

ご利用について
医療専門家向けの本PDQがん情報要約では、小児軟部肉腫の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児軟部肉腫に関する一般情報
-
小児および青年のがん患者の生存において、劇的な改善が達成されている。1975年から2010年の間に、小児がんの死亡率は50%以上低下した。[ 1 ]小児および青年のがん生存者では、治療から数ヵ月または数年経過後もがん療法の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。(小児および青年のがん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)に関するPDQ要約を参照のこと。)
横紋筋の腫瘍である横紋筋肉腫は0~14歳の小児に最もよくみられる軟部肉腫であり、この年齢群にみられる腫瘍の50%を占める。[ 2 ](詳しい情報については、小児横紋筋肉腫の治療に関するPDQ要約を参照のこと。)小児科では、残りの軟部肉腫は一般に非横紋筋肉腫性軟部肉腫と呼ばれ、全小児腫瘍の約3%を占める。[ 3 ]この混成の腫瘍群には以下の新生物が含まれる:[ 4 ]
年齢別および組織型別の軟部肉腫の分布。
小児軟部肉腫は、原始間葉組織由来の悪性腫瘍からなる混成群であり、全小児腫瘍の7%を占める(横紋筋肉腫、4%;他の軟部肉腫、3%)。[ 5 ]
2000年から2015年のSurveillance, Epidemiology, and End Results(SEER)の情報を基にした組織型および年齢別の軟部肉腫の分布を表1に記載している。年齢ごとの組織学的亜型の分布は、図2にも示されている。
表1.0~19歳の小児における軟部肉腫の年齢分布(SEER 2000~2015年)a 5歳未満 5~9歳 10~14歳 15~19歳 20歳未満 すべての年齢(成人を含む) pPNET = 末梢性原始神経外胚葉性腫瘍;SEER = Surveillance, Epidemiology, and End Results。 