal chromosome 6q loss.[ 17 ]
The same study of 556 patients with high-risk neuroblastoma that identified poor prognosis for patients with distal 6q loss also evaluated amplifications of regions not encompassing the MYCN locus. Regions of non-MYCN amplification were detected in 18% of the patients and were associated with a 10-year survival rate of 5.8%.[ 16 ]
MYCN gene amplification
MYCN amplification is detected in 16% to 25% of neuroblastoma tumors.[ 18 ] Among patients with high-risk neuroblastoma, 40% to 50% of cases show MYCN amplification.[ 19 ]
In all stages of disease, amplification of the MYCN gene strongly predicts a poorer prognosis, in both time to tumor progression and OS, in almost all multivariate regression analyses of prognostic factors.[ 1 ][ 2 ] In the ANBL00B1 (NCT00904241) study of 4,832 newly diagnosed patients enrolled between 2007 to 2017, the 5-year EFS and OS rates were 77% and 87%, respectively, for patients whose tumors were MYCN nonamplified (n = 3,647; 81%). In comparison, the 5-year EFS and OS rates were 51% and 57%, respectively, for patients whose tumors were MYCN amplified (n = 827; 19%).[ 9 ]
Within the localized-tumor MYCN-amplified cohort, patients with hyperdiploid tumors have better outcomes than patients with diploid tumors.[ 20 ] However, patients with hyperdiploid tumors with MYCN amplification or any SCAs do relatively poorly, compared with patients with hyperdiploid tumors without MYCN amplification.[ 3 ]
Most unfavorable clinical and pathobiological features are associated, to some degree, with MYCN amplification. In a multivariable logistic regression analysis of 7,102 patients in the International Neuroblastoma Risk Group (INRG) study, pooled SCAs and gains of 17q were poor prognostic features, even when not associated with MYCN amplification. However, another poor prognostic feature, SCAs at 11q, are almost entirely mutually exclusive of MYCN amplification.[ 21 ][ 22 ]
In a cohort of 6,223 patients from the INRG database with known MYCN status, the OS hazard ratio (HR) associated with MYCN amplification was 6.3 (95% confidence interval [CI], 5.7–7.0; P < .001). The greatest adverse prognostic impact of MYCN amplification for OS was in the youngest patients (aged <18 months: HR, 19.6; aged ≥18 months: HR, 3.0). Patients whose outcome was most impacted by MYCN status were those with otherwise favorable features, including age younger than 18 months, high mitosis-karyorrhexis index, and low ferritin.[ 23 ][Level of evidence C1]
Intratumoral heterogeneous MYCN amplification (hetMNA) refers to the coexistence of MYCN-amplified cells (as a cluster or as single scattered cells) and non–MYCN-amplified tumor cells. HetMNA has been reported infrequently. It can occur spatially within the tumor as well as between the tumor and the metastasis at the same time or temporally during the disease course. The International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) biology group investigated the prognostic significance of this neuroblastoma subtype. Tumor tissue from 99 patients identified as having hetMNA and diagnosed between 1991 and 2015 was analyzed to elucidate the prognostic significance of MYCN-amplified clones in otherwise non-MYCN–amplified neuroblastomas. Patients younger than 18 months showed a better outcome in all stages compared with older patients. The genomic background correlated significantly with relapse frequency and OS. No relapses occurred in cases of only numerical chromosomal aberrations. This study suggests that hetMNA tumors be evaluated in the context of the genomic tumor background in combination with the clinical pattern, including the patient's age and disease stage. Future studies are needed in patients younger than 18 months who have localized disease with hetMNA.[ 24 ]
Genomic alterations promoting telomere maintenance
Lengthening of telomeres, the tips of chromosomes, promotes cell survival. Telomeres otherwise shorten with each cell replication, eventually resulting in the cell’s inability to replicate. Patients whose tumors lack telomere maintenance mechanisms have an excellent prognosis, while patients whose tumors harbored telomere maintenance mechanisms have a substantially worse prognosis.[ 25 ] Low-risk neuroblastoma tumors, as defined by clinical/biological features, have little telomere lengthening activity. Aberrant genetic mechanisms for telomere lengthening have been identified in high-risk neuroblastoma tumors.[ 25 ][ 26 ][ 27 ][ 28 ] Thus far, the following three mechanisms, which appear to be mutually exclusive, have been described:
FOXR2 activation
FOXR2 gene expression is observed in approximately 8% of neuroblastoma cases. FOXR2 gene expression is normally absent postnatally, with the exception of male reproductive tissues.[ 34 ] FOXR2 expression is also observed in a subset of central nervous system (CNS) primitive neuroectodermal tumors, termed CNS NB-FOXR2.[ 35 ] FOXR2 overexpression was virtually mutually exclusive in neuroblastoma tumors with both elevated MYC and MYCN expression. Although MYCN gene expression was not elevated in neuroblastoma with FOXR2 activation, the gene expression profile for the FOXR2 expressing cases closely resembled that of MYCN-amplified neuroblastoma. FOXR2 binds MYCN and appears to stabilize the MYCN protein, leading to high levels of MYCN protein in neuroblastoma with FOXR2 activation. This finding provides an explanation for the similar gene expression profiles for neuroblastoma with FOXR2 activation and neuroblastoma with MYCN amplification.
Neuroblastoma with FOXR2 activation is observed at comparable rates in high-risk and non–high-risk cases.[ 34 ] Among high-risk cases, outcomes for patients whose tumors showed FOXR2 activation were similar to those for cases with MYCN amplification. In a multivariable analysis, FOXR2 activation was significantly associated with inferior OS, along with INSS stage 4, age 18 months or older, and MYCN amplification.
CDK4 and MDM2 amplification
CDK4 and MDM2 amplification are observed together in 1% to 2% of neuroblastoma cases, and these cases have distinctive biological and clinical features:[ 36 ][ 37 ]
Exonic variants in neuroblastoma (including ALK variants and amplification)
Compared with adult cancers, pediatric neuroblastoma tumors show a low number of variants per genome that affect protein sequence (10–20 per genome).[ 39 ] The most common gene variant is ALK, which is altered in approximately 10% of patients (see below). Other genes with even lower frequencies of variants include ATRX, PTPN11, ARID1A, and ARID1B.[ 26 ][ 27 ][ 30 ][ 40 ][ 41 ][ 42 ][ 43 ] As shown in Figure 2, most neuroblastoma cases lack variants in genes that are altered in a recurrent manner.
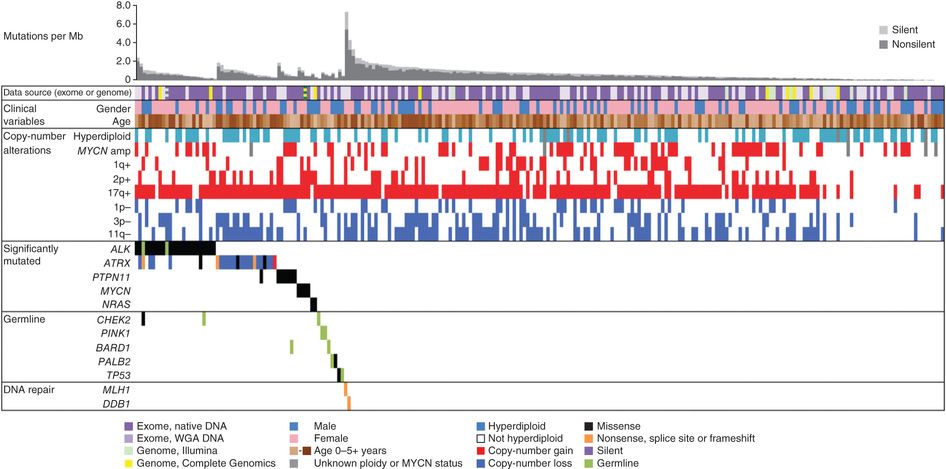
Figure 2. Data tracks (rows) facilitate the comparison of clinical and genomic data across cases with neuroblastoma (columns). The data sources and sequencing technology used were whole-exome sequencing (WES) from whole-genome amplification (WGA) (light purple), WES from native DNA (dark purple), Illumina WGS (green), and Complete Genomics WGS (yellow). Striped blocks indicate cases analyzed using two approaches. The clinical variables included were sex (male, blue; female, pink) and age (brown spectrum). Copy number alterations indicates ploidy measured by flow cytometry (with hyperdiploid meaning DNA index >1) and clinically relevant copy number alterations derived from sequence data. Significantly mutated genes are those with statistically significant mutation counts given the background mutation rate, gene size, and expression in neuroblastoma. Germline indicates genes with significant numbers of germline ClinVar variants or loss-of-function cancer gene variants in our cohort. DNA repair indicates genes that may be associated with an increased mutation frequency in two apparently hypermutated tumors. Predicted effects of somatic mutations are color coded according to the legend. Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics (Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013), copyright (2013). The ALK gene provides instructions for making a cell surface receptor tyrosine kinase, expressed at significant levels only in developing embryonic and neonatal brains. ALK is the exonic variant found most commonly in neuroblastoma. Germline pathogenic variants in ALK have been identified as the major cause of hereditary neuroblastoma. Somatically acquired ALK-activating exonic variants are also found as oncogenic drivers in neuroblastoma.[ 42 ]
Two large cohort studies examined the clinical correlates and prognostic significance of ALK alterations. One study from the COG examined ALK status in 1,596 diagnostic neuroblastoma samples across all risk groups.[ 42 ] Another study from SIOPEN evaluated 1,092 patients with high-risk neuroblastoma.[ 44 ]
In a study that compared the genomic data of primary diagnostic neuroblastomas originating in the adrenal gland (n = 646) with that of neuroblastomas originating in the thoracic sympathetic ganglia (n = 118), 16% of thoracic tumors harbored ALK variants.[ 45 ]
Small-molecule ALK kinase inhibitors such as lorlatinib (added to conventional therapy) are being tested in patients with recurrent ALK-altered neuroblastoma (NCT03107988) and in patients with newly diagnosed high-risk neuroblastoma with activated ALK (COG ANBL1531).[ 42 ] For more information, see the sections on Treatment of High-Risk Neuroblastoma and Treatment of Recurrent or Refractory Neuroblastoma in Neuroblastoma Treatment.
Genomic evolution of exonic variants
There are limited data regarding the genomic evolution of exonic variants from diagnosis to relapse for neuroblastoma. Whole-genome sequencing was applied to 23 paired diagnostic and relapsed neuroblastoma tumor samples to define somatic genetic alterations associated with relapse,[ 46 ] while a second study evaluated 16 paired diagnostic and relapsed specimens.[ 47 ] Both studies identified an increased number of variants in the relapsed samples compared with the samples at diagnosis. This has been confirmed in a study of neuroblastoma tumor samples sent for NGS.[ 48 ]
Given the widespread metastatic nature of high-risk and relapsed neuroblastoma, use of circulating tumor DNA (ctDNA) technologies may reveal additional genomic alterations not found in conventional tumor biopsies. Moreover, these approaches have demonstrated the ability to detect resistant variants in patients with neuroblastoma who were treated with ALK inhibitors.[ 50 ][Level of evidence C1] In one analysis of serial ctDNA samples from patients treated with lorlatinib, ALK VAF tracked with disease burden in most but not all patients.[ 51 ] In subsets of patients who progressed while taking lorlatinib, second compound variants in ALK or variants in other genes, including RAS pathway genes and TP53, have been reported.[ 51 ][ 52 ]
In a deep-sequencing study, 276 neuroblastoma samples (comprised of all stages and from patients of all ages at diagnosis) underwent very deep (33,000X) sequencing of just two amplified ALK variant hot spots, which revealed 4.8% clonal variants and an additional 5% subclonal variants. This finding suggests that subclonal ALK gene variants are common.[ 53 ] Thus, deep sequencing can reveal the presence of variants in tiny subsets of neuroblastoma tumor cells that may be able to survive during treatment and grow to constitute a relapse.
