

ご利用について
医療専門家向けの本PDQがん情報要約では、小児中枢神経系非定型奇形腫様およびラブドイド腫瘍の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児中枢神経系(CNS)非定型奇形腫様/ラブドイド腫瘍に関する一般情報
-
非定型奇形腫様/ラブドイド腫瘍を含む原発性脳腫瘍は、小児の最も一般的な充実性腫瘍をともに構成する多様な疾病からなる1つのグループである。PDQ小児脳腫瘍の治療要約は主に、神経系腫瘍に関する世界保健機関の分類に従って構成されている。[ 1 ][ 2 ]脳腫瘍はその組織像によって分類されるが、腫瘍の診断と分類には、免疫組織化学的分析、細胞遺伝学的ならびに分子遺伝学的所見、および細胞分裂能の測定が用いられることが多くなっている。腫瘍存在部位およびその拡がりは治療および予後を左右する重要な因子である。神経系腫瘍の分類の詳しい説明と各種の脳腫瘍に対応する治療要約へのリンクについては、小児脳腫瘍および脊髄腫瘍の治療の概要に関するPDQ要約を参照のこと。
CNS非定型奇形腫様/ラブドイド腫瘍(AT/RT)は、3歳以下の小児が最もしばしば罹患するまれな臨床的に侵攻性の腫瘍であるが、より年長の小児や成人にも起こりうる。AT/RTの約半数が後頭蓋窩に発生する。[ 3 ]診断的評価には、脳脊髄軸の磁気共鳴画像法(MRI)および腰椎の脳脊髄液検査が挙げられる。AT/RTは、腫瘍抑制遺伝子であるSMARCB1および、頻度は少ないがSMARCA4の体細胞変異および生殖細胞変異に関連している。[ 4 ]現在、AT/RTの小児に対する標準治療は存在しない。手術、化学療法、および放射線療法で構成された集学的治療が評価段階にある。
現状の生物学的知識に基づくと、AT/RTは、さらに大きなラブドイド腫瘍ファミリーの1つである。本要約では、AT/RTという用語はCNS腫瘍のみを指し、ラブドイド腫瘍という用語はCNS腫瘍および非CNS腫瘍の両者の可能性を示している。本文内で特に断らない限り、本要約ではCNS AT/RTを対象にしている。
小児および青年がん生存者には、治療から数ヵ月または数年経過後もがん療法の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。(小児および青年がん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)に関するPDQ要約を参照のこと。)
発生率
小児CNS AT/RTについては、この腫瘍が1996年に認識されるようになったばかりであることから、正確な発生率の確定が困難である。[ 5 ]
より年齢の高い患者における発生率は不明である。しかしながら、Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry(AT/RTレジストリー)では、患者42人中12人(29%)が診断時に生後36ヵ月を超えていた。[ 9 ]
解剖学
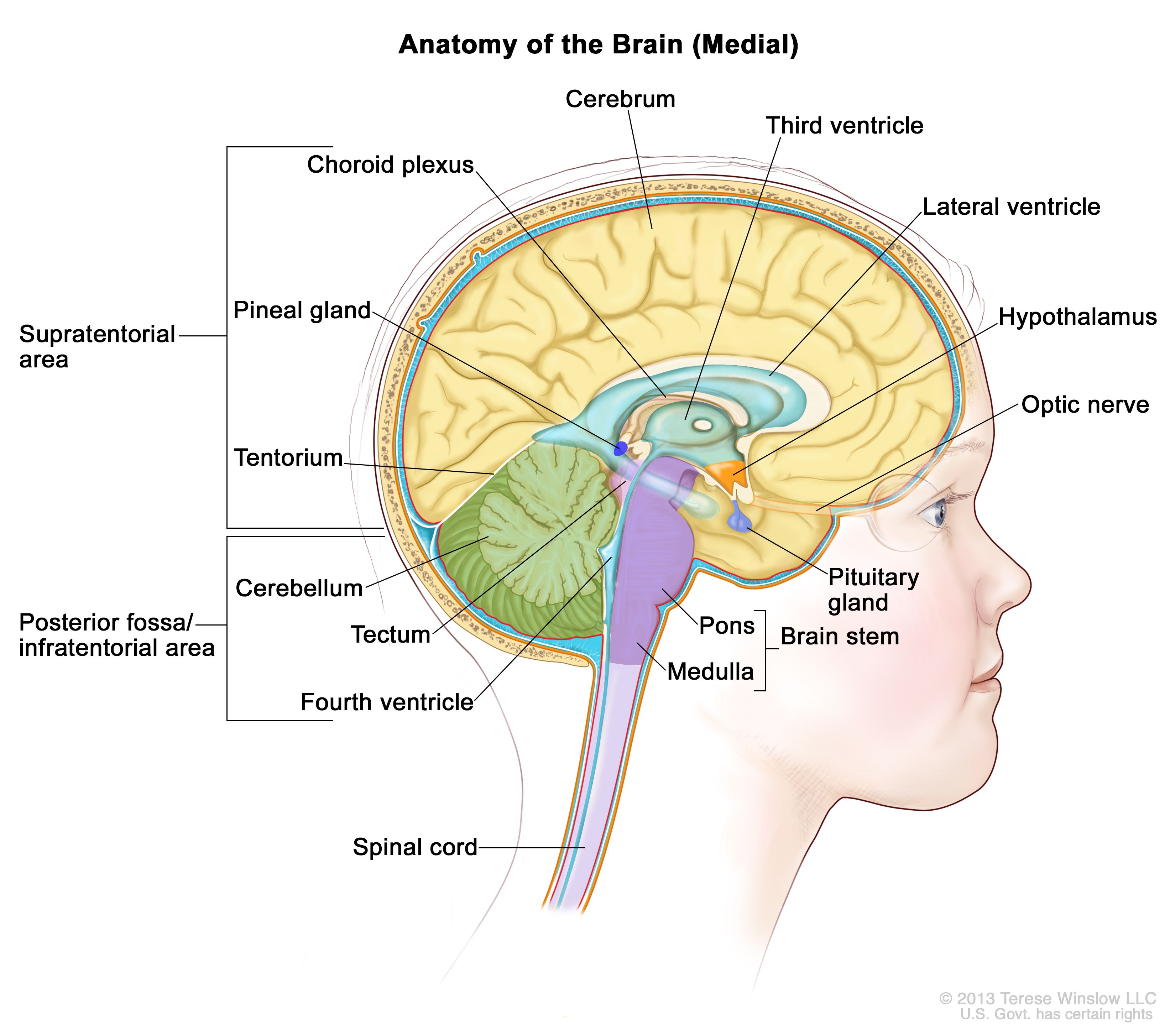
脳内部の解剖図で、松果体、下垂体、視神経、脳室(脳脊髄液を水色で示している)など、脳の構成要素が示されている。小脳テントは大脳と小脳を区切っている。テント下(後頭蓋窩)は小脳テントの下部に位置する領域で、脳幹、小脳、第四脳室が含まれている。テント上は小脳テント上部の大脳が含まれる領域を指す。 臨床像
小児AT/RTは臨床的に侵攻性の腫瘍であり、主に3歳未満の小児に発生するが、3歳以上の小児に発生することもあり、成人でも報告されている。[ 10 ][ 11 ]
腫瘍は、CNSのあらゆる部位に発生する可能性があるが、すべてのAT/RTの約半数は後頭蓋窩に発生する。[ 3 ][ 6 ]後頭蓋窩の腫瘍は、小脳橋角またはより正中線寄りに発生しうる。脳神経単独の病変が確認されている。
AT/RTは急速に増殖するため、患者は典型的に数日から数週間というかなり短期に進行する症状を有する。徴候および症状は腫瘍の位置によって異なる。後頭蓋窩腫瘍を有する若年患者は通常、水頭症に関係した以下のような症状を呈する:
運動失調または運動技能の退行を来すこともある。
レジストリーのデータから、約20%の患者が播種性病変を示すことが示唆される。[ 9 ][ 12 ]播種は典型的に軟髄膜を介して脊椎や脳の他の領域に播種する。患者の最大35%が生殖細胞変異を呈し、同時性、多病巣性腫瘍の傾向がみられる。同時性腎ラブドイド腫瘍およびCNS AT/RTを有する患者のまれな報告もある。[ 13 ][ 14 ][ 15 ]
診断的評価
小児AT/RTが疑われる患者はすべて、脳および脊椎のMRIを受けるべきである。医学的に禁忌でなければ、患者は腫瘍の証拠がないか、腰椎の脳脊髄液の検査も受けるべきである。また、同時性腫瘍を発見するための腎臓超音波検査を受けてもよい。