a出典:SEER database.[ 6 ] すべての軟部肉腫およびその他の骨外性肉腫 1,124 773 1,201 1,558 4,656 横紋筋肉腫 668 417 382 327 1,794 線維肉腫、末梢神経、およびその他の線維の新生物 137 64 112 181 494 線維芽細胞性および筋線維芽細胞性腫瘍 114 33 41 77 265 4,228 神経鞘腫瘍 23 31 70 102 226 2,303 その他の線維の新生物 0 0 1 2 3 114 カポジ肉腫 2 1 2 10 15 その他の特定の軟部肉腫 237 238 559 865 1,899 軟部組織のユーイング腫瘍およびアスキン腫瘍 37 36 72 113 258 596 軟部組織のpPNET 24 23 42 56 145 402 腎外性ラブドイド腫瘍 75 8 9 4 96 205 脂肪肉腫 4 6 37 79 126 10,749 線維組織球性腫瘍 43 73 142 223 481 13,531 平滑筋肉腫 11 14 19 41 85 14,107 滑膜肉腫 12 39 141 210 402 2,608 血管腫瘍 12 9 11 32 64 4,238 軟部組織の骨性および軟骨性新生物 1 6 16 14 37 1,018 胞巣状軟部肉腫 4 5 22 33 64 211 その他の軟部肉腫 14 19 48 60 141 1,339 未特定の軟部肉腫 80 53 146 175 454 非横紋筋肉腫性軟部肉腫は、青年および成人に多くみられ[ 4 ]、これより若年の患者における疾患の治療および自然経過に関する情報のほとんどは、成人を対象とした研究に基づいている。病期(図1)、組織学的亜型(図2)、腫瘍の部位(図3)に応じた年齢ごとのこれらの腫瘍の分布が、以下に示されている。[ 7 ]
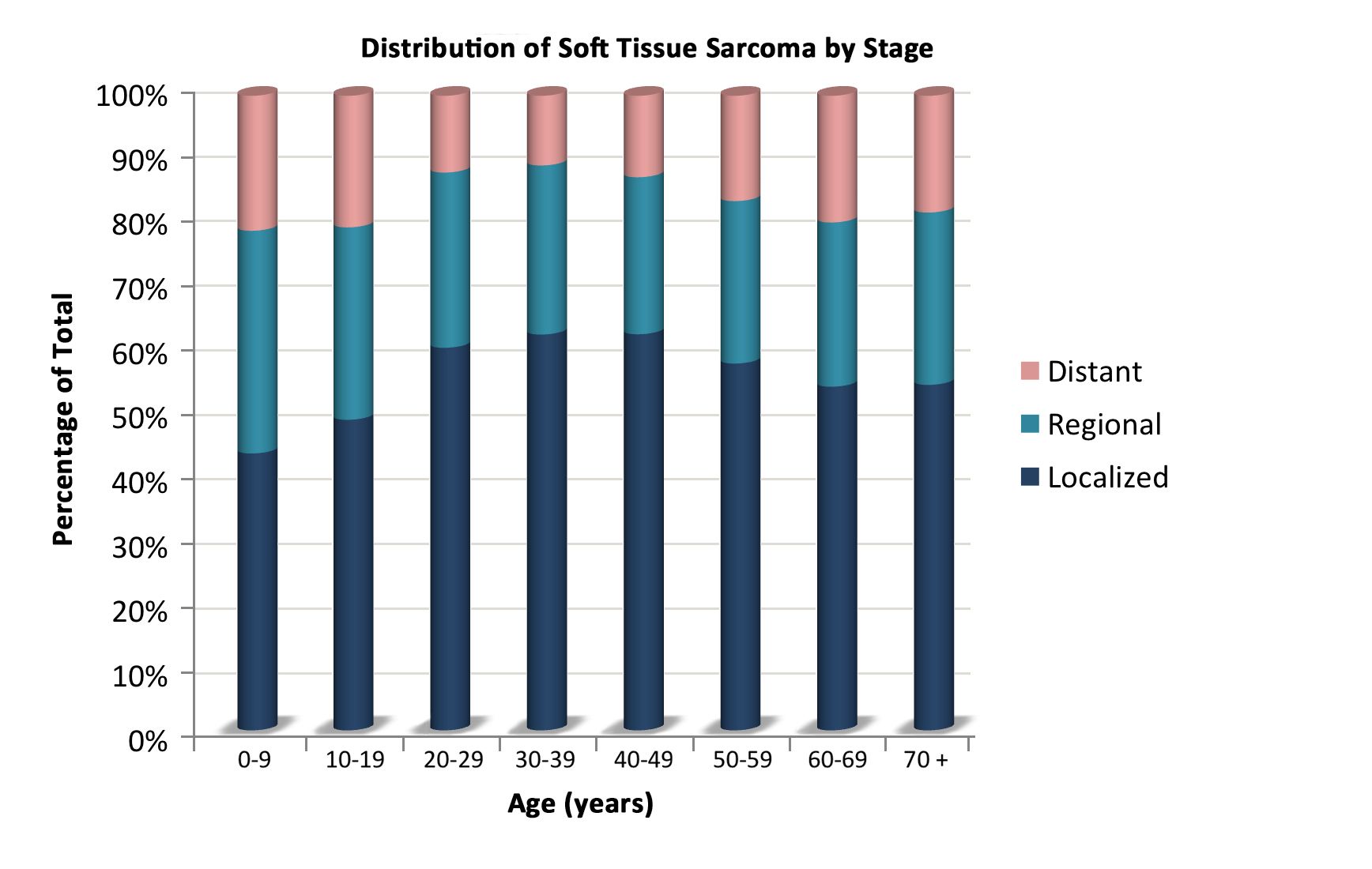