Additional biological factors associated with prognosis
MYC and MYCN expression
Immunostaining for MYC and MYCN proteins on a restricted subset of 357 undifferentiated/poorly differentiated neuroblastoma tumors demonstrated that elevated MYC/MYCN protein expression is prognostically significant.[ 54 ] Sixty-eight tumors (19%) highly expressed the MYCN protein, and 81 were MYCN amplified. Thirty-nine tumors (10.9%) expressed MYC highly and were mutually exclusive of high MYCN expression. In the MYC-expressing tumors, MYC or MYCN gene amplification was not seen. SCAs were not examined in this study.[ 54 ]
参考文献- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.[PUBMED Abstract]
- Schleiermacher G, Mosseri V, London WB, et al.: Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer 107 (8): 1418-22, 2012.[PUBMED Abstract]
- Janoueix-Lerosey I, Schleiermacher G, Michels E, et al.: Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27 (7): 1026-33, 2009.[PUBMED Abstract]
- Schleiermacher G, Michon J, Ribeiro A, et al.: Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105 (12): 1940-8, 2011.[PUBMED Abstract]
- Carén H, Kryh H, Nethander M, et al.: High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A 107 (9): 4323-8, 2010.[PUBMED Abstract]
- Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al.: Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28 (19): 3122-30, 2010.[PUBMED Abstract]
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.[PUBMED Abstract]
- Bagatell R, Park JR, Acharya S, et al.: Neuroblastoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 22 (6): 413-433, 2024.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Ambros IM, Tonini GP, Pötschger U, et al.: Age Dependency of the Prognostic Impact of Tumor Genomics in Localized Resectable MYCN-Nonamplified Neuroblastomas. Report From the SIOPEN Biology Group on the LNESG Trials and a COG Validation Group. J Clin Oncol 38 (31): 3685-3697, 2020.[PUBMED Abstract]
- Pinto N, Naranjo A, Ding X, et al.: Impact of Genomic and Clinical Factors on Outcome of Children ≥18 Months of Age with Stage 3 Neuroblastoma with Unfavorable Histology and without MYCN Amplification: A Children's Oncology Group (COG) Report. Clin Cancer Res 29 (8): 1546-1556, 2023.[PUBMED Abstract]
- Djos A, Thombare K, Vaid R, et al.: Telomere Maintenance Mechanisms in a Cohort of High-Risk Neuroblastoma Tumors and Its Relation to Genomic Variants in the TERT and ATRX Genes. Cancers (Basel) 15 (24): , 2023.[PUBMED Abstract]
- Yu Y, Zhang M, Yao X, et al.: Translational practice of fluorescence in situ hybridisation to identify neuroblastic tumours with TERT rearrangements. J Pathol Clin Res 9 (6): 475-487, 2023.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.[PUBMED Abstract]
- Depuydt P, Boeva V, Hocking TD, et al.: Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J Natl Cancer Inst 110 (10): 1084-1093, 2018.[PUBMED Abstract]
- Ognibene M, Morini M, Garaventa A, et al.: Identification of a minimal region of loss on chromosome 6q27 associated with poor survival of high-risk neuroblastoma patients. Cancer Biol Ther 21 (5): 391-399, 2020.[PUBMED Abstract]
- Ambros PF, Ambros IM, Brodeur GM, et al.: International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100 (9): 1471-82, 2009.[PUBMED Abstract]
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.[PUBMED Abstract]
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.[PUBMED Abstract]
- Plantaz D, Vandesompele J, Van Roy N, et al.: Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer 91 (5): 680-6, 2001.[PUBMED Abstract]
- Maris JM, Hogarty MD, Bagatell R, et al.: Neuroblastoma. Lancet 369 (9579): 2106-20, 2007.[PUBMED Abstract]
- Campbell K, Shyr D, Bagatell R, et al.: Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 66 (8): e27819, 2019.[PUBMED Abstract]
- Berbegall AP, Bogen D, Pötschger U, et al.: Heterogeneous MYCN amplification in neuroblastoma: a SIOP Europe Neuroblastoma Study. Br J Cancer 118 (11): 1502-1512, 2018.[PUBMED Abstract]
- Ackermann S, Cartolano M, Hero B, et al.: A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362 (6419): 1165-1170, 2018.[PUBMED Abstract]
- Peifer M, Hertwig F, Roels F, et al.: Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526 (7575): 700-4, 2015.[PUBMED Abstract]
- Valentijn LJ, Koster J, Zwijnenburg DA, et al.: TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47 (12): 1411-4, 2015.[PUBMED Abstract]
- Roderwieser A, Sand F, Walter E, et al.: Telomerase is a prognostic marker of poor outcome and a therapeutic target in neuroblastoma. JCO Precis Oncol 3: 1-20, 2019.[PUBMED Abstract]
- Mac SM, D'Cunha CA, Farnham PJ: Direct recruitment of N-myc to target gene promoters. Mol Carcinog 29 (2): 76-86, 2000.[PUBMED Abstract]
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.[PUBMED Abstract]
- Hartlieb SA, Sieverling L, Nadler-Holly M, et al.: Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun 12 (1): 1269, 2021.[PUBMED Abstract]
- Koneru B, Lopez G, Farooqi A, et al.: Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res 80 (12): 2663-2675, 2020.[PUBMED Abstract]
- Meeser A, Bartenhagen C, Werr L, et al.: Reliable assessment of telomere maintenance mechanisms in neuroblastoma. Cell Biosci 12 (1): 160, 2022.[PUBMED Abstract]
- Schmitt-Hoffner F, van Rijn S, Toprak UH, et al.: FOXR2 Stabilizes MYCN Protein and Identifies Non-MYCN-Amplified Neuroblastoma Patients With Unfavorable Outcome. J Clin Oncol 39 (29): 3217-3228, 2021.[PUBMED Abstract]
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.[PUBMED Abstract]
- Amoroso L, Ognibene M, Morini M, et al.: Genomic coamplification of CDK4/MDM2/FRS2 is associated with very poor prognosis and atypical clinical features in neuroblastoma patients. Genes Chromosomes Cancer 59 (5): 277-285, 2020.[PUBMED Abstract]
- Martinez-Monleon A, Kryh Öberg H, Gaarder J, et al.: Amplification of CDK4 and MDM2: a detailed study of a high-risk neuroblastoma subgroup. Sci Rep 12 (1): 12420, 2022.[PUBMED Abstract]
- Gundem G, Levine MF, Roberts SS, et al.: Clonal evolution during metastatic spread in high-risk neuroblastoma. Nat Genet 55 (6): 1022-1033, 2023.[PUBMED Abstract]
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.[PUBMED Abstract]
- Molenaar JJ, Koster J, Zwijnenburg DA, et al.: Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483 (7391): 589-93, 2012.[PUBMED Abstract]
- Sausen M, Leary RJ, Jones S, et al.: Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45 (1): 12-7, 2013.[PUBMED Abstract]
- Bresler SC, Weiser DA, Huwe PJ, et al.: ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26 (5): 682-94, 2014.[PUBMED Abstract]
- Janoueix-Lerosey I, Lequin D, Brugières L, et al.: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455 (7215): 967-70, 2008.[PUBMED Abstract]
- Bellini A, Pötschger U, Bernard V, et al.: Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 39 (30): 3377-3390, 2021.[PUBMED Abstract]
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.[PUBMED Abstract]
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.[PUBMED Abstract]
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.[PUBMED Abstract]
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.[PUBMED Abstract]
- Rosswog C, Fassunke J, Ernst A, et al.: Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer 128 (8): 1559-1571, 2023.[PUBMED Abstract]
- Bosse KR, Giudice AM, Lane MV, et al.: Serial Profiling of Circulating Tumor DNA Identifies Dynamic Evolution of Clinically Actionable Genomic Alterations in High-Risk Neuroblastoma. Cancer Discov 12 (12): 2800-2819, 2022.[PUBMED Abstract]
- Berko ER, Witek GM, Matkar S, et al.: Circulating tumor DNA reveals mechanisms of lorlatinib resistance in patients with relapsed/refractory ALK-driven neuroblastoma. Nat Commun 14 (1): 2601, 2023.[PUBMED Abstract]
- Bobin C, Iddir Y, Butterworth C, et al.: Sequential Analysis of cfDNA Reveals Clonal Evolution in Patients with Neuroblastoma Receiving ALK-Targeted Therapy. Clin Cancer Res 30 (15): 3316-3328, 2024.[PUBMED Abstract]
- Bellini A, Bernard V, Leroy Q, et al.: Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin Cancer Res 21 (21): 4913-21, 2015.[PUBMED Abstract]
- Wang LL, Teshiba R, Ikegaki N, et al.: Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer 113 (1): 57-63, 2015.[PUBMED Abstract]
- Special Considerations for the Treatment of Children With Cancer
-
Cancer in children and adolescents is rare, although the overall incidence has slowly increased since 1975.[ 1 ] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation to achieve optimal survival and quality of life:
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[ 2 ] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
参考文献- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.[PUBMED Abstract]
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.[PUBMED Abstract]
- Treatment Option Overview for Neuroblastoma
-
Generally, treatment of neuroblastoma is based on whether the tumor is classified as non-high risk (low or intermediate risk) or high risk. Because 5-year survival rates are generally 90% or higher, the goal of treatment for non–high-risk disease is to cure the disease with minimal toxicity. Outcomes for patients with high-risk disease have improved over time with the use of increasingly intensive multimodal therapy, but they remain suboptimal.
Table 6 summarizes the treatment options for patients with low-risk, intermediate-risk, and high-risk disease.
Table 6. Treatment Options for Neuroblastoma COG Risk-Group Assignment Treatment Options COG = Children's Oncology Group; GM-CSF = granulocyte-macrophage colony-stimulating factor; HSCT = hematopoietic stem cell transplant. Low-Risk Neuroblastoma Surgery followed by observation. Observation with or without biopsy. Chemotherapy with or without surgery (for symptomatic disease or unresectable progressive disease after surgery). Radiation therapy (only for emergency therapy). Intermediate-Risk Neuroblastoma Chemotherapy with or without surgery. Surgery and observation (in infants). Radiation therapy (for progressive disease, if needed). High-Risk Neuroblastoma A regimen of chemotherapy, surgery, tandem cycles of myeloablative therapy and HSCT, radiation therapy, and dinutuximab, with GM-CSF and isotretinoin. Stage 4S Neuroblastoma Observation with supportive care (for asymptomatic patients with favorable tumor biology). Chemotherapy (for symptomatic patients, those with unfavorable tumor biology, and infants aged <3 months). Surgery (rarely, for patients with hepatomegaly that compromises the kidney or other abdominal organs). Radiation therapy (rarely, for patients with symptoms related to hepatomegaly from metastatic disease). Principles of Surgery
In patients without metastatic disease, the standard of care is to perform an initial surgery. This surgery aims to accomplish the following, based on the disease stage and the risk group:
The COG reported that expectant observation in infants younger than 6 months with small (L1) adrenal masses resulted in an excellent EFS and OS while avoiding surgical intervention in a large majority of patients.[ 10 ] According to the surgical guidelines described in the intermediate-risk neuroblastoma clinical trial (ANBL0531 [NCT00499616]), the primary tumor is not routinely resected in patients with 4S neuroblastoma. German studies of selected groups of patients have biopsied tissue and observed infants with both L1 and L2 tumors without MYCN amplification, avoiding additional surgery and chemotherapy in most patients.[ 11 ]
Whether there is any advantage to gross-total resection (>90%) of the primary tumor mass after chemotherapy in patients older than 18 months with stage 4 disease remains controversial.[ 12 ][ 13 ][ 14 ][ 15 ][ 16 ][ 17 ] A meta-analysis of patients with stage 3 versus stage 4 neuroblastoma, at all ages combined, found an advantage for gross-total resection (>90%) over subtotal resection in stage 3 neuroblastoma only.[ 18 ] A small study suggested that after neoadjuvant chemotherapy, completeness of resection was affected by the number of IDRFs remaining.[ 19 ] When an experienced surgeon performed the procedure, a 90% or greater resection of the primary tumor in stage 4 neuroblastoma resulted in a higher local control rate, but it did not have a statistically significant impact on OS.[ 20 ]
For more information about IDRFs, see the International Neuroblastoma Risk Group Staging System (INRGSS) section.
Principles of Radiation Therapy
In the current treatment paradigm, radiation therapy for patients with low-risk or intermediate-risk neuroblastoma is reserved for symptomatic life-threatening or organ-threatening tumor bulk that did not respond rapidly enough to chemotherapy. Common situations in which radiation therapy is used in these patients include the following:
Radiation therapy has become part of the standard of care for patients with high-risk disease and is usually delivered after high-dose chemotherapy and stem cell rescue. For more information, see the Treatment of High-Risk Neuroblastoma section.
Limiting the use of radiation therapy in infants with neuroblastoma (who generally have non–high-risk disease) is supported by long-term follow-up data from the Childhood Cancer Survivor Study. This study demonstrated higher rates of second malignant neoplasms and significant chronic health conditions in infants who were treated with radiation therapy.[ 23 ][Level of evidence C1]
Treatment of Spinal Cord Compression
Spinal cord compression is considered a medical emergency. Patients receive immediate treatment because neurological recovery is more likely when symptoms are present for a relatively short time before diagnosis and treatment. Recovery also depends on the severity of neurological defects (weakness vs. paralysis). Neurological outcome appears to be similar whether cord compression is treated with chemotherapy, radiation therapy, or surgery, although radiation therapy is used less frequently than in the past.
The completed COG neuroblastoma clinical trials recommended immediate chemotherapy for cord compression in low-risk or intermediate-risk patients.[ 22 ][ 24 ][ 25 ] In a single study in this setting looking at the effect of glucocorticoids on neurological outcome, treatment was associated with improved early symptom relief. However, glucocorticoids did not prevent late residual impairment.[ 25 ]
Children with severe spinal cord compression that does not promptly improve or those with worsening symptoms may benefit from neurosurgical intervention. Laminectomy may result in later kyphoscoliosis and may not eliminate the need for chemotherapy.[ 22 ][ 24 ][ 25 ] Osteoplastic laminotomy, a procedure that does not remove bone, was thought to lessen spinal deformity. Osteoplastic laminotomy may be associated with a lower incidence of progressive spinal deformity requiring fusion, but there is no evidence that functional neurological deficit is improved with laminoplasty.[ 26 ]
The burden of long-term health problems in survivors of neuroblastoma with intraspinal extension is high. In a systematic review of 28 studies of treatment and outcome of patients with intraspinal extension, the severity of the symptoms at diagnosis and the treatment modalities were most associated with the presence of long-term health problems. In particular, the severity of neurological motor deficits was most likely to predict neurological outcome.[ 27 ] The severity of motor deficits at diagnosis is associated with spinal deformity and sphincter dysfunction at the end of follow-up, while sphincter dysfunction at diagnosis was correlated with long-term sphincter problems.[ 28 ] This supports the initiation of treatment before symptoms have deteriorated to complete loss of neurological function.
In a series of 34 infants with symptomatic epidural spinal cord compression, both surgery and chemotherapy provided unsatisfactory results once paraplegia had been established. The frequency of grade 3 motor deficits and bowel dysfunction increased with a longer symptom duration interval. Most infants with symptomatic epidural spinal cord compression developed sequelae, which were severe in about one-half of patients.[ 29 ]
An analysis of patients with intermediate-risk disease treated in the COG ANBL0531 [NCT00499616] study included 92 patients with intraspinal disease.[ 30 ] Of these patients, 42 (46%) were symptomatic. Among patients who were symptomatic, motor symptoms and bowel/bladder symptoms resolved completely in 73% and 88% of patients, respectively. Laminectomy or laminoplasty was performed in 22 of 42 symptomatic patients and was not significantly associated with improvements in symptoms.
Surveillance During and After Treatment
Although the role of surveillance imaging for detection of neuroblastoma relapse has not been well studied, most patients will undergo regular imaging tests after completing therapy. Many patients who relapse are asymptomatic, and relapse is detected on surveillance evaluations. Factors such as risk stratification, disease sites, biomolecular markers, and cumulative radiation dose may be considered in surveillance after treatment.[ 31 ][ 32 ][ 33 ]
One series included 154 patients with high-risk neuroblastoma who had a complete or very good partial response and subsequently had relapsed disease. The study found that 113 of the patients (73%) had asymptomatic relapse, while only 41 (27%) presented with symptoms. Metaiodobenzylguanidine (MIBG) scans were the most reliable study to detect asymptomatic relapse.[ 32 ]
In another series of 183 patients diagnosed with neuroblastoma, 50 patients experienced recurrence or progression. Relapsed disease was detected in most patients by symptoms/examination, MIBG scan, urinary catecholamines, and/or x-rays or ultrasonography.[ 33 ]
Cross-sectional imaging with CT scans is controversial because of the amount of radiation received and the low proportion of relapses detected with this modality.[ 33 ]
Evaluation of Disease Response
Evaluation of response is critical for the management of individual patients, but it is also necessary for comparing results of clinical trials. Given the complexities of a disease with propensity for bone and bone marrow metastasis, international consensus criteria have been developed and refined over the last several decades. The current version of these International Neuroblastoma Response Criteria (INRC) is presented below.
Revised International Neuroblastoma Response Criteria (INRC)
INRC is used to assess response to treatment.[ 34 ][ 35 ][ 36 ] Overall response in the revised INRC integrates tumor response in three components: primary tumor, soft tissue and bone metastases, and bone marrow. Primary and metastatic soft tissue sites are assessed using Response Evaluation Criteria in Solid Tumors (RECIST) and iodine I 123 (123I) MIBG scans or fluorine F 18-fludeoxyglucose (18F-FDG) positron emission tomography (PET) scans if the tumor is MIBG nonavid. 123I-MIBG scans, or 18F-FDG PET scans for MIBG-nonavid disease, replaced Technetium Tc 99m (99mTc) diphosphonate bone scintigraphy for osteomedullary metastasis assessment. Bone marrow is assessed by histology with or without immunohistochemistry and cytology or immunocytology. Bone marrow with 5% or less tumor involvement is classified as minimal disease. Urinary catecholamine levels are not included in response assessment. Overall response is defined as complete response, partial response, minor response, stable disease, or progressive disease.[ 36 ]
The overall INRC response criteria are defined as follows:[ 34 ][ 35 ]
Care should be taken in interpreting the development of metastatic disease in an infant who was initially considered to have stage 1 or 2 disease. If the pattern of metastases in such a patient is consistent with a 4S pattern of disease (involvement of skin, liver, and/or bone marrow, the latter less than 10% involved), these patients are not classified as having progressive/metastatic disease, which would typically be a criterion for removal from protocol therapy. Instead, these patients are managed as stage 4S patients.
Controversy exists regarding the necessity of measuring the primary tumor response in all three dimensions or whether the single longest dimension, as in RECIST tumor response determination, is equally useful.[ 37 ] The latter has been adopted for use in the INRC.