AT/RTと他の悪性脳腫瘍とを臨床歴またはX線の評価のみに基づいて確実に識別することはできない。組織を入手し、診断を確定するために手術が必要である。診断を確定するためにSMARCB1蛋白発現の消失について免疫染色が用いられる。[ 16 ][ 17 ]
予後
AT/RT患者の生存率に影響を及ぼす予後因子は十分に概説されていない。
不良な転帰と関連する既知の因子には以下のものがある:
AT/RT患者の転帰について公表されているほとんどのデータは、小規模シリーズからであり、実際はレトロスペクティブなデータである。初期のレトロスペクティブ研究では、診断からわずかに約12ヵ月の平均生存期間が報告された。[ 5 ][ 6 ][ 10 ][ 20 ][ 21 ]あるレトロスペクティブ報告では、2年全生存率は肉眼的全切除を受けた患者の方が亜全切除を受けた患者よりも良好であった。ただし、この研究では生存に対する放射線療法の効果は明らかにならなかった。[ 20 ]
長期生存者の報告がある。[ 22 ]注目すべきこととして、生存の改善は集中的な集学的治療法を受けた患者で報告されている。[ 12 ][ 15 ]
参考文献- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. Lyon, France: IARC Press, 2016.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Dho YS, Kim SK, Cheon JE, et al.: Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst 31 (8): 1305-11, 2015.[PUBMED Abstract]
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.[PUBMED Abstract]
- Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.[PUBMED Abstract]
- Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.[PUBMED Abstract]
- Ho DM, Hsu CY, Wong TT, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99 (5): 482-8, 2000.[PUBMED Abstract]
- Woehrer A, Slavc I, Waldhoer T, et al.: Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116 (24): 5725-32, 2010.[PUBMED Abstract]
- Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.[PUBMED Abstract]
- Burger PC, Yu IT, Tihan T, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22 (9): 1083-92, 1998.[PUBMED Abstract]
- Lutterbach J, Liegibel J, Koch D, et al.: Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol 52 (1): 49-56, 2001.[PUBMED Abstract]
- Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.[PUBMED Abstract]
- Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.[PUBMED Abstract]
- Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.[PUBMED Abstract]
- Seeringer A, Reinhard H, Hasselblatt M, et al.: Synchronous congenital malignant rhabdoid tumor of the orbit and atypical teratoid/rhabdoid tumor--feasibility and efficacy of multimodal therapy in a long-term survivor. Cancer Genet 207 (9): 429-33, 2014.[PUBMED Abstract]
- Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.[PUBMED Abstract]
- Margol AS, Judkins AR: Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet 207 (9): 358-64, 2014.[PUBMED Abstract]
- Kordes U, Gesk S, Frühwald MC, et al.: Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer 49 (2): 176-81, 2010.[PUBMED Abstract]
- Dufour C, Beaugrand A, Le Deley MC, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118 (15): 3812-21, 2012.[PUBMED Abstract]
- Lafay-Cousin L, Hawkins C, Carret AS, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48 (3): 353-9, 2012.[PUBMED Abstract]
- Athale UH, Duckworth J, Odame I, et al.: Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 31 (9): 651-63, 2009.[PUBMED Abstract]
- Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.[PUBMED Abstract]
- Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.[PUBMED Abstract]
- Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.[PUBMED Abstract]
- 小児CNS非定型奇形腫様/ラブドイド腫瘍に関する腫瘍の生物学
-
小児中枢神経系(CNS)非定型奇形腫様/ラブドイド腫瘍(AT/RT)は、その特有の病理的および遺伝的特徴に基づいて、別個の疾患実体として、1987年に初めて報告された。[ 1 ]それ以前は、髄芽腫、CNS原始神経外胚葉性腫瘍(CNS PNET)、または脈絡叢がんとして最もしばしば分類されていた。世界保健機関(WHO)は、AT/RTを1993年に胎児性悪性度IV新生物(embryonal grade IV neoplasm)として分類し始めた。[ 2 ]
組織学的にみると、AT/RTは形態学的に不均一で、典型的には好酸性細胞質が豊富な大型類上皮細胞層、および散在したラブドイド細胞層を含み、ほとんどの場合、原始神経外胚葉細胞(小円形青色細胞)、間葉細胞、および/またはグリア細胞のような成分を伴っている。[ 3 ]
上皮マーカー(サイトケラチンまたは上皮膜抗原)、グリア線維酸性蛋白、シナプトフィジン(またはニューロフィラメント)、および平滑筋(デスミン)の免疫組織化学染色は、分化の不均一性の特定に有用な場合があるが、細胞組成によって変動する。[ 4 ]ラブドイド細胞は、すべてのAT/RTに存在するわけではないが、ビメンチン、上皮膜抗原、および平滑筋アクチンを発現している。
SMARCB1蛋白の免疫組織化学検査は、AT/RTの診断確定に有用である。腫瘍細胞ではSMARCB1染色陰性であるが、非腫瘍細胞(例、血管内皮細胞)では染色は陽性を保っている。[ 5 ][ 6 ][ 7 ]
AT/RTは、50~100%のMIB-1 labeling indexを有しうる急速に増殖する腫瘍である。[ 8 ]
CNS非定型奇形腫様/ラブドイド腫瘍(AT/RT)に関するゲノム学
SMARCB1遺伝子
AT/RTは、腫瘍抑制遺伝子候補のSMARCB1(以前はINI1およびhSNF5として知られていた)が同定された初めての原発性小児脳腫瘍であった。[ 9 ]SMARCB1は、CNS、腎、および腎外のラブドイド悪性腫瘍を含むほとんどのラブドイド腫瘍でゲノム的に変化している。[ 9 ]SMARCB1/SMARCA4染色喪失はAT/RTの決定マーカーである。SMARCB1関連AT/RT患者における他の遺伝子の追加のゲノム変化(変異と増加/欠失)はきわめてまれである。まれに、SMARCA4陰性(SMARCB1が保持されている)腫瘍が報告されている。[ 10 ]AT/RTにおいて反復的な変異を生じる他の遺伝子はない。[ 11 ][ 12 ][ 13 ]