図1.病期に応じた年齢ごとの非横紋筋肉腫性軟部肉腫の分布。 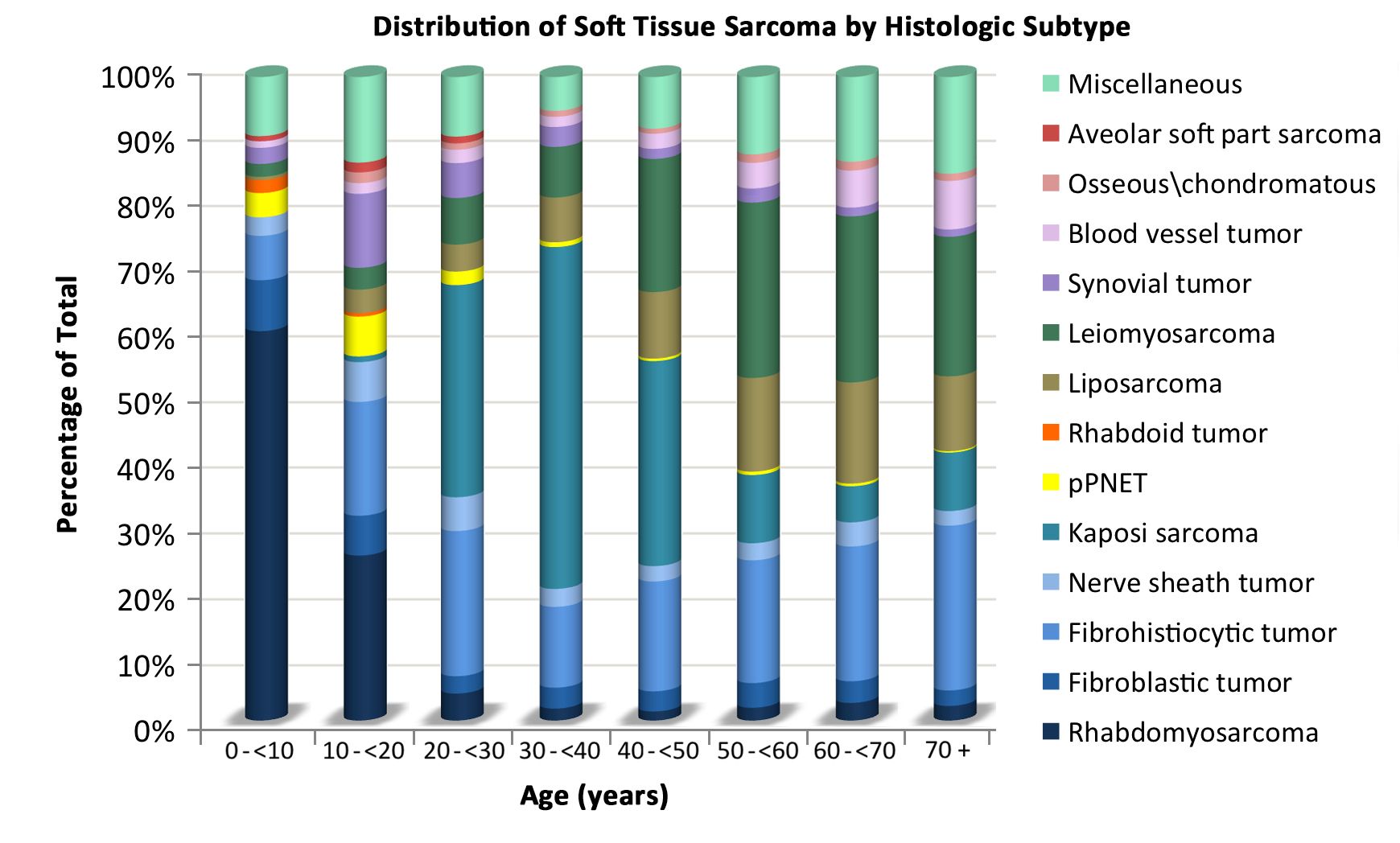
図2.組織学的亜型に応じた年齢ごとの非横紋筋肉腫性軟部肉腫の分布。 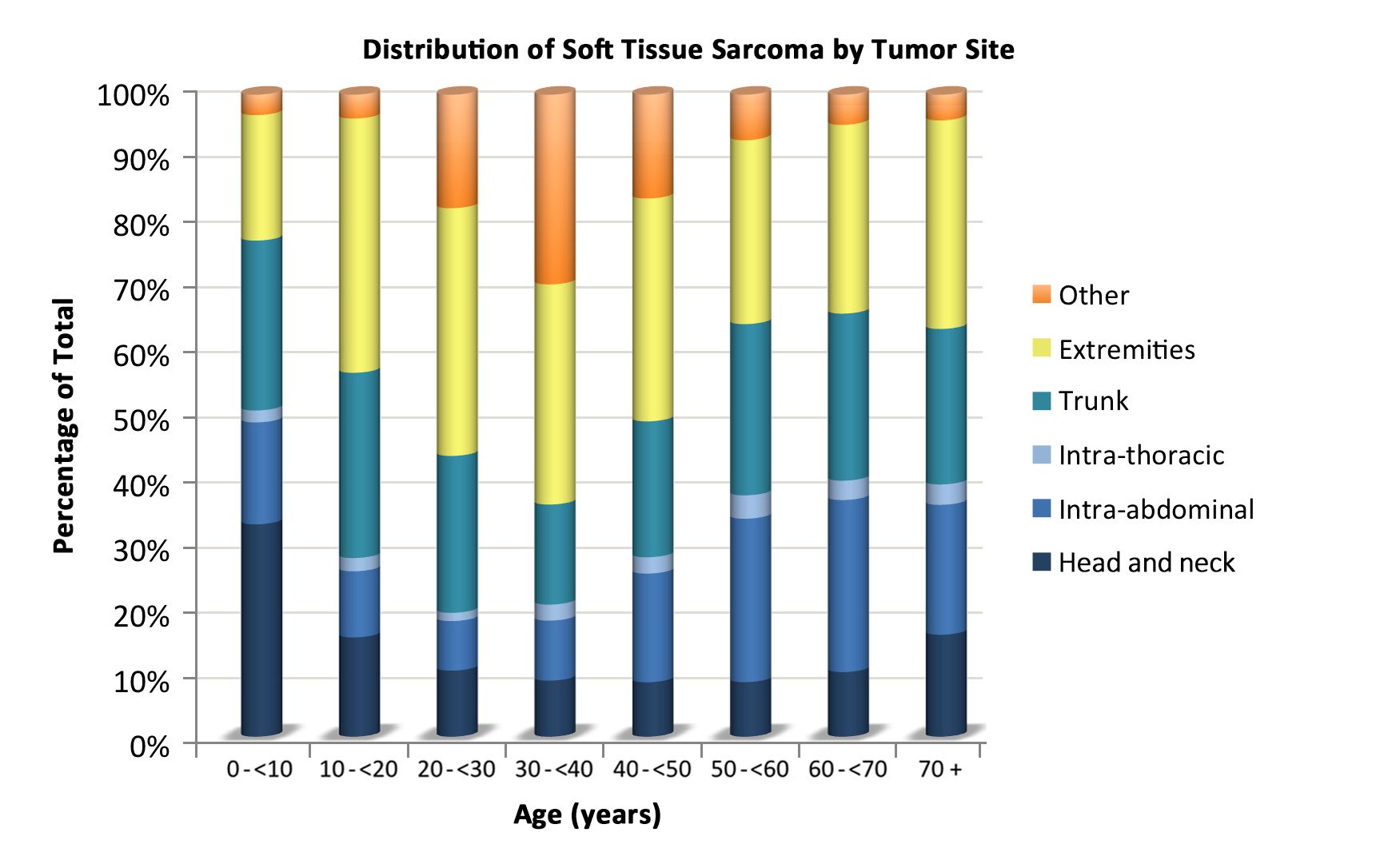
図3.腫瘍の部位に応じた年齢ごとの非横紋筋肉腫性軟部肉腫の分布。 危険因子
いくつかの遺伝的因子および外部曝露が、非横紋筋肉腫性軟部肉腫の発症に関連しており、その中には以下のものがある:
臨床症状
非横紋筋肉腫性軟部肉腫は身体のあらゆる部位に生じうるが、体幹および四肢に最も好発する。[ 27 ][ 28 ][ 29 ]このような腫瘍は、最初に無症候性の充実性腫瘤として出現することもあれば、隣接する解剖学的組織への局所浸潤のために、症候性であることもある。まれではあるが、これらの腫瘍は脳組織に発生することがあり、組織型に応じて治療される。[ 30 ]
全身症状(例えば、発熱、体重減少、および寝汗)はまれである。血管周皮腫(現在では、改訂世界保健機関分類システムにおいて孤立性線維性腫瘍と確認されている)の症例では低血糖と低リン血症性くる病が報告されているのに対して、肺線維肉腫患者では高血糖が報告されている。[ 31 ]
診断的評価および病期評価
疑わしい病変が特定された場合、完全な検査の後に十分な生検を実施することがきわめて重要である。何らかの介入を開始する前に、以下の検査を用いて病変が画像化される:
一部の腫瘍の画像所見の特徴からこの診断が強く示唆される場合がある。例えば、小児低悪性度線維粘液性肉腫および胞巣状軟部肉腫の画像所見の特徴が報告されており、これらのまれな新生物の診断に有用となりうる。[ 35 ]
生検戦略
非横紋筋肉腫性軟部腫瘍は、病理学的に横紋筋肉腫やユーイング肉腫と異なるが、小児の非横紋筋肉腫性軟部肉腫の種類の分類はしばしば困難となる。非横紋筋肉腫性軟部肉腫の診断には、コア針生検、切開生検、または切除生検を用いることができる。可能な場合は、根治的切除を行う予定の外科医が生検の決定に関与する必要がある。切開生検または針生検の位置が不適切だと、切除断端陰性の達成に悪影響が生じる場合がある。