参考文献- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.[PUBMED Abstract]
- Park JR, Kreissman SG, London WB, et al.: Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 322 (8): 746-755, 2019.[PUBMED Abstract]
- Newman EA, Nuchtern JG: Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg 25 (5): 257-264, 2016.[PUBMED Abstract]
- Overman RE, Kartal TT, Cunningham AJ, et al.: Optimization of percutaneous biopsy for diagnosis and pretreatment risk assessment of neuroblastoma. Pediatr Blood Cancer 67 (5): e28153, 2020.[PUBMED Abstract]
- Gabra HO, Irtan S, Cross K, et al.: Minimally invasive surgery for neuroblastic tumours: A SIOPEN multicentre study: Proposal for guidelines. Eur J Surg Oncol 48 (1): 283-291, 2022.[PUBMED Abstract]
- Zenitani M, Yoshida M, Matsumoto S, et al.: Feasibility and safety of laparoscopic tumor resection in children with abdominal neuroblastomas. Pediatr Surg Int 39 (1): 91, 2023.[PUBMED Abstract]
- Chang S, Lin Y, Yang S, et al.: Safety and feasibility of laparoscopic resection of abdominal neuroblastoma without image-defined risk factors: a single-center experience. World J Surg Oncol 21 (1): 113, 2023.[PUBMED Abstract]
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.[PUBMED Abstract]
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.[PUBMED Abstract]
- Adkins ES, Sawin R, Gerbing RB, et al.: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39 (6): 931-6, 2004.[PUBMED Abstract]
- Castel V, Tovar JA, Costa E, et al.: The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37 (11): 1574-8, 2002.[PUBMED Abstract]
- La Quaglia MP, Kushner BH, Su W, et al.: The impact of gross total resection on local control and survival in high-risk neuroblastoma. J Pediatr Surg 39 (3): 412-7; discussion 412-7, 2004.[PUBMED Abstract]
- Simon T, Häberle B, Hero B, et al.: Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31 (6): 752-8, 2013.[PUBMED Abstract]
- Englum BR, Rialon KL, Speicher PJ, et al.: Value of surgical resection in children with high-risk neuroblastoma. Pediatr Blood Cancer 62 (9): 1529-35, 2015.[PUBMED Abstract]
- von Allmen D, Davidoff AM, London WB, et al.: Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol 35 (2): 208-216, 2017.[PUBMED Abstract]
- Mullassery D, Farrelly P, Losty PD: Does aggressive surgical resection improve survival in advanced stage 3 and 4 neuroblastoma? A systematic review and meta-analysis. Pediatr Hematol Oncol 31 (8): 703-16, 2014.[PUBMED Abstract]
- Irtan S, Brisse HJ, Minard-Colin V, et al.: Image-defined risk factor assessment of neurogenic tumors after neoadjuvant chemotherapy is useful for predicting intra-operative risk factors and the completeness of resection. Pediatr Blood Cancer 62 (9): 1543-9, 2015.[PUBMED Abstract]
- Wolden SL, Gollamudi SV, Kushner BH, et al.: Local control with multimodality therapy for stage 4 neuroblastoma. Int J Radiat Oncol Biol Phys 46 (4): 969-74, 2000.[PUBMED Abstract]
- Hsu LL, Evans AE, D'Angio GJ: Hepatomegaly in neuroblastoma stage 4s: criteria for treatment of the vulnerable neonate. Med Pediatr Oncol 27 (6): 521-8, 1996.[PUBMED Abstract]
- Katzenstein HM, Kent PM, London WB, et al.: Treatment and outcome of 83 children with intraspinal neuroblastoma: the Pediatric Oncology Group experience. J Clin Oncol 19 (4): 1047-55, 2001.[PUBMED Abstract]
- Friedman DN, Goodman PJ, Leisenring WM, et al.: Long-Term Morbidity and Mortality Among Survivors of Neuroblastoma Diagnosed During Infancy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (8): 1565-1576, 2023.[PUBMED Abstract]
- De Bernardi B, Pianca C, Pistamiglio P, et al.: Neuroblastoma with symptomatic spinal cord compression at diagnosis: treatment and results with 76 cases. J Clin Oncol 19 (1): 183-90, 2001.[PUBMED Abstract]
- Simon T, Niemann CA, Hero B, et al.: Short- and long-term outcome of patients with symptoms of spinal cord compression by neuroblastoma. Dev Med Child Neurol 54 (4): 347-52, 2012.[PUBMED Abstract]
- McGirt MJ, Chaichana KL, Atiba A, et al.: Incidence of spinal deformity after resection of intramedullary spinal cord tumors in children who underwent laminectomy compared with laminoplasty. J Neurosurg Pediatr 1 (1): 57-62, 2008.[PUBMED Abstract]
- Kraal K, Blom T, van Noesel M, et al.: Treatment and outcome of neuroblastoma with intraspinal extension: A systematic review. Pediatr Blood Cancer 64 (8): , 2017.[PUBMED Abstract]
- Angelini P, Plantaz D, De Bernardi B, et al.: Late sequelae of symptomatic epidural compression in children with localized neuroblastoma. Pediatr Blood Cancer 57 (3): 473-80, 2011.[PUBMED Abstract]
- De Bernardi B, Quaglietta L, Haupt R, et al.: Neuroblastoma with symptomatic epidural compression in the infant: the AIEOP experience. Pediatr Blood Cancer 61 (8): 1369-75, 2014.[PUBMED Abstract]
- Voeller J, Katzenstein HM, Naranjo A, et al.: Outcomes of patients with intermediate-risk neuroblastoma presenting with motor deficits relating to intraspinal tumor extension: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 72 (1): e31407, 2025.[PUBMED Abstract]
- Papathanasiou ND, Gaze MN, Sullivan K, et al.: 18F-FDG PET/CT and 123I-metaiodobenzylguanidine imaging in high-risk neuroblastoma: diagnostic comparison and survival analysis. J Nucl Med 52 (4): 519-25, 2011.[PUBMED Abstract]
- Kushner BH, Kramer K, Modak S, et al.: Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. J Clin Oncol 27 (7): 1041-6, 2009.[PUBMED Abstract]
- Owens C, Li BK, Thomas KE, et al.: Surveillance imaging and radiation exposure in the detection of relapsed neuroblastoma. Pediatr Blood Cancer 63 (10): 1786-93, 2016.[PUBMED Abstract]
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.[PUBMED Abstract]
- Brodeur GM, Seeger RC, Barrett A, et al.: International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6 (12): 1874-81, 1988.[PUBMED Abstract]
- Park JR, Bagatell R, Cohn SL, et al.: Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 35 (22): 2580-2587, 2017.[PUBMED Abstract]
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.[PUBMED Abstract]
- Treatment of Non–High-Risk Neuroblastoma
-
Approximately one-half of all newly diagnosed patients with neuroblastoma have non–high-risk disease (i.e., low and intermediate risk).[ 1 ] Since these patients have excellent survival, with 5-year survival rates higher than 95% for patients with low-risk disease and between 90% and 95% for patients with intermediate-risk disease, the goal of therapy for these patients is to cure the disease with minimal toxicity.
The staging system, risk classification system, and response criteria definitions for neuroblastoma have evolved over the past 20 years. As a result, published results from clinical trials for patients with non–high-risk neuroblastoma from the past used different staging systems (International Neuroblastoma Staging System) and response criteria or protocol-specific response criteria, making it difficult to compare trial results.
Low-Risk Neuroblastoma
The success of previous Children's Oncology Group (COG) clinical trials has contributed to the continued reduction in therapy for select patients with neuroblastoma. According to the COG risk categorization, patients with low-risk disease generally have low-stage disease (International Neuroblastoma Risk Group [INRG] stage L1) and the tumors are MYCN-nonamplified, hyperdiploid, and have favorable histology (FH). For more information about the COG risk categories, see Table 3.
For more information about low-stage disease, see the Treatment of INSS Stage 4S and INRG Stage MS Neuroblastoma section.
Treatment options for low-risk neuroblastoma
Surgery, by an experienced surgeon, is the treatment of choice for patients with low-risk, INRG stage L1 tumors. The exception is for patients who are younger than 6 months with isolated adrenal masses with maximum diameter smaller than 3.1 cm if solid, or 5 cm if at least 25% of the mass is cystic. For these patients, observation without biopsy is the recommended approach. If the biology is confirmed to be favorable, residual disease after surgery is not considered a risk factor for relapse and chemotherapy is not indicated. Several studies have shown that patients with favorable biology and residual disease have excellent outcomes, with event-free survival (EFS) rates exceeding 90% and overall survival (OS) rates ranging from 99% to 100%.[ 2 ][ 3 ]
In patients with INRG stage MS disease who are asymptomatic and have tumors with favorable biology, observation is the preferred approach.
Some patients with presumed neuroblastoma have been observed without biopsy. The COG is studying this strategy further in the ANBL1232 (NCT02176967) trial (closed to accrual).[ 4 ][ 5 ]
Treatment options for low-risk neuroblastoma include the following:
- Surgery followed by observation (INRG stage L1 or MS).
- Observation with or without biopsy.
- Chemotherapy with or without surgery (for symptomatic disease or unresectable progressive disease after surgery).
- Radiation therapy (only for emergency therapy).
Surgery followed by observation
Treatment for patients categorized as low risk may be surgery alone. For more information, see Table 3.
Evidence (surgery followed by observation):
- Results from the COG-P9641 (NCT00003119) study showed that surgery alone, even without complete resection, can cure nearly all patients with stage 1 neuroblastoma and the vast majority of patients with asymptomatic, favorable-biology, and International Neuroblastoma Staging System (INSS) stage 2A or stage 2B disease.[ 3 ]
- Similar outcomes were seen in a nonrandomized clinical trial in Japan.[ 6 ]
Observation with or without biopsy
Observation without biopsy has been used to treat perinatal neuroblastoma with small adrenal tumors.
A COG study determined that selected small INSS stage 1 or stage 2 adrenal masses, presumed to be neuroblastoma, detected in infants younger than 6 months by screening or incidental ultrasonography, may safely be observed without obtaining a definitive histological diagnosis and without surgical intervention. This technique avoids potential complications of surgery in newborn patients.[ 4 ] Patients are observed frequently to detect any tumor growth or spread, indicating a need for intervention. Additional studies, including an expansion of criteria allowing observation without surgery, are under way in the COG ANBL1232 (NCT02176967) study (closed to accrual).
Evidence (observation without biopsy):
- The COG-ANBL00P2 (NCT00445718) study reported that expectant observation is safe in patients younger than 6 months with solid adrenal tumors smaller than 3.1 cm (or cystic tumors smaller than 5 cm) and INSS stage 1 disease.[ 4 ]
Controversy exists about the need to attempt resection, at the time of diagnosis or later, in asymptomatic infants aged 12 months or younger with apparent stage 2B and stage 3 MYCN-nonamplified and favorable-biology disease. In a German clinical trial, some of these patients were observed after biopsy or partial resection without chemotherapy or radiation therapy. Many patients did not progress locally and never underwent a first or additional resection.[ 5 ] In the COG ANBL1232 (NCT02176967) study (closed to accrual), infants younger than 18 months who have L2 tumors with favorable biology are being observed after tumor biopsy.
Chemotherapy with or without surgery
Chemotherapy with or without surgery is used to treat the following:
Evidence (for removal of chemotherapy):
- The COG-P9641 study was one of the first COG studies to test risk stratification based on consensus-derived factors. In this phase III nonrandomized trial, 915 infants and children with INSS stage 2A and 2B disease underwent an initial operation to obtain tissue for diagnosis and biology studies and for maximal safe primary tumor resection. Chemotherapy was reserved for patients with, or at risk of, symptomatic disease, with less than 50% tumor resection at diagnosis, or with unresectable progressive disease after surgery alone.[
3
]
- Stage 1:
- Stage 2A and 2B:
- Outcome of asymptomatic patients at diagnosis who were observed after initial operation and patients treated with chemotherapy postoperatively: Of the initial 915 patients, 800 were asymptomatic at diagnosis and observed after their initial operations. Within this group, 11% of patients experienced recurrent or progressive disease. Of the 115 patients who underwent surgery followed by immediate chemotherapy (median, 4 cycles; range, 1–8 cycles), 81% of the patients had a very good partial response or better. After chemotherapy, 10% of the patients had disease recurrence or progression.
- MYCN amplification: The impact of MYCN-amplified tumors was analyzed in patients with stage 1 disease.
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Intermediate-Risk Neuroblastoma
According to the 2021 COG risk classifier, intermediate risk includes the following for localized, INRG stage L2 tumors (no MYCN amplification):[ 1 ]
Stage M (no MYCN amplification):[ 1 ]
Stage MS (no MYCN amplification):[ 1 ]
For infants with stage MS tumors who are too unstable to undergo biopsy before starting treatment, chemotherapy is initiated, and a biopsy is obtained when safe.
For more information about the COG risk categories, see Table 3.
For more information about stage 4S and MS tumors, see the Treatment of INSS Stage 4S and INRG Stage MS Neuroblastoma section.
Treatment options for intermediate-risk neuroblastoma
Treatment options for intermediate-risk neuroblastoma include the following:
- Chemotherapy with or without surgery.
- Surgery and observation (in infants).
- Radiation therapy (for progressive disease, if needed).
Chemotherapy with or without surgery
Patients categorized as intermediate risk have been successfully treated with complete surgical resection and two, four, or eight cycles of neoadjuvant chemotherapy. The chemotherapy regimen consists of carboplatin, cyclophosphamide, doxorubicin, and etoposide. The cumulative dose of each agent is kept low to minimize long-term effects from the chemotherapy regimen (ANBL0531 [NCT00499616]). As a rule, patients whose tumors had unfavorable biology received eight cycles of chemotherapy, and patients whose tumors had favorable biology received either two or four cycles of chemotherapy. Favorable biological features include FH, DI higher than 1, and no SCAs.
Tumor response assessment is measured with a single dimension, as per Response Evaluation Criteria in Solid Tumors (RECIST). After the number of assigned cycles of chemotherapy (based on disease stage, age, and biological features), if greater than a partial response has not been obtained, then a multidisciplinary discussion should occur to discuss the role of surgery versus additional chemotherapy. Patients who achieve a partial response or greater will enter surveillance. Surgical resection should be considered if chemotherapy has resulted in less than 50% reduction in tumor size. For patients unable to undergo surgical resection, and additional chemotherapy is given, response should be re-evaluated after every two cycles of therapy. Another biopsy to look for histological differentiation may be necessary to assess a residual mass that did not shrink sufficiently with chemotherapy.[ 7 ]
In cases of abdominal neuroblastoma thought to involve a kidney, nephrectomy is not undertaken before a course of chemotherapy has been given.[ 8 ] Nephrectomy should be avoided in all cases.
Cyclophosphamide and topotecan were used in the ANBL0531 (NCT00499616) study as additional treatment in patients who had received eight cycles of intermediate-risk chemotherapy and did not achieve the targeted response.[ 9 ][ 10 ]
Whether initial chemotherapy is indicated for all intermediate-risk infants with localized neuroblastoma requires further study.