SMARCB1は、Switch(SWI)型およびSucrose non-fermenting(SNF)型のアデノシン三リン酸依存性クロマチン再構築因子複合体の成分である。[ 14 ]SMARCB1を発現しているが、SMARCB1変異を認めない、まれなラブドイド腫瘍の家族性症例では、SWI/SNFクロマチン再構築因子複合体の別のメンバーであるSMARCA4/BRG1の生殖細胞変異との関連も認められている。[ 7 ][ 15 ]
2016年WHO分類では、SMARCB1またはSMARCA4のいずれかの変化の存在によりAT/RTが定義される。AT/RTの組織学的特徴を示し、これらのゲノム変化がみられない腫瘍は、ラブドイドの特徴を示すCNS胚芽腫と呼ばれる。[ 2 ]
SMARCB1(および、さらにまれに、他のSWI/SNF複合体メンバー)のほかに反復性のゲノム変化が認められないにもかかわらず、生物学的に異なるAT/RTサブセットが同定されている。[ 16 ][ 17 ]150例のAT/RT腫瘍に対するDNAメチル化アレイおよび67例のAT/RT腫瘍に対する遺伝子発現アレイを用いて、以下の3つの異なるAT/RTサブセットが同定された:[ 17 ]
かなりの割合のAT/RT患者で、体細胞変異に加えて、SMARCB1における生殖細胞変異が報告されている。[ 9 ][ 19 ]ラブドイド腫瘍の小児65人を対象とした研究では、23人(35%)に生殖細胞変異および/またはSMARCB1の欠失が認められたことが明らかにされた。[ 5 ]SMARCB1に生殖細胞変異を認める小児は、散発例より若い年齢(年齢中央値、生後約5ヵ月 vs 18ヵ月)で発症し、同時性、多病巣性腫瘍を示す場合が多かった。[ 5 ]生殖細胞変異を示す評価可能症例22人中7人は、片親がSMARCB1生殖細胞系異常のキャリアであり、そのキャリアの親のうち4人は、SMARCB1関連がんに罹患していなかったことが明らかにされた。[ 5 ]これは、AT/RTが不完全浸透性の常染色体優性遺伝パターンを示すことを指している。
複数の同胞がAT/RTに罹患しており、同一のSMARCB1変異が認められるが、両親ともSMARCB1変異/欠失が認められない家系があることから明らかなように、性腺モザイク現象も観察されている。[ 5 ][ 6 ]AT/RTと診断された小児をSMARCB1の生殖細胞変異についてスクリーニングすることで、その子のAT/RT診断の遺伝的関係に関する家族のカウンセリングに有用な情報が得られる場合がある。[ 5 ]
SMARCB1またはSMARCA4の蛋白発現喪失は、この喪失によりがん細胞がEZH2活性に依存するようになるため、治療的意義がある。[ 20 ]前臨床研究により、SMARCB1が喪失した一部のAT/RT異種移植系はEZH2阻害薬に反応し、腫瘍増殖の阻害およびときに腫瘍の退縮が認められることが示されている。[ 21 ][ 22 ]EZH2阻害薬、タゼメトスタットの研究では、SMARCB1またはSMARCA4のいずれかが喪失した腫瘍(非CNS悪性ラブドイド腫瘍および類上皮肉腫)の成人患者において客観的奏効が観察された。[ 23 ](詳しい情報については、本要約の再発小児CNS非定型奇形腫様/ラブドイド腫瘍の治療のセクションを参照のこと。)
ラブドイド腫瘍素因症候群(RTPS)
RTPSは、主にSMARCB1の生殖細胞変異に関連しており、明確に定義されている。[ 9 ][ 19 ]RTPSは、頭蓋外悪性ラブドイド腫瘍(腎または軟部組織)とAT/RTを同時発症した患者、両腎悪性ラブドイド腫瘍の患者、または悪性ラブドイド腫瘍の同胞が2人以上いる患者で強く示唆される。
この症候群では、悪性ラブドイド腫瘍が幼児および年少児に著しく発生しやすい傾向が認められる。AT/RTの3分の1までが、RTPSにおいて生じると考えられ、これらのほとんどが生後1年以内に発生する。最もよくみられるRTPSの非CNS悪性腫瘍は、腎臓の悪性ラブドイド腫瘍であり、幼児でも認められる。
(RTPSに関する詳しい情報については、ウィルムス腫瘍とその他の小児腎腫瘍の治療に関するPDQ要約のラブドイド素因症候群のセクションを参照のこと。)
篩状神経上皮腫瘍
篩状神経上皮腫瘍は組織学的および臨床的にAT/RTと異なるが、AT/RT TYRと非常に類似したゲノム的およびエピゲノム的特徴を有する。[ 18 ]AT/RTと同様に、篩状神経上皮腫瘍は若年小児(年齢中央値、1~2歳)に発生し、腫瘍細胞にSMARCB1発現は認められない。組織学的に、篩状神経上皮腫瘍は篩状の撚りひもやリボンの存在により特徴付けられるが、好酸性細胞質が豊富なラブドイド腫瘍細胞は認められない。AT/RT TYRと同様に、チロシナーゼの発現が一般的に観察される。篩状神経上皮腫瘍を有する患者の治療成績は、AT/RT TYRを有する患者の治療成績よりも良好であり、篩状神経上皮腫瘍を有する小児10人で死亡の報告は1例のみであった。[ 18 ]