転座および他の分子的変化の診断的意義を考慮して、従来の組織学的検査、免疫細胞化学的検査のほか、光学および電子顕微鏡による観察、細胞遺伝学的検査、蛍光in situハイブリダイゼーション、分子病理学的検査などを可能とするには、十分な量の腫瘍組織を採取するコア針生検または小切開生検がきわめて重要となる。[ 36 ][ 37 ]針生検法では適切な組織サンプルの採取を確実に行う必要がある。組織について複数コアの採取が必要となる場合がある。(主に成人患者において)腫瘍が疑われてコア針生検を受けた530の軟部組織の腫瘤のうち、426(80%)が軟部組織腫瘍であることが明らかにされ、そのうち225(52.8%)が悪性であった。コア針生検は、感度96.3%および特異度99.4%で軟部肉腫と良性病変を鑑別できた。腫瘍サブタイプは良性病変の89.5%および軟部肉腫の88%で正確に割り付けられた。合併症発生率は0.4%であった。[ 38 ]生検方法に関して考慮すべき事項には以下のものがある:
予定外の切除
非横紋筋肉腫性軟部肉腫を予定外に切除した小児では、多くの患児で再切除した標本中に腫瘍が認められるため、しばしば一次再切除が推奨される。[ 50 ][ 51 ]青年および成人を対象にした単一施設の解析で、軟部肉腫の予定外の切除を受けた患者が病期をマッチさせた対照と比較された。このレトロスペクティブ解析では、軟部肉腫の予定外の初回切除により、局所再発、転移、および死亡のリスクが増加したが、この増加は高悪性度の腫瘍で最も大きかった。[ 52 ][証拠レベル:3iiA]この場合、二次切除が予定される。
染色体異常
多くの非横紋筋肉腫性軟部肉腫は染色体異常により特徴付けられる。そうした染色体転座の中には、2つの離れた遺伝子の融合を引き起こすものがある。この結果生じた融合転写産物は、ポリメラーゼ連鎖反応を基にした方法を用いることによって容易に検出できるため、転座を有する腫瘍の診断が容易となる。