Evidence (chemotherapy with or without surgery):
- The goal of the ANBL0531 (NCT00499616) study was to reduce therapy for subsets of patients with intermediate-risk neuroblastoma (MYCN-nonamplified, age and stage defined). Treatment duration (two, four, or eight cycles of moderate-dose neoadjuvant chemotherapy) was assigned according to clinical features and a tumor biology (including allelic status of 1p36 and 11q23) and response-based algorithm. The 10-year EFS and OS rates for the entire study cohort (N = 404) were 82.0% and 94.7%, respectively.[ 10 ] Treatment duration and intensity was reduced for several subsets of patients. The study added stage 4 patients with favorable biology who were aged 12 to 18 months.[ 9 ]
- A German prospective clinical trial enrolled 340 infants aged 1 year or younger whose tumors were stage 1, 2, or 3; histologically verified; and lacked MYCN amplification. Chemotherapy was given at diagnosis to 57 infants with organs threatened by the tumor. The tumor was completely resected or nearly so in 190 infants who underwent low-risk surgery. A total of 93 infants whose tumors were not resectable without high-risk surgery because of age or organ involvement were observed without chemotherapy.[ 5 ]
- Moderate-dose chemotherapy has been shown to be effective in the prospective Infant Neuroblastoma European Study (EURO-INF-NB-STUDY-1999-99.1). About one-half of the infants with unresectable, nonmetastatic neuroblastoma and no MYCN amplification underwent a safe surgical resection and avoided long-term adverse effects.[ 12 ][Level of evidence C1]
- In two European prospective trials of infants with disseminated neuroblastoma without MYCN gene amplification, infants with INSS stage 3 primary or positive skeletal scintigraphy without radiological bone metastasis (identified mostly by MIBG scan, but a few with just technetium Tc 99m bone scan) were not administered chemotherapy unless life-threatening or organ-threatening symptoms developed. When given, chemotherapy consisted of short and standard doses.[ 13 ]
- A retrospective analysis from the COG evaluated patients aged 12 to 18 months with metastatic disease and favorable biological features. In legacy trials, these patients were treated with high-risk disease regimens.[ 14 ]
- A prospective International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) trial treated infants with MYCN-nonamplified, stage 2 or stage 3 unresectable neuroblastoma, as well as those aged 12 to 18 months who had favorable INPC.[ 15 ][Level of evidence C2]
Surgery and observation (in infants)
The need for chemotherapy in all asymptomatic infants with stage 3 or stage 4 disease is controversial, as some European studies have shown favorable outcomes with surgery and observation.[ 13 ]
Evidence (surgery and observation in infants):
- A French study classified infants as stage 4 because of a primary tumor infiltrating across the midline (INSS stage 3 primary with metastases limited to 4S category) or positive bone scintigraphy not associated with changes in the cortical bone documented on plain radiographs and/or CT.[ 16 ]
- Building on the French study, SIOPEN conducted a prospective trial of 125 infants (n = 41 with INSS 3 primary tumors or positive scintigraphy) with disseminated neuroblastoma without MYCN amplification to determine whether these patients could be observed in the absence of symptoms. However, treating physicians did not always follow the wait-and-see strategy.[ 13 ]
- A German prospective clinical trial enrolled 340 infants aged 1 year or younger whose tumors were stage 1, 2, or 3; verified histologically; and lacked MYCN amplification. Of the 190 infants who underwent resection, 8 had stage 3 disease. A total of 93 infants whose tumors were not resectable without high-risk surgery, because of age or organ involvement, were observed without chemotherapy, which included 21 patients with stage 3 disease. Fifty-seven infants, including 41 with stage 3 disease, were treated with chemotherapy to control threatening symptoms.[ 5 ]
Radiation therapy
Radiation therapy for children with intermediate-risk disease is reserved for patients with progressive disease during treatment with chemotherapy or progressive unresectable disease after treatment with chemotherapy.
In a prospective randomized COG trial that tested reduced-intensity chemotherapy for patients with intermediate-risk neuroblastoma, only 12 of 479 patients (2.5%) received local radiation therapy (21 Gy). One patient had stage 4S disease, five patients had stage 3 disease, and six patients had stage 4 disease. Radiation therapy was administered for clinical deterioration despite initial therapy (eight patients), residual macroscopic disease and unfavorable biological features (three patients), or relapse after therapy (one patient).[ 2 ][ 11 ][ 17 ]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
参考文献- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Matthay KK, Perez C, Seeger RC, et al.: Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children's Cancer Group study. J Clin Oncol 16 (4): 1256-64, 1998.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.[PUBMED Abstract]
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.[PUBMED Abstract]
- Iehara T, Hamazaki M, Tajiri T, et al.: Successful treatment of infants with localized neuroblastoma based on their MYCN status. Int J Clin Oncol 18 (3): 389-95, 2013.[PUBMED Abstract]
- Bagatell R, Park JR, Acharya S, et al.: Neuroblastoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 22 (6): 413-433, 2024.[PUBMED Abstract]
- Shamberger RC, Smith EI, Joshi VV, et al.: The risk of nephrectomy during local control in abdominal neuroblastoma. J Pediatr Surg 33 (2): 161-4, 1998.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.[PUBMED Abstract]
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.[PUBMED Abstract]
- Rubie H, De Bernardi B, Gerrard M, et al.: Excellent outcome with reduced treatment in infants with nonmetastatic and unresectable neuroblastoma without MYCN amplification: results of the prospective INES 99.1. J Clin Oncol 29 (4): 449-55, 2011.[PUBMED Abstract]
- De Bernardi B, Gerrard M, Boni L, et al.: Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol 27 (7): 1034-40, 2009.[PUBMED Abstract]
- Bender HG, Irwin MS, Hogarty MD, et al.: Survival of Patients With Neuroblastoma After Assignment to Reduced Therapy Because of the 12- to 18-Month Change in Age Cutoff in Children's Oncology Group Risk Stratification. J Clin Oncol 41 (17): 3149-3159, 2023.[PUBMED Abstract]
- Kohler JA, Rubie H, Castel V, et al.: Treatment of children over the age of one year with unresectable localised neuroblastoma without MYCN amplification: results of the SIOPEN study. Eur J Cancer 49 (17): 3671-9, 2013.[PUBMED Abstract]
- Minard V, Hartmann O, Peyroulet MC, et al.: Adverse outcome of infants with metastatic neuroblastoma, MYCN amplification and/or bone lesions: results of the French society of pediatric oncology. Br J Cancer 83 (8): 973-9, 2000.[PUBMED Abstract]
- Kim C, Choi YB, Lee JW, et al.: Excellent treatment outcomes in children younger than 18 months with stage 4 MYCN nonamplified neuroblastoma. Korean J Pediatr 61 (2): 53-58, 2018.[PUBMED Abstract]
- Treatment of High-Risk Neuroblastoma
-
Patients most at risk for disease progression and mortality are older than 18 months, have metastatic disease or localized disease with unfavorable biology such as MYCN amplification, or have unfavorable histology. For more information about the Children's Oncology Group (COG) risk categories, see Table 3.
Approximately 8% to 10% of infants with stage MS disease have MYCN-amplified tumors and are usually treated using high-risk protocols. The 5-year event-free survival (EFS) and overall survival (OS) rates were 60% and 64%, respectively, for the infants with stage MS disease and MYCN amplification (n = 23), among the 5,000 patients enrolled in the COG ANBL00B1 (NCT00904241) trial.[ 1 ]
For children with high-risk neuroblastoma who received current treatments, the 5-year OS rate was about 60% for patients diagnosed between 2007 and 2017.[ 1 ] Children with aggressively treated, high-risk neuroblastoma may develop late recurrences, some more than 5 years after completion of therapy.[ 2 ][ 3 ]
A study from the International Neuroblastoma Risk Group (INRG) database found 146 patients with distant metastases limited to lymph nodes, termed stage 4N, who tended to have favorable-biology disease and a good outcome (5-year OS rate, 85%). This finding suggests that for this very rare, special subgroup of high-risk, stage 4 patients, less-intensive therapy might be considered.[ 4 ] These more favorable outcomes were confirmed in a single-institution study of 51 patients.[ 5 ]
Treatment Options for High-Risk Neuroblastoma
Outcomes for patients with high-risk neuroblastoma remain poor despite recent improvements in survival in randomized trials.
Treatment options for high-risk neuroblastoma typically include the following:
Chemotherapy, surgery, tandem cycles of myeloablative therapy and HSCT, radiation therapy, and dinutuximab, with GM-CSF and isotretinoin
Treatment for patients with high-risk disease is generally divided into the following three phases:
Induction phase
The backbone of the most commonly used induction therapy includes dose-intensive cycles of cisplatin and etoposide alternating with vincristine, cyclophosphamide, and doxorubicin.[ 6 ] Topotecan and cyclophosphamide were added to this regimen based on the antineuroblastoma activity seen in patients with relapsed disease.[ 7 ] Response to therapy after four cycles of chemotherapy or at the end of induction chemotherapy correlates with EFS at the completion of high-risk therapy.[ 8 ][ 9 ][ 10 ]
Evidence (induction chemotherapy with or without additional treatments):
- In one study, the addition of anti-GD2 antibody therapy with GM-CSF and low-dose interleukin-2 (IL-2), given with each induction chemotherapy course, had encouraging outcomes in 42 children with newly diagnosed stage 4 disease.[ 11 ]
- A European prospective randomized controlled trial investigated extended induction therapy in 422 patients with newly diagnosed high-risk neuroblastoma. Patients were randomly assigned to receive either standard induction chemotherapy with six chemotherapy courses or experimental induction chemotherapy that began with two additional courses of topotecan, cyclophosphamide, and etoposide, followed by standard induction chemotherapy.[ 12 ][Level of evidence B3]
- European investigators completed another randomized study of induction regimens for patients with high-risk neuroblastoma. A total of 630 patients were randomly assigned to receive either cisplatin, vincristine, carboplatin, etoposide, and cyclophosphamide (rCOJEC regimen; n = 313) or the Memorial Sloan Kettering Cancer Center N5 induction regimens (MSKCC-N5; n = 317).[ 13 ][Level of evidence B1]
After a response to induction chemotherapy, resection of the primary tumor is recommended by most treatment protocols. Whether a gross-total resection is beneficial is controversial.[ 14 ]
Evidence (extent of resection of the primary tumor):
- The COG A3973 (NCT00004188) study had central surgical review of 220 patients who underwent attempted gross-total resection after induction chemotherapy. By the surgeon’s estimate, the degree of resection was determined to be 90% or greater versus less than 90%, but only 63% concordance with central review of imaging was found.[ 15 ][Level of evidence C1]
- A single-center retrospective study of 87 children with high-risk neuroblastoma demonstrated no significant benefit of gross-total resection compared with near-total (>90%) resection.[ 16 ][Level of evidence C2]
The potential benefit of aggressive surgical approaches in high-risk patients with metastatic disease to achieve complete tumor resection, either at the time of diagnosis or after chemotherapy, has not been unequivocally demonstrated. Several studies have reported that complete resection of the primary tumor at diagnosis improved survival. However, the outcome in these patients may be more dependent on the biology of the tumor, which itself may determine resectability, than on the extent of surgical resection.[ 17 ][ 18 ][ 19 ]
In patients older than 18 months with stage 4 neuroblastoma, controversy exists about whether there is any advantage to gross-total resection of the primary tumor after chemotherapy.[ 15 ][ 18 ][ 19 ][ 20 ] In some studies, patients who underwent incomplete resections fared less well than those who underwent complete resections.[ 21 ] These outcomes could have resulted from either the biology of unresectable tumors or reduction of tumor bulk.[ 22 ][Level of evidence B1] Complete resection that requires nephrectomy is not recommended because of the nephrotoxic nature of standard chemotherapy and unproven effect of complete resection on outcome.
In most group studies, surgical resection of the primary tumor is performed during the induction phase. However, the JN-H-11 trial evaluated the feasibility of delayed resection after high-dose chemotherapy with stem cell rescue, with the goal of prioritizing systemic therapy.[ 23 ] Rates of complete or greater-than-90% resection were similar to those seen in a previous trial, in which surgery was performed during induction. The rate of nephrectomy was nominally lower (5.8% vs. 17%), and the 3-year cumulative incidence of local failure rate was nominally higher (17.3% vs. 11.6%), compared with the rates found in the previous trial.
At the end of induction therapy, patients with high-risk disease typically undergo a full disease evaluation. Management of patients with residual disease at the end of conventional induction therapy is not standardized. A retrospective study analyzed 201 patients with high-risk disease who had a partial response or less at the end of induction therapy. Patients were selected to immediately receive either high-dose chemotherapy (cohort 1), bridging therapy (usually chemoimmunotherapy or iodine I 131-metaiodobenzylguanidine [MIBG]) followed by high-dose chemotherapy (cohort 2), or additional therapy but not high-dose chemotherapy (cohort 3).[ 24 ]
These retrospective data suggest a role for bridging therapy in patients with incomplete response to conventional induction therapy.
Consolidation phase
The consolidation phase of high-risk regimens involves myeloablative chemotherapy and HSCT, which attempts to eradicate minimal residual disease (MRD) using otherwise lethal doses of ablative chemotherapy rescued by autologous stem cells (collected during induction chemotherapy) to repopulate the bone marrow. Several large randomized controlled studies showed improved 3-year EFS rates for treatment with HSCT (31%–47%) versus conventional chemotherapy (22%–31%).[ 25 ][ 26 ][ 27 ] Previously, total-body irradiation had been used in HSCT conditioning regimens. Most current protocols in North America use tandem cycles of chemotherapy and HSCT with cyclophosphamide/thiotepa and carboplatin/etoposide/melphalan.[ 28 ][Level of evidence C1] In Europe, clinical trials have also evaluated busulfan/melphalan and HSCT.
Evidence (myeloablative chemotherapy and stem cell rescue):
- A large European multicenter trial of consolidation therapy randomly assigned patients who had completed a multidrug induction regimen (cisplatin, carboplatin, cyclophosphamide, vincristine, and etoposide with or without topotecan, vincristine, and doxorubicin) and achieved an adequate response to receive either busulfan/melphalan or carboplatin/etoposide/melphalan.[ 29 ][Level of evidence A1]
- A randomized clinical study (COG-ANBL0532) tested the efficacy of two cycles versus one cycle of myeloablative chemotherapy with stem cell rescue.[ 30 ][Level of evidence A1] Children older than 18 months with stage 4 neuroblastoma who had received six cycles of induction chemotherapy were then randomly assigned to receive a single autologous HSCT with carboplatin/etoposide/melphalan or tandem transplants with cyclophosphamide/thiotepa followed by reduced-dose carboplatin/etoposide/melphalan. After tumor bed radiation therapy, most patients were randomly assigned to a second separate trial to receive isotretinoin alone or isotretinoin with dinutuximab and immune enhancement.
- An updated Cochrane review evaluated three randomized clinical trials comparing autologous bone marrow transplant (BMT) with standard chemotherapy.[ 25 ][ 26 ][ 27 ][ 31 ][ 32 ]
- A review of 147 allogeneic transplant cases submitted to the Center for International Blood and Marrow Transplant Research found no advantage for allogeneic transplant over autologous transplant, even if the allogeneic transplant recipient had received a previous autologous transplant.[ 33 ]
In a separate prospective randomized study, there was no advantage to purging harvested autologous stem cells of neuroblastoma cells before transplant.[ 34 ]
For more information about transplant, see Pediatric Autologous Hematopoietic Stem Cell Transplant and Pediatric Hematopoietic Stem Cell Transplant and Cellular Therapy for Cancer.
Radiation to the primary tumor site (whether or not a complete excision was obtained) is indicated after myeloablative therapy.[ 35 ][ 36 ]; [ 37 ][Level of evidence C1] Boost radiation therapy for gross-residual disease did not show improved local control when studied prospectively in the ANBL0532 (NCT00567567) trial.[ 38 ][Level of evidence C1] The optimal dose of radiation therapy has not been determined.[ 39 ]
Evidence (radiation therapy with a boost vs. radiation therapy without a boost for incomplete resection):
- Because of the high rates of local recurrence after incomplete surgical resection, the COG ANBL0532 (NCT00567567) trial prospectively evaluated the potential benefit of boost radiation therapy for patients with gross-residual tumor and compared the results with the preceding COG clinical trial for high-risk neuroblastoma (A3973 [NCT00004188]), in which patients did not receive boost radiation therapy. All patients on the ANBL0532 trial received 21.6 Gy of radiation to the preoperative primary tumor volume after induction chemotherapy.[ 38 ][Level of evidence C1]
Extensive lymph node irradiation, regardless of the extent of surgical resection preceding HSCT, did not benefit patients for local progression or OS.[ 40 ][Level of evidence C1]
A detailed retrospective multicenter review of locoregional recurrences demonstrated that 48.4% were in-field recurrences and another 19.4% were marginal recurrences.[ 41 ] These findings suggest that additional optimization of radiation therapy approaches are still needed.