参考文献- Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.[PUBMED Abstract]
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.[PUBMED Abstract]
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. Lyon, France: IARC Press, 2016.[PUBMED Abstract]
- McLendon RE, Adekunle A, Rajaram V, et al.: Embryonal central nervous system neoplasms arising in infants and young children: a pediatric brain tumor consortium study. Arch Pathol Lab Med 135 (8): 984-93, 2011.[PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.[PUBMED Abstract]
- Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.[PUBMED Abstract]
- Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.[PUBMED Abstract]
- Kleihues P, Louis DN, Scheithauer BW, et al.: The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 61 (3): 215-25; discussion 226-9, 2002.[PUBMED Abstract]
- Biegel JA, Tan L, Zhang F, et al.: Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8 (11): 3461-7, 2002.[PUBMED Abstract]
- Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.[PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.[PUBMED Abstract]
- Kieran MW, Roberts CW, Chi SN, et al.: Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer 59 (7): 1155-7, 2012.[PUBMED Abstract]
- Hasselblatt M, Isken S, Linge A, et al.: High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 52 (2): 185-90, 2013.[PUBMED Abstract]
- Biegel JA, Kalpana G, Knudsen ES, et al.: The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62 (1): 323-8, 2002.[PUBMED Abstract]
- Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.[PUBMED Abstract]
- Torchia J, Picard D, Lafay-Cousin L, et al.: Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16 (5): 569-82, 2015.[PUBMED Abstract]
- Johann PD, Erkek S, Zapatka M, et al.: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3): 379-93, 2016.[PUBMED Abstract]
- Johann PD, Hovestadt V, Thomas C, et al.: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 27 (4): 411-418, 2017.[PUBMED Abstract]
- Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.[PUBMED Abstract]