非横紋筋肉腫性軟部肉腫で最も頻繁にみられる染色体異常の一部を表2に示す。
表2.非横紋筋肉腫性軟部肉腫に高頻度にみられる染色体異常a 組織型 染色体異常 関係する遺伝子 a出典:Sandberg[ 53 ]、Slater et al.[ 54 ]、Mertens et al.[ 55 ]、Romeo[ 56 ]、およびSchaefer et al.。[ 57 ] 胞巣状軟部肉腫 t(x;17)(p11.2;q25) ASPL/TFE3 [ 58 ][ 59 ][ 60 ] 血管腫様線維性組織球腫 t(12;16)(q13;p11)、t(2;22)(q33;q12)、t(12;22)(q13;q12) FUS/ATF1、EWSR1/CREB1[ 61 ]、EWSR1/ATF1 BCOR再構成肉腫 inv(X)(p11.4;p11.2) BCOR/CCNB3 CIC再構成肉腫 t(4;19)(q35;q13)、t(10;19)(q26;q13) CIC-DUX4 明細胞肉腫 t(12;22)(q13;q12)、t(2;22)(q33;q12) ATF1/EWSR1、EWSR1/CREB1[ 62 ] 先天性(乳児性)線維肉腫/中胚葉性腎腫 t(12;15)(p13;q25) ETV-NTRK3 隆起性皮膚線維肉腫 t(17;22)(q22;q13) COL1A1/PDGFB デスモイド線維腫症 8または20トリソミー、5q21欠失 CTNNB1またはAPCの突然変異 線維形成性小円形細胞腫瘍 t(11;22)(p13;q12) EWSR1/WT1[ 63 ][ 64 ] 類上皮血管内皮腫 t(1;3)(p36;q25)[ 65 ] WWTR1/CAMTA1 類上皮肉腫 SMARCB1の不活性化 SMARCB1 骨外性粘液型軟骨肉腫 t(9;22)(q22;q12)、t(9;17)(q22;q11)、t(9;15)(q22;q21)、t(3;9)(q11;q22) EWSR1/NR4A3、TAF2N/NR4A3、TCF12/NR4A3、TGF/NR4A3 血管周皮腫 