Treatment of bony metastatic disease, delivered at the time of primary tumor bed irradiation, is also considered to maximize disease control. Radiation therapy to metastatic disease sites is determined on an individual basis or according to protocol guidelines for patients enrolled in studies. Many children present with widespread bony metastases. Because it is not feasible to irradiate all initial sites, the current practice is to treat the sites that have not responded, as assessed by MIBG before HSCT.[ 42 ][ 43 ][ 44 ] Metastatic sites identified at diagnosis that did not receive radiation during frontline therapy appeared to have a higher risk of involvement at first relapse relative to previously irradiated metastatic sites.[ 42 ] In one single-institution study, 17 of 24 patients with residual MIBG-avid skeletal uptake at the end of front-line therapy without metastatic-site radiation therapy had disease recurrence. Of the 17 patients, 13 (76.5%) had disease recurrence at sites of prior skeletal disease.[ 45 ]
In a retrospective series of 159 children with high-risk stage M neuroblastoma, focal irradiation was delivered to all metastatic sites, regardless of response to chemotherapy, unless metastases were too numerous.[ 46 ]
These observations support the current paradigm of irradiating metastases that persist by MIBG uptake after induction chemotherapy in high-risk patients. Irradiation of more than 50% of the bone marrow is not advised.[ 46 ]
In cases where diffuse bone metastases remain after induction chemotherapy, high-dose chemotherapy is followed by reassessment before deciding on consolidative radiation therapy.
Preliminary outcomes of proton radiation therapy to treat patients with high-risk neuroblastoma primary tumors have been published, demonstrating acceptable efficacy and toxicity.[ 47 ]
Postconsolidation phase
Postconsolidation therapy is designed to treat potential MRD after HSCT.[ 31 ] For high-risk patients in remission after HSCT, dinutuximab combined with GM-CSF given together with isotretinoin demonstrated improved EFS.[ 48 ][ 49 ]
Evidence (all treatments):
- A randomized study compared high-dose therapy and purged autologous BMT with three cycles of intensive consolidation chemotherapy. In addition, after the completion of either chemotherapy or autologous BMT, patients were randomly assigned to stop therapy or to receive 6 months of isotretinoin. The EFS and OS results described below reflect outcome from the time of each randomization.[ 25 ]; [ 31 ][Level of evidence A1]
- A retrospective, single-institution, nonrandomized trial compared patients who received GM-CSF and 3F8 anti-GD2 antibody therapy after either autologous HSCT or conventional chemotherapy.[ 50 ] The patients were a mixture of those referred for initial treatment or further therapy, and included patients with refractory and relapsed disease, some of whom had received autologous HSCT at referring institutions. In the autologous HSCT group, there was a significantly longer time from first chemotherapy or from autologous HSCT to initiation of GM-CSF and 3F8 anti-GD2 antibody treatment. The autologous HSCT group also had significantly more ultra–high-risk patients.
- In a COG phase III trial (ANBL0032 [NCT00026312]), participants who had previously undergone HSCT were randomly assigned to receive dinutuximab administered with GM-CSF and IL-2 in conjunction with isotretinoin, versus isotretinoin alone.[ 48 ]
- A European study compared dinutuximab-beta (dinutuximab manufactured in hamster cells instead of mouse cells) to dinutuximab-beta plus subcutaneous (SQ) IL-2 administered as maintenance therapy after high-dose chemotherapy with autologous HSCT. All patients additionally received isotretinoin.[ 56 ]
- A second SIOPEN trial reported the following:[ 56 ]
- A third randomized, phase II, SIOPEN trial compared treatment with IL-2 to treatment without IL-2.[ 57 ]
Based on the SIOPEN data, the COG removed IL-2 from standard postconsolidation immunotherapy.
Radioactive MIBG therapy has been used to treat recurrent neuroblastoma with some success. This therapy has been shown to be safe and feasible to incorporate into the treatment regimen for children with newly diagnosed high-risk neuroblastoma.[ 58 ] A randomized trial (ANBL1531 [NCT03126916]) incorporating radioactive MIBG therapy into the complex therapy for newly diagnosed high-risk neuroblastoma has completed accrual.
A multi-institution, phase II clinical trial of children with high-risk neuroblastoma evaluated 2 years of continuation therapy using eflornithine (previously known as difluoromethylornithine [DFMO]), an oral ornithine decarboxylase inhibitor.[ 59 ] Although the study concluded that survival was improved compared with a subset of patients who were previously treated in the ANBL0032 (NCT00026312) trial, the historical comparison and potential patient selection bias limit the validity of this finding. An updated report describes the results of a propensity matching analysis that compared patients who were treated with eflornithine with patients in the ANBL0032 trial who were not treated with eflornithine.[ 60 ] Propensity matching generally balanced differences in available patient characteristics. In the matched analysis, patients in the eflornithine cohort had statistically significantly higher EFS and OS, compared with patients in the non-eflornithine cohort (4-year EFS rates, 84% vs. 73% and 4-year OS rates, 96% vs. 84%). The authors noted that uncontrolled confounders may exist in this nonrandomized comparison. Based on these results, the FDA approved the use of eflornithine as continuation therapy in December 2023.
A GD2/GD3 ganglioside vaccine has been studied for patients in first remission after completion of standard therapy. In a randomized trial that mainly included patients in first remission, early introduction of beta-glucan along with a GD2/GD3 vaccine increased GD2/GD3 antibody titers without increasing toxicity. Progression-free survival (PFS) rates were similar for patients in both randomized treatment arms. However, patients with higher titers had more favorable PFS rates, regardless of the treatment arm.[ 61 ][Level of evidence B1]
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
参考文献- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Cotterill SJ, Pearson AD, Pritchard J, et al.: Late relapse and prognosis for neuroblastoma patients surviving 5 years or more: a report from the European Neuroblastoma Study Group "Survey". Med Pediatr Oncol 36 (1): 235-8, 2001.[PUBMED Abstract]
- Mertens AC, Yasui Y, Neglia JP, et al.: Late mortality experience in five-year survivors of childhood and adolescent cancer: the Childhood Cancer Survivor Study. J Clin Oncol 19 (13): 3163-72, 2001.[PUBMED Abstract]
- Morgenstern DA, London WB, Stephens D, et al.: Metastatic neuroblastoma confined to distant lymph nodes (stage 4N) predicts outcome in patients with stage 4 disease: A study from the International Neuroblastoma Risk Group Database. J Clin Oncol 32 (12): 1228-35, 2014.[PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Cardenas FI, et al.: Stage 4N neuroblastoma before and during the era of anti-GD2 immunotherapy. Int J Cancer 153 (12): 2019-2031, 2023.[PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Bonilla MA, et al.: Highly effective induction therapy for stage 4 neuroblastoma in children over 1 year of age. J Clin Oncol 12 (12): 2607-13, 1994.[PUBMED Abstract]
- Park JR, Scott JR, Stewart CF, et al.: Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high-risk neuroblastoma: a Children's Oncology Group study. J Clin Oncol 29 (33): 4351-7, 2011.[PUBMED Abstract]
- Decarolis B, Schneider C, Hero B, et al.: Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol 31 (7): 944-51, 2013.[PUBMED Abstract]
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.[PUBMED Abstract]
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.[PUBMED Abstract]
- Furman WL, McCarville B, Shulkin BL, et al.: Improved Outcome in Children With Newly Diagnosed High-Risk Neuroblastoma Treated With Chemoimmunotherapy: Updated Results of a Phase II Study Using hu14.18K322A. J Clin Oncol 40 (4): 335-344, 2022.[PUBMED Abstract]
- Berthold F, Faldum A, Ernst A, et al.: Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: results of the randomized open-label GPOH trial NB2004-HR. Ann Oncol 31 (3): 422-429, 2020.[PUBMED Abstract]
- Garaventa A, Poetschger U, Valteau-Couanet D, et al.: Randomized Trial of Two Induction Therapy Regimens for High-Risk Neuroblastoma: HR-NBL1.5 International Society of Pediatric Oncology European Neuroblastoma Group Study. J Clin Oncol 39 (23): 2552-2563, 2021.[PUBMED Abstract]
- De Ioris MA, Crocoli A, Contoli B, et al.: Local control in metastatic neuroblastoma in children over 1 year of age. BMC Cancer 15: 79, 2015.[PUBMED Abstract]
- von Allmen D, Davidoff AM, London WB, et al.: Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol 35 (2): 208-216, 2017.[PUBMED Abstract]
- Englum BR, Rialon KL, Speicher PJ, et al.: Value of surgical resection in children with high-risk neuroblastoma. Pediatr Blood Cancer 62 (9): 1529-35, 2015.[PUBMED Abstract]
- DeCou JM, Bowman LC, Rao BN, et al.: Infants with metastatic neuroblastoma have improved survival with resection of the primary tumor. J Pediatr Surg 30 (7): 937-40; discussion 940-1, 1995.[PUBMED Abstract]
- Castel V, Tovar JA, Costa E, et al.: The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37 (11): 1574-8, 2002.[PUBMED Abstract]
- Simon T, Häberle B, Hero B, et al.: Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31 (6): 752-8, 2013.[PUBMED Abstract]
- Adkins ES, Sawin R, Gerbing RB, et al.: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39 (6): 931-6, 2004.[PUBMED Abstract]
- Seemann NM, Erker C, Irwin MS, et al.: Survival effect of complete surgical resection of the primary tumor in patients with metastatic, high-risk neuroblastoma in a large Canadian cohort. Pediatr Blood Cancer 70 (6): e30286, 2023.[PUBMED Abstract]
- Holmes K, Pötschger U, Pearson ADJ, et al.: Influence of Surgical Excision on the Survival of Patients With Stage 4 High-Risk Neuroblastoma: A Report From the HR-NBL1/SIOPEN Study. J Clin Oncol 38 (25): 2902-2915, 2020.[PUBMED Abstract]
- Yoneda A, Shichino H, Hishiki T, et al.: A nationwide phase II study of delayed local treatment for children with high-risk neuroblastoma: The Japan Children's Cancer Group Neuroblastoma Committee Trial JN-H-11. Pediatr Blood Cancer 71 (6): e30976, 2024.[PUBMED Abstract]
- Desai AV, Applebaum MA, Karrison TG, et al.: Efficacy of post-induction therapy for high-risk neuroblastoma patients with end-induction residual disease. Cancer 128 (15): 2967-2977, 2022.[PUBMED Abstract]
- Matthay KK, Villablanca JG, Seeger RC, et al.: Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med 341 (16): 1165-73, 1999.[PUBMED Abstract]
- Berthold F, Boos J, Burdach S, et al.: Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol 6 (9): 649-58, 2005.[PUBMED Abstract]
- Pritchard J, Cotterill SJ, Germond SM, et al.: High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatr Blood Cancer 44 (4): 348-57, 2005.[PUBMED Abstract]
- Elborai Y, Hafez H, Moussa EA, et al.: Comparison of toxicity following different conditioning regimens (busulfan/melphalan and carboplatin/etoposide/melphalan) for advanced stage neuroblastoma: Experience of two transplant centers. Pediatr Transplant 20 (2): 284-9, 2016.[PUBMED Abstract]
- Ladenstein R, Pötschger U, Pearson ADJ, et al.: Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol 18 (4): 500-514, 2017.[PUBMED Abstract]
- Park JR, Kreissman SG, London WB, et al.: Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 322 (8): 746-755, 2019.[PUBMED Abstract]
- Matthay KK, Reynolds CP, Seeger RC, et al.: Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J Clin Oncol 27 (7): 1007-13, 2009.[PUBMED Abstract]
- Yalçin B, Kremer LC, Caron HN, et al.: High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst Rev 8: CD006301, 2013.[PUBMED Abstract]
- Hale GA, Arora M, Ahn KW, et al.: Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience. Bone Marrow Transplant 48 (8): 1056-64, 2013.[PUBMED Abstract]
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.[PUBMED Abstract]
- Haas-Kogan DA, Swift PS, Selch M, et al.: Impact of radiotherapy for high-risk neuroblastoma: a Children's Cancer Group study. Int J Radiat Oncol Biol Phys 56 (1): 28-39, 2003.[PUBMED Abstract]
- Casey DL, Kushner BH, Cheung NK, et al.: Local Control With 21-Gy Radiation Therapy for High-Risk Neuroblastoma. Int J Radiat Oncol Biol Phys 96 (2): 393-400, 2016.[PUBMED Abstract]
- Gatcombe HG, Marcus RB, Katzenstein HM, et al.: Excellent local control from radiation therapy for high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 74 (5): 1549-54, 2009.[PUBMED Abstract]
- Liu KX, Naranjo A, Zhang FF, et al.: Prospective Evaluation of Radiation Dose Escalation in Patients With High-Risk Neuroblastoma and Gross Residual Disease After Surgery: A Report From the Children's Oncology Group ANBL0532 Study. J Clin Oncol 38 (24): 2741-2752, 2020.[PUBMED Abstract]
- Casey DL, Kushner BH, Cheung NV, et al.: Dose-escalation is needed for gross disease in high-risk neuroblastoma. Pediatr Blood Cancer 65 (7): e27009, 2018.[PUBMED Abstract]
- Braunstein SE, London WB, Kreissman SG, et al.: Role of the extent of prophylactic regional lymph node radiotherapy on survival in high-risk neuroblastoma: A report from the COG A3973 study. Pediatr Blood Cancer 66 (7): e27736, 2019.[PUBMED Abstract]
- Liu KX, Shaaban SG, Chen JJ, et al.: Patterns of recurrence after radiotherapy for high-risk neuroblastoma: Implications for radiation dose and field. Radiother Oncol 198: 110384, 2024.[PUBMED Abstract]
- Polishchuk AL, Li R, Hill-Kayser C, et al.: Likelihood of bone recurrence in prior sites of metastasis in patients with high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 89 (4): 839-45, 2014.[PUBMED Abstract]
- Li R, Polishchuk A, DuBois S, et al.: Patterns of Relapse in High-Risk Neuroblastoma Patients Treated With and Without Total Body Irradiation. Int J Radiat Oncol Biol Phys 97 (2): 270-277, 2017.[PUBMED Abstract]
- Mazloom A, Louis CU, Nuchtern J, et al.: Radiation therapy to the primary and postinduction chemotherapy MIBG-avid sites in high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 90 (4): 858-62, 2014.[PUBMED Abstract]
- Rossillon L, Edeline V, Agrigoroaie L, et al.: Rationale for irradiation of persisting oligo-skeletal metastases to improve survival of metastatic neuroblastoma patients with a poor response to chemotherapy: A retrospective study. Pediatr Blood Cancer 72 (1): e31350, 2025.[PUBMED Abstract]
- Casey DL, Pitter KL, Kushner BH, et al.: Radiation Therapy to Sites of Metastatic Disease as Part of Consolidation in High-Risk Neuroblastoma: Can Long-term Control Be Achieved? Int J Radiat Oncol Biol Phys 100 (5): 1204-1209, 2018.[PUBMED Abstract]
- Hattangadi JA, Rombi B, Yock TI, et al.: Proton radiotherapy for high-risk pediatric neuroblastoma: early outcomes and dose comparison. Int J Radiat Oncol Biol Phys 83 (3): 1015-22, 2012.[PUBMED Abstract]
- Yu AL, Gilman AL, Ozkaynak MF, et al.: Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363 (14): 1324-34, 2010.[PUBMED Abstract]
- Cheung NK, Cheung IY, Kushner BH, et al.: Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 30 (26): 3264-70, 2012.[PUBMED Abstract]
- Kushner BH, Ostrovnaya I, Cheung IY, et al.: Lack of survival advantage with autologous stem-cell transplantation in high-risk neuroblastoma consolidated by anti-GD2 immunotherapy and isotretinoin. Oncotarget 7 (4): 4155-66, 2016.[PUBMED Abstract]
- Yu AL, Gilman AL, Ozkaynak MF, et al.: Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin Cancer Res 27 (8): 2179-2189, 2021.[PUBMED Abstract]
- Desai AV, Gilman AL, Ozkaynak MF, et al.: Outcomes Following GD2-Directed Postconsolidation Therapy for Neuroblastoma After Cessation of Random Assignment on ANBL0032: A Report From the Children's Oncology Group. J Clin Oncol 40 (35): 4107-4118, 2022.[PUBMED Abstract]
- Forlenza CJ, Boudreau JE, Zheng J, et al.: KIR3DL1 Allelic Polymorphism and HLA-B Epitopes Modulate Response to Anti-GD2 Monoclonal Antibody in Patients With Neuroblastoma. J Clin Oncol 34 (21): 2443-51, 2016.[PUBMED Abstract]
- Venstrom JM, Zheng J, Noor N, et al.: KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin Cancer Res 15 (23): 7330-4, 2009.[PUBMED Abstract]
- Erbe AK, Wang W, Carmichael L, et al.: Neuroblastoma Patients' KIR and KIR-Ligand Genotypes Influence Clinical Outcome for Dinutuximab-based Immunotherapy: A Report from the Children's Oncology Group. Clin Cancer Res 24 (1): 189-196, 2018.[PUBMED Abstract]
- Ladenstein R, Pötschger U, Valteau-Couanet D, et al.: Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 19 (12): 1617-1629, 2018.[PUBMED Abstract]
- Ladenstein RL, Poetschger U, Valteau-Couanet D, et al.: Randomization of dose-reduced subcutaneous interleukin-2 (scIL2) in maintenance immunotherapy (IT) with anti-GD2 antibody dinutuximab beta (DB) long-term infusion (LTI) in front–line high-risk neuroblastoma patients: Early results from the HR-NBL1/SIOPEN trial. [Abstract] J Clin Oncol 37 (suppl 15): A-10013, 2019. Also available online. Last accessed August 21, 2023.[PUBMED Abstract]
- Weiss BD, Yanik G, Naranjo A, et al.: A safety and feasibility trial of 131 I-MIBG in newly diagnosed high-risk neuroblastoma: A Children's Oncology Group study. Pediatr Blood Cancer 68 (10): e29117, 2021.[PUBMED Abstract]
- Sholler GLS, Ferguson W, Bergendahl G, et al.: Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci Rep 8 (1): 14445, 2018.[PUBMED Abstract]
- Oesterheld J, Ferguson W, Kraveka JM, et al.: Eflornithine as Postimmunotherapy Maintenance in High-Risk Neuroblastoma: Externally Controlled, Propensity Score-Matched Survival Outcome Comparisons. J Clin Oncol 42 (1): 90-102, 2024.[PUBMED Abstract]
- Cheung IY, Mauguen A, Modak S, et al.: Effect of Oral β-Glucan on Antibody Response to Ganglioside Vaccine in Patients With High-Risk Neuroblastoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 9 (2): 242-250, 2023.[PUBMED Abstract]
- Treatment of INSS Stage 4S and INRG Stage MS Neuroblastoma
-
International Neuroblastoma Staging System (INSS) stage 4S patients are younger than 12 months and have an INSS stage 1 or stage 2 primary tumor. International Neuroblastoma Risk Group (INRG) stage MS patients are younger than 18 months with any stage of primary tumor. Both staging systems have the same definition of limited pattern of metastases.