- Wilson BG, Wang X, Shen X, et al.: Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 (4): 316-28, 2010.[PUBMED Abstract]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.[PUBMED Abstract]
- Kurmasheva RT, Sammons M, Favours E, et al.: Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 64 (3): , 2017.[PUBMED Abstract]
- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.[PUBMED Abstract]
- 小児CNS非定型奇形腫様/ラブドイド腫瘍の病期情報
小児中枢神経系非定型奇形腫様/ラブドイド腫瘍に対して定義された病期分類システムは存在しない。患者は治療目的で、新規診断例または再発例、さらに脳脊髄軸播種例または非播種例で分類される。
- 新たに診断された小児CNS非定型奇形腫様/ラブドイド腫瘍の治療
-
中枢神経系(CNS)非定型奇形腫様/ラブドイド腫瘍(AT/RT)の小児に対する標準治療はまだ明確にされていない。この腫瘍の非常に侵攻性の性質を考慮して、ほとんどの患者は集中的な集学的治療法で治療される。しかしながら、ほとんどの患者が若年であることが、治療、特に放射線療法の程度を制限する。
新たに診断されたCNS AT/RTの治療法選択肢には、以下が挙げられる:
手術、化学療法、および放射線療法(集学的治療法)
外科的切除範囲は、生存に影響しうる。Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry(AT/RTレジストリー)のデータによると、外科的完全切除は、腫瘍が浸潤性であるためしばしば困難であるが、完全切除された患者は、生存期間中央値が長いことが示唆されている。[ 1 ]
化学療法は、非常に若年のAT/RT小児に対する主要な補助療法となっている。生後36ヵ月未満の小児を含む共同グループ研究により、標準化学療法レジメン単独で治療した場合は、生存が不良なことが実証された。[ 2 ]Children's Cancer Groupは、多剤化学療法で治療した生後36ヵ月未満の小児28人では、2年イベントフリー生存率(EFS)が14%であったことを報告した。[ 3 ]
大量化学療法[ 4 ][証拠レベル:3iA];[ 5 ][ 6 ][証拠レベル:3iiiDi]、髄腔内化学療法、および放射線療法のような、さまざまな併用療法を用いる強化レジメンにより、一部の患者に生存期間の延長がもたらされている。AT/RTレジストリーの患者13人が、初期治療の一環として造血幹細胞救助を伴う大量化学療法による治療を受けた。[ 1 ]これらの患者の4人(このうち2人は放射線も受けた)は、最後の報告で診断から21.5~90ヵ月経過後に疾患が進行することなく生存していた。Head Start III化学療法プロトコルで治療されていた評価可能な小児15人(全員が診断時に生後32ヵ月未満であった)中、2人は47ヵ月以上生存した。[ 7 ][証拠レベル:3iA]
放射線療法はAT/RT患者の生存にプラスの影響をもたらすようである。
証拠(放射線療法):
- AT/RTレジストリーの患者42人中13人(31%)が初期治療の一環として化学療法に加えて放射線療法を受けた。[ 1 ]照射野は、9人の小児が原発腫瘍床、4人の小児が腫瘍床および頭蓋脊髄軸であった。登録患者全体の生存期間中央値は16.75ヵ月であったのに対し、これらの患者の生存期間中央値は48ヵ月であった。
- St. Jude Children's Research HospitalのAT/RT患者31人に対するレトロスペクティブ・シリーズでは、3歳を超える患者の2年EFS率は78%で、これは3歳未満の患者のEFS率(11%)よりもかなり良好であった。[ 8 ]年長グループの生存患者では、1人を除くすべての患者(8人中7人)が、頭蓋脊髄照射に加え、造血幹細胞移植を伴う強化化学療法を受けた;年少患者では、何らかの形の放射線療法を受けたのは22人中わずか3人で、このうち2人は無病状態である。
- Surveillance Epidemiology and End Results登録によるレビューでは、3歳未満の小児において放射線療法は生存率の改善と関連しているとされた。[ 9 ]