The decision by the INRG Task Force to replace the category of 4S disease with that of the new MS definition was based on reports in which small numbers of infants with L2 primary tumors and 4S metastatic patterns, including patients aged 12 to 18 months, had favorable outcomes.[ 1 ][ 2 ] A subsequent study of the actual INRG data found that a number of biological characteristics predicted poor outcome of patients aged 12 to 18 months with stage MS disease, and that only those infants with favorable biology had long-term outcomes similar to those with the traditional 4S diagnosis.[ 2 ]
Infants with INRG stage MS disease have more favorable biology and superior outcomes despite receiving less aggressive therapy. The 5-year event-free survival (EFS) rate was 86%, and the overall survival (OS) rate was 95%. For patients with MYCN-amplified tumors, the 5-year EFS rate was 60%, and the OS rate was 65%.[ 3 ]
Many patients with stage 4S/MS neuroblastoma do not require therapy. However, tumors with unfavorable biology or patients who are symptomatic because of evolving hepatomegaly and organ compromise are at increased risk of death and are treated with low-dose to moderate-dose chemotherapy. Eight percent to 10% of these patients will have MYCN amplification and are treated with high-risk treatment regimens.[ 4 ]
For more information about the Children's Oncology Group (COG) classification schema for stage 4S/MS neuroblastoma, see Table 3.
Treatment Options for Stage 4S/MS Neuroblastoma
There is no standard approach for the treatment of stage 4S/MS neuroblastoma.
Treatment options for stage 4S/MS neuroblastoma include the following:
- Observation with supportive care (for asymptomatic patients with favorable tumor biology).
- Chemotherapy (for symptomatic patients or patients with unfavorable tumor biology).
- Surgery (rarely, for patients with hepatomegaly that compromises the kidney or other abdominal organs).
- Radiation therapy (rarely, for patients with symptoms related to hepatomegaly from metastatic disease).
Resection of the primary tumor is not associated with improved outcome.[ 5 ][ 6 ][ 7 ] Rarely, infants with massive hepatic 4S/MS neuroblastoma develop cirrhosis from the chemotherapy and/or radiation therapy that is used to control the disease and may benefit from orthotopic liver transplant.[ 8 ]
Observation with supportive care
Observation with supportive care is used to treat asymptomatic patients with favorable tumor biology.
The treatment of children with stage 4S/MS disease depends on clinical presentation.[ 5 ][ 6 ] Most patients do not require therapy unless bulky disease causes organ compromise and risk of death.
Chemotherapy
Chemotherapy is used to treat symptomatic patients or patients with unfavorable tumor biology. Patients with evidence of rapid tumor growth in the first several weeks of life require immediate intervention with chemotherapy to avoid potentially irreversible abdominal compartment syndrome and hepatic and/or renal failure.[ 9 ]
Infants diagnosed with INSS stage 4S/MS neuroblastoma, particularly those with hepatomegaly or those younger than 2 months with high-risk features or hepatomegaly, have the potential for rapid clinical deterioration and may benefit from early initiation of therapy.[ 9 ] It has been difficult to identify infants with stage 4S disease who will benefit from chemotherapy.
A scoring system to measure signs and symptoms of deterioration or compromise was developed to better assess this group of stage 4S patients.[ 10 ] This scoring system has been evaluated retrospectively, was predictive of the clinical course, and has been applied prospectively to guide the management of patients with INSS stage 4S disease.[ 10 ][ 11 ] The scoring system has been modified based on the ANBL0531 (NCT00499616) study results in the youngest infants discussed above to guide chemotherapeutic intervention for 4S/MS in infants.[ 9 ]
Various chemotherapy regimens (cyclophosphamide alone, carboplatin/etoposide, cyclophosphamide/doxorubicin/vincristine) have been used to treat symptomatic patients. The approach is to administer the chemotherapy only as long as symptoms persist to avoid toxicity, which contributes to poorer survival. Additionally, lower doses of chemotherapy are often recommended for very young or low-weight infants, along with granulocyte colony-stimulating factors after each cycle of chemotherapy.
Evidence (chemotherapy for 4S/MS disease):
- The COG ANBL0531 (NCT00499616) trial prospectively studied a subset of 4S patients who had MYCN-nonamplified tumors with impaired or impending organ dysfunction or unfavorable biology (unfavorable histology and/or diploid DNA index [DI]). Forty-nine patients were enrolled, 41 of whom were symptomatic and 28 of whom had unfavorable biology. Patients were assigned to receive two, four, or eight cycles of chemotherapy based on the tumor biology, age of the patient, and symptoms.[ 9 ][Level of evidence C1]
- Eighty patients with stage 4S disease were enrolled on the COG-P9641 (NCT00003119) trial. Forty-one patients with asymptomatic stage 4S neuroblastoma were treated with surgery or biopsy alone, and 39 patients were treated with surgery and chemotherapy.[ 12 ]
- Also, on the COG-P9641 trial, asymptomatic infants with biologically favorable (MYCN-nonamplified) INSS stage 4S disease did not receive chemotherapy until the development of progressive disease or clinical symptoms.[ 12 ]
- For the COG-ANBL0531 trial, treatment was allocated based on symptoms, patient age, and tumor biology.[ 9 ]
- A prospective study was performed in 125 infants with stage 4S MYCN-nonamplified tumors or INSS stage 3 primary tumors and/or positive bone scintigraphy not associated with changes in the cortical bone (documented on plain radiographs and/or computed tomography).[
11
] A pretreatment symptom score was used to determine initial treatment. Observation was recommended for infants with low symptom scores (n = 86), and chemotherapy was recommended for infants with high symptom scores (n = 37).
The chemotherapy for patients with high symptom scores included two to four 3-day courses of carboplatin and etoposide. If symptoms persisted or progressive disease developed, up to four 5-day courses of cyclophosphamide, doxorubicin, and vincristine were administered. One-half of the patients underwent complete or partial resection of the primary tumor.
Surgery
Occasionally, if the liver becomes too large and is compromising the kidney and other abdominal organs, a decompressive laparotomy may be necessary,[ 13 ][ 14 ] although this would typically be an indication for chemotherapy as well. Likewise, emergent surgical abdominal decompression can be used to avoid respiratory deterioration and improve ventilation.[ 13 ][ 14 ]
Radiation therapy
In rare cases of marked hepatomegaly in symptomatic MS (4S) infants with neuroblastoma who were unresponsive to chemotherapy, very low-dose radiation therapy has been used. In a series of 41 symptomatic infants with MS disease, radiation therapy was administered to five infants, three of whom died.[ 9 ]
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
参考文献- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.[PUBMED Abstract]
- Taggart DR, London WB, Schmidt ML, et al.: Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol 29 (33): 4358-64, 2011.[PUBMED Abstract]
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.[PUBMED Abstract]
- Canete A, Gerrard M, Rubie H, et al.: Poor survival for infants with MYCN-amplified metastatic neuroblastoma despite intensified treatment: the International Society of Paediatric Oncology European Neuroblastoma Experience. J Clin Oncol 27 (7): 1014-9, 2009.[PUBMED Abstract]
- Guglielmi M, De Bernardi B, Rizzo A, et al.: Resection of primary tumor at diagnosis in stage IV-S neuroblastoma: does it affect the clinical course? J Clin Oncol 14 (5): 1537-44, 1996.[PUBMED Abstract]
- Katzenstein HM, Bowman LC, Brodeur GM, et al.: Prognostic significance of age, MYCN oncogene amplification, tumor cell ploidy, and histology in 110 infants with stage D(S) neuroblastoma: the pediatric oncology group experience--a pediatric oncology group study. J Clin Oncol 16 (6): 2007-17, 1998.[PUBMED Abstract]
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.[PUBMED Abstract]
- Steele M, Jones NL, Ng V, et al.: Successful liver transplantation in an infant with stage 4S(M) neuroblastoma. Pediatr Blood Cancer 60 (3): 515-7, 2013.[PUBMED Abstract]
- Twist CJ, Naranjo A, Schmidt ML, et al.: Defining Risk Factors for Chemotherapeutic Intervention in Infants With Stage 4S Neuroblastoma: A Report From Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (2): 115-124, 2019.[PUBMED Abstract]
- Hsu LL, Evans AE, D'Angio GJ: Hepatomegaly in neuroblastoma stage 4s: criteria for treatment of the vulnerable neonate. Med Pediatr Oncol 27 (6): 521-8, 1996.[PUBMED Abstract]
- De Bernardi B, Gerrard M, Boni L, et al.: Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol 27 (7): 1034-40, 2009.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Keene DJ, Minford J, Craigie RJ, et al.: Laparostomy closure in stage 4S neuroblastoma. J Pediatr Surg 46 (1): e1-4, 2011.[PUBMED Abstract]
- Harper L, Perel Y, Lavrand F, et al.: Surgical management of neuroblastoma-related hepatomegaly: do material and method really count? Pediatr Hematol Oncol 25 (4): 313-7, 2008.[PUBMED Abstract]
- Treatment of Recurrent or Refractory Neuroblastoma
-
Tumor growth resulting from maturation should be differentiated from tumor progression by performing a biopsy and reviewing histology. Patients may have persistent maturing disease with metaiodobenzylguanidine (MIBG) uptake that does not affect outcome, particularly patients with low-risk and intermediate-risk disease.[ 1 ] An analysis of 23 paired MIBG and positron emission tomography (PET) scans in 14 patients with refractory or recurrent high-risk neuroblastoma treated with iodine I 131-MIBG (131I-MIBG) found that the MIBG scan was more sensitive than fluorine F 18-fludeoxyglucose (18F-FDG) PET for detecting metastatic bone lesions, although there was a trend for 18F-FDG PET to be more sensitive for soft tissue lesions.[ 2 ]
Subclonal ALK variants or other MAPK pathway lesions may be present at diagnosis, with subsequent clonal expansion at relapse. Consequently, serial sampling of progressive tumors may lead to the identification of potentially actionable variants.[ 3 ][ 4 ] Modern comprehensive molecular analysis comparing primary and relapsed neuroblastoma from the same patients revealed extensive clonal enrichment and several newly discovered variants, with many tumors showing new or clonal-enriched variants in the RAS-MAPK pathway. This was true for patients with both high-risk and low-risk tumors at diagnosis.[ 5 ][ 6 ] For more information, see the Genomic and Biological Features of Neuroblastoma section.
Sequencing of recurrent and refractory neuroblastoma tumors from pediatric (n = 59) and young adult patients (n = 1) enrolled in the NCI-COG Pediatric MATCH trial revealed genomic alterations that were considered actionable for treatment in MATCH study arms in 27 of 60 tumors (45%).[ 7 ] Hotspot variants in ALK were most frequent, reported in 19 of 60 tumors (31.7%). MAPK pathway variants (NF1, NRAS) were detected in 4 of 60 tumors (6.7%), and FGFR1 variants were detected in 3 of 60 tumors (5%).
If neuroblastoma recurs in a child originally diagnosed with high-risk disease, the prognosis is usually poor despite additional intensive therapy.[ 8 ][ 9 ][ 10 ][ 11 ] However, it is often possible to gain many additional months of life for these patients with alternative chemotherapy regimens.[ 12 ][ 13 ] Clinical trials are appropriate for these patients and may be offered. Information about ongoing clinical trials is available from the NCI website.
Prognostic Factors in Patients With Recurrent Neuroblastoma
A comprehensive analysis of the patterns of relapse was conducted using the International Neuroblastoma Risk Group (INRG) database on patients diagnosed/enrolled between 1989 and 2017.[ 14 ][Level of evidence C1]
The INRG database was used to examine clinical and biological features that are prognostic of survival after relapse or progression of INRG Staging System (INRGSS) stage MS pattern neuroblastoma. Of the 1,511 patients diagnosed between 1984 and 2021 who met the eligibility criteria, 209 patients were identified as having an event. Eligibility criteria included patients younger than 365 days at initial diagnosis with INRGSS stage MS disease or with International Neuroblastoma Staging System (INSS) stage 4S, or patients aged 365 to 546 days with INSS stage 4 disease and metastasis limited to the liver, skin, and/or bone marrow.[ 15 ][Level of evidence C1]
The International Neuroblastoma Risk Group Project performed a survival-tree analysis of clinical and biological characteristics (defined at diagnosis) associated with survival after relapse in 2,266 patients with neuroblastoma entered in large clinical trials in well-established clinical trials groups around the world.[ 8 ] The survival-tree analysis revealed the following:
Significant prognostic factors determined at diagnosis for postrelapse survival include the following:[ 8 ]
The Children’s Oncology Group (COG) experience with recurrence in patients with low-risk and intermediate-risk neuroblastoma showed that most patients can be salvaged. The COG reported a 3-year event free survival (EFS) rate of 88% and an OS rate of 96% in intermediate-risk patients and a 5-year EFS rate of 89% and OS rate of 97% in low-risk patients.[ 16 ][ 17 ] Moreover, in most patients originally diagnosed with low-risk or intermediate-risk disease, local recurrence or recurrence in the 4S pattern may be treated successfully with observation alone, surgery alone, or with moderate-dose chemotherapy, without myeloablative therapy and stem cell transplant.
The OS after recurrence in children presenting with high-risk neuroblastoma is generally extremely poor. However, such patients at first relapse after complete remission or minimal residual disease (MRD) in whom relapse was a single site of soft tissue mass (a few children also had bone marrow or bone disease at relapse) had a 5-year OS rate of 35% in one single-institution study. All patients underwent surgical resection of the soft tissue disease. MYCN amplification and multifocal soft tissue disease were associated with a worse postprogression survival.[ 18 ] Older children with local recurrence, with either unfavorable International Neuroblastoma Pathology Classification at diagnosis or MYCN gene amplification, have a poor prognosis and may be treated with surgery or aggressive combination chemotherapy, or they may be offered entry into a clinical trial.
Table 7 summarizes the treatment options for recurrent neuroblastoma by INSS-based risk group.
Table 7. Treatment Options for Recurrent Neuroblastoma COG Risk-Group Assignment Treatment Options COG = Children's Oncology Group; 131I-MIBG = iodine I 131-metaiodobenzylguanidine. Locoregional recurrence in patients initially classified as low risk Surgery followed by observation or chemotherapy. Chemotherapy that may be followed by surgery. Metastatic recurrence in patients initially classified as low risk Observation (if metastatic disease is in a 4S pattern in an infant). Chemotherapy. Surgery followed by chemotherapy. Locoregional recurrence in patients initially classified as intermediate risk Surgery (complete resection). Surgery (incomplete resection) followed by chemotherapy. Radiation therapy (only for patients with disease progression after chemotherapy and second-look surgery). Metastatic recurrence in patients initially classified as intermediate risk High-risk therapy. Recurrence in patients initially classified as high risk Chemotherapy combined with immunotherapy. 131I-MIBG alone, in combination with other therapy, followed by stem cell rescue. Novel therapies, including ALK inhibitors for those patients with ALK variants. Chemotherapy. Immunotherapy. Recurrence in the central nervous system Surgery and radiation therapy. Chemotherapy in combination with surgery and radiation therapy. Novel therapeutic approaches. Recurrent Neuroblastoma in Patients Initially Classified as Low Risk
Locoregional recurrence
Treatment options for locoregional recurrent neuroblastoma initially classified as low risk include the following:
- Surgery followed by observation or chemotherapy.
- Chemotherapy that may be followed by surgery.
Local or regional recurrent cancer is resected if possible.
Patients with favorable biology and regional recurrence more than 3 months after completion of planned treatment are observed if resection of the recurrence is total or near total (≥90% resection). Those with favorable biology and a less-than-near-total resection are treated with chemotherapy.[ 16 ][ 17 ][ 19 ]
Infants younger than 1 year at the time of locoregional recurrence whose tumors have any unfavorable biological properties are observed if resection is total or near total. If the resection is less than near total, these infants are treated with chemotherapy. Chemotherapy may consist of moderate doses of carboplatin, cyclophosphamide, doxorubicin, and etoposide, or cyclophosphamide and topotecan. The cumulative dose of each agent is kept low to minimize long-term effects, as used in previous COG trials (COG-P9641 and COG-A3961).[ 16 ][ 17 ][ 19 ]
Evidence (surgery followed by observation or chemotherapy):
- A COG study of low-risk patients with stages 1, 2A, 2B, and 4S neuroblastoma enrolled 915 patients, 800 of whom were asymptomatic and treated with surgery alone followed by observation. The other patients received chemotherapy with or without surgery.[ 17 ]
Metastatic recurrence or disease refractory to standard treatment
Treatment options for metastatic recurrent neuroblastoma initially classified as low risk include the following:
- Observation.
- Chemotherapy (based on age of patient, tumor biology, and prior treatment; treatment may include intermediate-risk or high-risk therapies, as used at initial diagnosis).
- Surgery followed by chemotherapy.
Metastatic recurrent or progressive neuroblastoma in an infant initially categorized as low risk and younger than 1 year at recurrence may be treated according to tumor biology, as defined in the previous COG trials (COG-P9641 and COG-A3961):
- If the biology is completely favorable, metastasis is in a 4S pattern, and the recurrence or progression is within 3 months of diagnosis, the patient is observed symptomatically.
- If the metastatic progression or recurrence occurs more than 3 months after diagnosis or not in a 4S pattern,
then the primary tumor is resected, if possible, and chemotherapy is given.
Chemotherapy may consist of moderate doses of carboplatin, cyclophosphamide, doxorubicin, and etoposide. The cumulative dose of each agent is kept low to minimize long-term effects, as used in previous COG trials (COG-P9641 and COG-A3961).
Any child initially categorized as low risk who is older than 18 months at the time of metastatic recurrent or progressive disease and whose recurrence is not in the stage 4S pattern usually has a poor prognosis and is treated as follows:
- High-risk therapy.
Patients with metastatic recurrent neuroblastoma are treated like patients with newly diagnosed high-risk neuroblastoma. For more information, see the Treatment Options for High-Risk Neuroblastoma section.
Recurrent Neuroblastoma in Patients Initially Classified as Intermediate Risk
The COG ANBL0531 (NCT00499616) study treated patients with newly diagnosed intermediate-risk neuroblastoma with chemotherapy consisting of carboplatin, etoposide, cyclophosphamide, and doxorubicin. Retrieval therapy was included in the protocol for patients who developed progressive nonmetastatic disease within 3 years of study enrollment. Up to six cycles of cyclophosphamide and topotecan could be given to patients. Of 29 patients who received cyclophosphamide and topotecan, 18 remained event free, 9 experienced relapse, and 2 died. Twenty patients who experienced an inadequate initial response to eight cycles of chemotherapy were treated with cyclophosphamide and topotecan. Of those 20 patients, 9 patients achieved a very good partial response or better; however, 6 patients developed progressive disease or experienced relapse, and 1 patient died. This suggests that more aggressive therapy is needed for patients who do not achieve the defined treatment end point after eight cycles of chemotherapy.[ 19 ]
A COG study for intermediate-risk neuroblastoma (COG-A3961) enrolled 479 patients, 42 of whom developed disease progression. The recurrence rate was 10% for those with favorable biology and 17% for those with unfavorable biology. Thirty patients had locoregional recurrences, 11 had metastatic recurrences, and 1 had both types of recurrent disease. Six of the 42 patients died of disease, while 36 patients responded to therapy. Thus, most patients with intermediate-risk neuroblastoma and disease progression may be salvaged.[ 16 ] It is not feasible to compare these results with the results of the other COG intermediate-risk study (ANBL0531) because of differences between the classification of patients for eligibility in the two studies.[ 19 ]
Locoregional recurrence
Treatment options for locoregional recurrent neuroblastoma initially classified as intermediate risk include the following:
- Surgery (complete resection).
- Surgery (incomplete resection) followed by chemotherapy.
- Radiation therapy. Radiation therapy is considered only for patients with disease progression after chemotherapy and second-look surgery.[ 16 ]
Locoregional recurrence of neuroblastoma with favorable biology that occurs more than 3 months after completion of chemotherapy may be treated surgically. If resection is less than near total, then additional chemotherapy may be given. Chemotherapy should be selected based on the previous chemotherapy received.[ 16 ]
Metastatic recurrence
Treatment options for metastatic recurrent neuroblastoma initially classified as intermediate risk include the following:
- High-risk therapy.
Patients with metastatic recurrent neuroblastoma are treated like patients with newly diagnosed high-risk neuroblastoma. For more information, see the Treatment Options for High-Risk Neuroblastoma section.
Recurrent or Refractory Neuroblastoma in Patients Initially Classified as High Risk
Any recurrence in patients initially classified as high risk signifies a very poor prognosis.[ 8 ] Clinical trials may be considered. Palliative care should also be considered as part of the patient's treatment plan.
An analysis of several trials included 383 patients with neuroblastoma whose tumor recurred or progressed in COG modern-era, early-phase trials. The 1-year progression-free survival (PFS) rate was 21%, and the 4-year PFS rate was 6%. The OS rates were 57% at 1 year and 20% at 4 years. Less than 10% of patients experienced no subsequent recurrence or progression. MYCN amplification predicted worse PFS and OS rates.[ 20 ] Although the OS after recurrence in children presenting with high-risk neuroblastoma is generally extremely poor, patients with high-risk neuroblastoma at first relapse after complete remission or MRD in whom relapse was a single site of soft tissue mass (a few children also had bone marrow or bone disease at relapse) had a 5-year OS rate of 35% in one single-institution study.[ 18 ]
Treatment options for recurrent or refractory neuroblastoma in patients initially classified as high risk include the following:
- Chemotherapy combined with immunotherapy.
- 131I-MIBG. 131I-MIBG alone, in combination with other therapy, followed by stem cell rescue.
- Novel therapies.
- Chemotherapy (phase I/II studies).
- Immunotherapy. Novel anti-GD2 drugs have been evaluated in patients with recurrent or refractory neuroblastoma. Hu14.18 anti-GD2 has been chemically linked with IL-2 and combined with granulocyte-macrophage colony-stimulating factor (GM-CSF), and a phase II trial of this regimen reported a few durable responses.[ 29 ]
Chemotherapy combined with immunotherapy produces the best response rate and response duration of treatments for high-risk patients with disease progression.
Evidence (chemotherapy combined with immunotherapy):
- The ANBL1221 (NCT01767194) trial was the first multicenter trial to evaluate anti-GD2 therapy combined with chemotherapy in a cohort of patients with relapsed or refractory neuroblastoma. Patients in first relapse or progression were randomly assigned to receive either irinotecan/temozolomide/dinutuximab/GM-CSF (I/T/DIN/GM-CSF) or temozolomide/irinotecan/temsirolimus.[ 21 ]; [ 30 ][Level of evidence C3]
- In a retrospective cohort study of 146 patients with high-risk neuroblastoma who received chemoimmunotherapy with I/T/DIN/GM-CSF in first relapse, the following results were reported:[ 31 ][Level of evidence C2]
- Limited data are available about the use of chemotherapy backbones other than irinotecan/temozolomide as part of a chemoimmunotherapy strategy. A registry study included 24 patients with relapsed or progressive high-risk neuroblastoma who were treated with topotecan/cyclophosphamide and dinutuximab.[ 32 ]
Evidence (131I-MIBG alone or in combination with other therapies):
- For children with recurrent or refractory neuroblastoma, 131I-MIBG is an effective palliative agent and may be considered alone or in combination with chemotherapy and stem cell support in a clinical research trial.[ 33 ][ 34 ][ 35 ][ 36 ][ 37 ][ 38 ]; [ 39 ][ 40 ][Level of evidence C1]
- A North American retrospective study of more than 200 patients treated with 131I-MIBG therapy compared children who had recurrence or progression of disease with children who had stable or persistent disease since diagnosis.[ 41 ]
- Tandem consolidation using 131I-MIBG, vincristine, and irinotecan with autologous hematopoietic stem cell transplant (HSCT) followed by busulfan/melphalan with autologous HSCT was retrospectively reported in eight patients.[ 40 ]
- Single autologous HSCT with 131I-MIBG and carboplatin/etoposide/melphalan was studied in additional patients. Patients were treated after completing induction chemotherapy.[ 42 ]
- A randomized phase II trial included 105 evaluable patients who were treated with either 131I-MIBG alone, 131I-MIBG with irinotecan and vincristine, or 131I-MIBG with vorinostat.[ 43 ]
- A single-arm, phase II trial included 30 patients with relapsed or refractory neuroblastoma who were treated with 131I-MIBG and topotecan.[ 44 ]
- 131I-MIBG is associated with risk of second malignancy. In one report of 563 5-year survivors of neuroblastoma, 15.5% had received prior 131I-MIBG therapy.[ 45 ]
Evidence (chemotherapy):
- Irinotecan and temozolomide.
- A retrospective study reported on 74 patients who received 92 cycles of ifosfamide, carboplatin, and etoposide. The study included 37 patients who received peripheral blood stem cell rescue after responding to this drug combination.[ 47 ]
- Topotecan in combination with cyclophosphamide alone or with etoposide has been used in patients with recurrent disease who did not receive topotecan initially.[ 28 ][Level of evidence A2]
- In the BEACON trial, patients were randomly assigned to receive either temozolomide or topotecan and temozolomide, with or without bevacizumab.[ 26 ]
- High-dose carboplatin, irinotecan, and/or temozolomide has been used to treat patients with refractory disease or new relapses (after treatment that included topotecan) occurring off therapy (68% objective response rate). However, this regimen is not used to treat patients whose disease progresses while on therapy.[ 48 ]
A range of other immunotherapy approaches have been used in patients with relapsed neuroblastoma. Single-agent anti-GD2 monoclonal antibody therapy has shown activity in this setting. For example, a phase II trial evaluated a 10-day, long-term infusion of dinutuximab in 40 children with relapsed or refractory high-risk neuroblastoma. The study reported an objective response rate of 26%. This approach was tolerable, with no grade 4 or grade 5 events.[ 49 ]
Allogeneic transplant has a historically low success rate in recurrent or progressive neuroblastoma. In a retrospective registry study, allogeneic HSCT after a previous autologous HSCT appeared to offer no benefit. Disease recurrence remains the most common cause of treatment failure.[ 50 ] A similar conclusion was reached in a multicenter phase II trial of reduced-intensity conditioning allogeneic HSCT in 51 patients, 44 of whom had relapsed or refractory high-risk neuroblastoma. The 5-year disease-free survival (DFS) rate was 11.8%.[ 51 ]
The use of GD2-directed therapy after haploidentical transplant may be a more promising strategy. In one trial of 68 patients with relapsed neuroblastoma, the use of dinutuximab and subcutaneous interleukin-2 after haploidentical transplant was feasible, with a low rate of graft-versus-host disease. The 5-year EFS rate was 43%. Superior outcomes were obtained for patients who had complete or partial responses at the start of dinutuximab therapy. Among patients with disease after transplant, the complete response rate to anti-GD2 immunotherapy was 35%.[ 52 ]
Clinical trials of vaccines designed to induce host antiganglioside antibodies that can replicate the antineoplastic activities of intravenously administered monoclonal antibodies are ongoing. Patients also receive a beta-glucan treatment, which has a broad range of immunostimulatory effects and synergizes with anti-GD2/GD3 monoclonal antibodies. In a phase I study of 15 children with high-risk neuroblastoma, the therapy was tolerated without any dose-limiting toxicity.[ 53 ] Long-term PFS has been reported in patients who achieve a second or later complete or very good partial remission followed by consolidation with anti-GD2 immunotherapy and isotretinoin with or without maintenance therapy. This includes patients who had previously received anti-GD2 immunotherapy and isotretinoin.[ 54 ]
In a phase I/II trial, the use of autologous chimeric antigen receptor (CAR)–expressing T cells directed against GD2 was feasible and safe in treating children with relapsed or refractory, high-risk neuroblastoma. This treatment resulted in a response rate of 63%.[ 55 ] These findings contrast with earlier reports that showed only modest activity of other GD2-directed CAR T-cell approaches in this same population.