- ラブドイド腫瘍シリーズについてのEuropean Registryでも、放射線療法は生存の改善に関連し、照射された患者における6年全生存(OS)率は66%(±0.1%)であった。[ 10 ][証拠レベル:3iA]
証拠(集学的治療法):
- Third Intergroup Rhabdomyosarcoma Study(IRS-III)では、放射線療法、髄腔内メトトレキサート、シタラビン、ヒドロコルチゾン、および全身多剤化学療法が用いられた。小規模のレトロスペクティブ・シリーズの結果は勇気付けられるものであり、このグループの患者における集学的治療に関する初のプロスペクティブ研究につながった。[ 11 ][ 12 ]
- 以前のパイロットシリーズに基づいて、新たに診断されたCNS AT/RTの小児に対してプロスペクティブ多施設試験が実施された。治療は次の5段階に分けられた:照射前療法、化学放射線療法、地固め療法、維持療法、および継続療法。髄腔内化学療法は、腰椎内と脳室内ルートから交互に実施された。放射線療法は、小児の年齢および診断時の疾患の範囲に応じて、局所(54Gy)または脳脊髄(36Gy、+原発巣へのブースト照射)のいずれかで実施された。[ 13 ]
- National Cancer Databaseを用いた報告で、診断時年齢が0~18歳で、2つの期間(2004~2008年および2009~2012年)に治療されたCNS AT/RTの小児361人の転帰が記述された。この報告で、研究期間中に3併用療法の使用が増えており、転帰が改善していることが確認された。[ 14 ]
AT/RTは放射線療法に反応を示すため、この治療法は多くの治療プロトコルに組み込まれる。[ 15 ]
年齢および治療程度がどのように生存に影響を及ぼすか、より深く理解するために、AT/RTを対象としたプロスペクティブな共同グループ臨床試験が強く求められている。
臨床評価段階にある治療法の選択肢
初期相の臨床試験が、特定の患者について利用できる場合がある。これらの試験は、小児腫瘍学グループ、Pediatric Brain Tumor Consortium、または他の団体を通して利用できる場合がある。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
参考文献- Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.[PUBMED Abstract]
- Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.[PUBMED Abstract]
- Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.[PUBMED Abstract]
- Nicolaides T, Tihan T, Horn B, et al.: High-dose chemotherapy and autologous stem cell rescue for atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 98 (1): 117-23, 2010.[PUBMED Abstract]
- Gardner SL, Asgharzadeh S, Green A, et al.: Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer 51 (2): 235-40, 2008.[PUBMED Abstract]
- Finkelstein-Shechter T, Gassas A, Mabbott D, et al.: Atypical teratoid or rhabdoid tumors: improved outcome with high-dose chemotherapy. J Pediatr Hematol Oncol 32 (5): e182-6, 2010.[PUBMED Abstract]
- Zaky W, Dhall G, Ji L, et al.: Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 61 (1): 95-101, 2014.[PUBMED Abstract]
- Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.[PUBMED Abstract]
- Buscariollo DL, Park HS, Roberts KB, et al.: Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118 (17): 4212-9, 2012.[PUBMED Abstract]
- Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.[PUBMED Abstract]
- Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.[PUBMED Abstract]
- Zimmerman MA, Goumnerova LC, Proctor M, et al.: Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol 72 (1): 77-84, 2005.[PUBMED Abstract]
- Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.[PUBMED Abstract]
- Fischer-Valuck BW, Chen I, Srivastava AJ, et al.: Assessment of the treatment approach and survival outcomes in a modern cohort of patients with atypical teratoid rhabdoid tumors using the National Cancer Database. Cancer 123 (4): 682-687, 2017.[PUBMED Abstract]
- De Amorim Bernstein K, Sethi R, Trofimov A, et al.: Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys 86 (1): 114-20, 2013.[PUBMED Abstract]
- 再発小児CNS非定型奇形腫様/ラブドイド腫瘍の治療
-
再発非定型奇形腫様/ラブドイド腫瘍(AT/RT)の小児に対する標準治療は存在しない。分子標的療法の試験が進行中である。SMARCB1またはSMARCA4が喪失した類上皮肉腫および非中枢神経系(CNS)悪性ラブドイド腫瘍の成人患者におけるEZH2阻害薬、タゼメトスタットの研究では、疾患の長期安定および客観的奏効が観察された。[ 1 ]AT/RTの小児におけるタゼメトスタットの活性が臨床評価段階にある。
臨床的に顕著な活性を示す標準的な薬剤がないため、疾患に向けた追加の治療を希望する患者または家族は、新たな治療アプローチの試験への参加を考慮すべきである。
進行時に疾患に向けた治療を行う決定がなされたかどうかに関係なく、緩和ケアは依然として管理の中心となっている。これにより、QOLを最大化しながら、末期疾患に関連する症状とストレスを緩和する試みが確保される。
臨床評価段階にある治療法の選択肢
初期相の臨床試験が、特定の患者について利用できる場合がある。これらの試験は、小児腫瘍学グループ(COG)、Pediatric Brain Tumor Consortium、または他の団体を通して利用できる場合がある。米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
参考文献- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.[PUBMED Abstract]
- 最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も、入手することができる。
- 本要約の変更点(12/17/2019)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
再発小児中枢神経系非定型奇形腫様/ラブドイド腫瘍の治療
本文に以下の記述が追加された;臨床的に顕著な活性を示す標準的な薬剤がないため、疾患に向けた追加の治療を希望する患者または家族は、新たな治療アプローチの試験への参加を考慮すべきである。
本文に以下の記述が追加された;進行時に疾患に向けた治療を行う決定がなされたかどうかに関係なく、緩和ケアは依然として管理の中心となっている。これにより、QOLを最大化しながら、末期疾患に関連する症状とストレスを緩和する試みが確保される。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児中枢神経系非定型奇形腫様およびラブドイド腫瘍の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/brain/hp/child-cns-atrt-treatment-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389426]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。