Recurrent Neuroblastoma in the Central Nervous System
Central nervous system (CNS) involvement, although rare at initial presentation, may occur in 3% to 10% of patients with recurrent neuroblastoma. CNS relapses represented 6% of all metastatic relapses in a series of 1,161 first relapses in 1,977 patients with stage 4 disease treated in a trial of patients with high-risk neuroblastoma.[ 56 ] Because up-front treatment for newly diagnosed patients does not adequately treat the CNS, the CNS has emerged as a sanctuary site leading to relapse.[ 56 ][ 57 ][ 58 ]
Significant risk factors for CNS relapse identified in the International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) trial were patient and disease features at diagnosis. These features included female sex (HR, 2.0; P = .016), MYCN amplification (HR, 2.4; P = .0008), hepatic disease (HR, 2.5; P = .01), or more than one metastatic system/compartment involvement (HR, 7.1; P = .047). Neither high-dose chemotherapy nor immunotherapy was associated with higher risk of recurrence. Investigators noted stable incidence of CNS relapse reported over time.[ 56 ]
CNS relapses are almost always fatal, with a median time to death of 6 months. The 1-year and 3-year postrelapse OS rates were 25% and 7%, respectively, in the SIOPEN trial.[ 56 ] Patients with isolated CNS relapses may be able to achieve long-term survival.[ 56 ]
Treatment options for recurrent neuroblastoma in the CNS include the following:
- Surgery and radiation therapy.
- Chemotherapy (including temozolomide-containing regimens) in combination with surgery and radiation therapy.
- Novel therapeutic approaches.
Current treatment approaches generally include eradicating bulky and microscopic residual disease in the CNS and minimal residual systemic disease that may herald further relapses. Neurosurgical interventions serve to decrease edema, control hemorrhage, and remove bulky tumor before starting therapy.
A single institution had some success while testing intraventricular compartmental radioimmunotherapy using intrathecal radioiodinated anti-B7H3 monoclonal antibodies, combined with 18 Gy or 21 Gy of craniospinal irradiation with boosts to gross CNS disease, in patients with recurrent metastatic CNS neuroblastoma.[ 13 ] The posttreatment 5-year CNS DFS rate was about 69%, and the 5-year OS rate was about 45%.[ 59 ][Level of evidence C2]
For patients who experience prolonged survival after an initial CNS relapse, some may develop a second relapse after cranial spinal irradiation (CSI). Published data for patients who experience a second CNS relapse are limited. A second CNS relapse indicates a poor prognosis.[ 57 ]
In a single-institution study that included 128 patients treated with CSI for first CNS relapse, 40 developed a second CNS relapse at a median of 6.3 months from the initial CSI treatment. Patient outcomes after second CNS relapse are poor, although treatment with radiation therapy at the time of second CNS relapse may be associated with longer OS.[ 60 ][Level of evidence C1]
Treatment Options Under Clinical Evaluation for Recurrent or Refractory Neuroblastoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
参考文献- Marachelian A, Shimada H, Sano H, et al.: The significance of serial histopathology in a residual mass for outcome of intermediate risk stage 3 neuroblastoma. Pediatr Blood Cancer 58 (5): 675-81, 2012.[PUBMED Abstract]
- Taggart DR, Han MM, Quach A, et al.: Comparison of iodine-123 metaiodobenzylguanidine (MIBG) scan and [18F]fluorodeoxyglucose positron emission tomography to evaluate response after iodine-131 MIBG therapy for relapsed neuroblastoma. J Clin Oncol 27 (32): 5343-9, 2009.[PUBMED Abstract]
- Schleiermacher G, Javanmardi N, Bernard V, et al.: Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 32 (25): 2727-34, 2014.[PUBMED Abstract]
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.[PUBMED Abstract]
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.[PUBMED Abstract]
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.[PUBMED Abstract]
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.[PUBMED Abstract]
- London WB, Castel V, Monclair T, et al.: Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol 29 (24): 3286-92, 2011.[PUBMED Abstract]
- Pole JG, Casper J, Elfenbein G, et al.: High-dose chemoradiotherapy supported by marrow infusions for advanced neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 9 (1): 152-8, 1991.[PUBMED Abstract]
- Castel V, Cañete A, Melero C, et al.: Results of the cooperative protocol (N-III-95) for metastatic relapses and refractory neuroblastoma. Med Pediatr Oncol 35 (6): 724-6, 2000.[PUBMED Abstract]
- Lau L, Tai D, Weitzman S, et al.: Factors influencing survival in children with recurrent neuroblastoma. J Pediatr Hematol Oncol 26 (4): 227-32, 2004.[PUBMED Abstract]
- Saylors RL, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001.[PUBMED Abstract]
- Kramer K, Kushner BH, Modak S, et al.: Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol 97 (3): 409-18, 2010.[PUBMED Abstract]
- Vo KT, DuBois SG, Neuhaus J, et al.: Pattern and predictors of sites of relapse in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 69 (9): e29616, 2022.[PUBMED Abstract]
- Campbell K, Kao PC, Naranjo A, et al.: Clinical and biological features prognostic of survival after relapse or progression of INRGSS stage MS pattern neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 70 (2): e30054, 2023.[PUBMED Abstract]
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.[PUBMED Abstract]
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.[PUBMED Abstract]
- Murphy JM, Lim II, Farber BA, et al.: Salvage rates after progression of high-risk neuroblastoma with a soft tissue mass. J Pediatr Surg 51 (2): 285-8, 2016.[PUBMED Abstract]
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.[PUBMED Abstract]
- London WB, Bagatell R, Weigel BJ, et al.: Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children's Oncology Group early-phase trials. Cancer 123 (24): 4914-4923, 2017.[PUBMED Abstract]
- Mody R, Naranjo A, Van Ryn C, et al.: Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol 18 (7): 946-957, 2017.[PUBMED Abstract]
- Foster JH, Voss SD, Hall DC, et al.: Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children's Oncology Group Study (ADVL0912). Clin Cancer Res 27 (13): 3543-3548, 2021.[PUBMED Abstract]
- Goldsmith KC, Park JR, Kayser K, et al.: Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med 29 (5): 1092-1102, 2023.[PUBMED Abstract]
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.[PUBMED Abstract]
- Cole KA, Ijaz H, Surrey LF, et al.: Pediatric phase 2 trial of a WEE1 inhibitor, adavosertib (AZD1775), and irinotecan for relapsed neuroblastoma, medulloblastoma, and rhabdomyosarcoma. Cancer 129 (14): 2245-2255, 2023.[PUBMED Abstract]
- Moreno L, Weston R, Owens C, et al.: Bevacizumab, Irinotecan, or Topotecan Added to Temozolomide for Children With Relapsed and Refractory Neuroblastoma: Results of the ITCC-SIOPEN BEACON-Neuroblastoma Trial. J Clin Oncol 42 (10): 1135-1145, 2024.[PUBMED Abstract]
- Corbacioglu S, Lode H, Ellinger S, et al.: Irinotecan and temozolomide in combination with dasatinib and rapamycin versus irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma (RIST-rNB-2011): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol 25 (7): 922-932, 2024.[PUBMED Abstract]
- London WB, Frantz CN, Campbell LA, et al.: Phase II randomized comparison of topotecan plus cyclophosphamide versus topotecan alone in children with recurrent or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol 28 (24): 3808-15, 2010.[PUBMED Abstract]
- Shusterman S, Naranjo A, Van Ryn C, et al.: Antitumor Activity and Tolerability of hu14.18-IL2 with GMCSF and Isotretinoin in Recurrent or Refractory Neuroblastoma: A Children's Oncology Group Phase II Study. Clin Cancer Res 25 (20): 6044-6051, 2019.[PUBMED Abstract]
- Mody R, Yu AL, Naranjo A, et al.: Irinotecan, Temozolomide, and Dinutuximab With GM-CSF in Children With Refractory or Relapsed Neuroblastoma: A Report From the Children's Oncology Group. J Clin Oncol 38 (19): 2160-2169, 2020.[PUBMED Abstract]
- Lerman BJ, Li Y, Carlowicz C, et al.: Progression-Free Survival and Patterns of Response in Patients With Relapsed High-Risk Neuroblastoma Treated With Irinotecan/Temozolomide/Dinutuximab/Granulocyte-Macrophage Colony-Stimulating Factor. J Clin Oncol 41 (3): 508-516, 2023.[PUBMED Abstract]
- Raiser P, Schleiermacher G, Gambart M, et al.: Chemo-immunotherapy with dinutuximab beta in patients with relapsed/progressive high-risk neuroblastoma: does chemotherapy backbone matter? Eur J Cancer 202: 114001, 2024.[PUBMED Abstract]
- DuBois SG, Groshen S, Park JR, et al.: Phase I Study of Vorinostat as a Radiation Sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for Patients with Relapsed or Refractory Neuroblastoma. Clin Cancer Res 21 (12): 2715-21, 2015.[PUBMED Abstract]
- Polishchuk AL, Dubois SG, Haas-Kogan D, et al.: Response, survival, and toxicity after iodine-131-metaiodobenzylguanidine therapy for neuroblastoma in preadolescents, adolescents, and adults. Cancer 117 (18): 4286-93, 2011.[PUBMED Abstract]
- Matthay KK, Yanik G, Messina J, et al.: Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol 25 (9): 1054-60, 2007.[PUBMED Abstract]
- Matthay KK, Tan JC, Villablanca JG, et al.: Phase I dose escalation of iodine-131-metaiodobenzylguanidine with myeloablative chemotherapy and autologous stem-cell transplantation in refractory neuroblastoma: a new approaches to Neuroblastoma Therapy Consortium Study. J Clin Oncol 24 (3): 500-6, 2006.[PUBMED Abstract]
- Matthay KK, Quach A, Huberty J, et al.: Iodine-131--metaiodobenzylguanidine double infusion with autologous stem-cell rescue for neuroblastoma: a new approaches to neuroblastoma therapy phase I study. J Clin Oncol 27 (7): 1020-5, 2009.[PUBMED Abstract]
- DuBois SG, Chesler L, Groshen S, et al.: Phase I study of vincristine, irinotecan, and ¹³¹I-metaiodobenzylguanidine for patients with relapsed or refractory neuroblastoma: a new approaches to neuroblastoma therapy trial. Clin Cancer Res 18 (9): 2679-86, 2012.[PUBMED Abstract]
- Johnson K, McGlynn B, Saggio J, et al.: Safety and efficacy of tandem 131I-metaiodobenzylguanidine infusions in relapsed/refractory neuroblastoma. Pediatr Blood Cancer 57 (7): 1124-9, 2011.[PUBMED Abstract]
- French S, DuBois SG, Horn B, et al.: 131I-MIBG followed by consolidation with busulfan, melphalan and autologous stem cell transplantation for refractory neuroblastoma. Pediatr Blood Cancer 60 (5): 879-84, 2013.[PUBMED Abstract]
- Zhou MJ, Doral MY, DuBois SG, et al.: Different outcomes for relapsed versus refractory neuroblastoma after therapy with (131)I-metaiodobenzylguanidine ((131)I-MIBG). Eur J Cancer 51 (16): 2465-72, 2015.[PUBMED Abstract]
- Yanik GA, Villablanca JG, Maris JM, et al.: 131I-metaiodobenzylguanidine with intensive chemotherapy and autologous stem cell transplantation for high-risk neuroblastoma. A new approaches to neuroblastoma therapy (NANT) phase II study. Biol Blood Marrow Transplant 21 (4): 673-81, 2015.[PUBMED Abstract]
- DuBois SG, Granger MM, Groshen S, et al.: Randomized Phase II Trial of MIBG Versus MIBG, Vincristine, and Irinotecan Versus MIBG and Vorinostat for Patients With Relapsed or Refractory Neuroblastoma: A Report From NANT Consortium. J Clin Oncol 39 (31): 3506-3514, 2021.[PUBMED Abstract]
- Sevrin F, Kolesnikov-Gauthier H, Cougnenc O, et al.: Phase II study of 131 I-metaiodobenzylguanidine with 5 days of topotecan for refractory or relapsed neuroblastoma: Results of the French study MIITOP. Pediatr Blood Cancer 70 (11): e30615, 2023.[PUBMED Abstract]
- Westerveld ASR, Tytgat GAM, van Santen HM, et al.: Long-Term Risk of Subsequent Neoplasms in 5-Year Survivors of Childhood Neuroblastoma: A Dutch Childhood Cancer Survivor Study-LATER 3 Study. J Clin Oncol 43 (2): 154-166, 2025.[PUBMED Abstract]
- Bagatell R, London WB, Wagner LM, et al.: Phase II study of irinotecan and temozolomide in children with relapsed or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol 29 (2): 208-13, 2011.[PUBMED Abstract]
- Kushner BH, Modak S, Kramer K, et al.: Ifosfamide, carboplatin, and etoposide for neuroblastoma: a high-dose salvage regimen and review of the literature. Cancer 119 (3): 665-71, 2013.[PUBMED Abstract]
- Kushner BH, Kramer K, Modak S, et al.: Differential impact of high-dose cyclophosphamide, topotecan, and vincristine in clinical subsets of patients with chemoresistant neuroblastoma. Cancer 116 (12): 3054-60, 2010.[PUBMED Abstract]
- Lode HN, Ehlert K, Huber S, et al.: Long-term, continuous infusion of single-agent dinutuximab beta for relapsed/refractory neuroblastoma: an open-label, single-arm, Phase 2 study. Br J Cancer 129 (11): 1780-1786, 2023.[PUBMED Abstract]
- Hale GA, Arora M, Ahn KW, et al.: Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience. Bone Marrow Transplant 48 (8): 1056-64, 2013.[PUBMED Abstract]
- Prete A, Lanino E, Saglio F, et al.: Phase II Study of Allogeneic Hematopoietic Stem Cell Transplantation for Children with High-Risk Neuroblastoma Using a Reduced-Intensity Conditioning Regimen: Results from the AIEOP Trial. Transplant Cell Ther 30 (5): 530.e1-530.e8, 2024.[PUBMED Abstract]
- Flaadt T, Ladenstein RL, Ebinger M, et al.: Anti-GD2 Antibody Dinutuximab Beta and Low-Dose Interleukin 2 After Haploidentical Stem-Cell Transplantation in Patients With Relapsed Neuroblastoma: A Multicenter, Phase I/II Trial. J Clin Oncol 41 (17): 3135-3148, 2023.[PUBMED Abstract]
- Kushner BH, Cheung IY, Modak S, et al.: Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res 20 (5): 1375-82, 2014.[PUBMED Abstract]
- Kushner BH, Ostrovnaya I, Cheung IY, et al.: Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-GD2 immunotherapy and isotretinoin: a prospective Phase II study. Oncoimmunology 4 (7): e1016704, 2015.[PUBMED Abstract]
- Del Bufalo F, De Angelis B, Caruana I, et al.: GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N Engl J Med 388 (14): 1284-1295, 2023.[PUBMED Abstract]
- Berlanga P, Pasqualini C, Pötschger U, et al.: Central nervous system relapse in high-risk stage 4 neuroblastoma: The HR-NBL1/SIOPEN trial experience. Eur J Cancer 144: 1-8, 2021.[PUBMED Abstract]
- Kramer K, Kushner B, Heller G, et al.: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91 (8): 1510-9, 2001.[PUBMED Abstract]
- Matthay KK, Brisse H, Couanet D, et al.: Central nervous system metastases in neuroblastoma: radiologic, clinical, and biologic features in 23 patients. Cancer 98 (1): 155-65, 2003.[PUBMED Abstract]
- Luo LY, Kramer K, Cheung NV, et al.: Reduced-dose craniospinal irradiation for central nervous system relapsed neuroblastoma. Pediatr Blood Cancer 67 (9): e28364, 2020.[PUBMED Abstract]
- Tringale KR, Wolden SL, Casey DL, et al.: Clinical outcomes of pediatric patients receiving multimodality treatment of second central nervous system relapse of neuroblastoma. Pediatr Blood Cancer 70 (2): e30075, 2023.[PUBMED Abstract]
- Latest Updates to This Summary (04/28/2025)
-
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was comprehensively reviewed and extensively revised.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
- About This PDQ Summary
-
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of neuroblastoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Neuroblastoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389190]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.