t(12;19)(q13;q13.3)およびt(13;22)(q22;q13.3) LMNA-NTRK1[ 66 ] 乳児型線維肉腫 t(12;15)(p13;q25) ETV6/NTRK3 炎症性筋線維芽細胞性腫瘍 t(1;2)(q23;q23)、t(2;19)(q23;q13)、t(2;17)(q23;q23)、t(2;2)(p23;q13)、t(2;11)(p23;p15)[ 67 ] TPM3/ALK、TPM4/ALK、CLTC/ALK、RANBP2/ALK、CARS/ALK、RAS 乳児筋線維腫症 PDGFRB[ 68 ] 低悪性度線維粘液性肉腫 t(7;16)(q33;p11)、t(11;16)(p11;p11) FUS/CREB3L2、FUS/CREB3L1 悪性末梢神経鞘腫瘍 17q11.2、10p、11q、17q、22qの欠失または再構成 NF1 間葉性軟骨肉腫 Del(8)(q13.3q21.1) HEY1/NCOA2 筋上皮腫 t(19;22)(q13;q12)、t(1;22)(q23;q12)、t(6;22)(p21;q12) EWSR1/ZNF44、EWSR1/PBX1、EWSR1/POU5F1 粘液型脂肪肉腫/円形細胞脂肪肉腫 t(12;16)(q13;p11)、t(12;22)(q13;q12) FUS/DD1T3、EWSR1/DD1T3 幼児期原発性粘液状間葉系腫瘍 BCOR遺伝子内縦列重複 ラブドイド腫瘍 SMARCB1の不活性化 SMARCB1 硬化性類上皮線維肉腫 EWSR1/CREB3L2 孤立性線維性腫瘍 inv(12)(q13q13) NAB2/STAT6 滑膜肉腫 t(x;18)(p11.2;q11.2) SYT/SSX 腱鞘巨細胞腫 t(1;2)(p13;q35) COL6A3/CSF1 予後および予後因子
非横紋筋肉腫性軟部肉腫の予後は以下の因子に応じて大きく異なる:[ 69 ][ 70 ][ 71 ]
成人非横紋筋肉腫性軟部肉腫の多数例のシリーズのレビューでは、四肢の表在性肉腫は深在性腫瘍より良好な予後を示した。このため、悪性度および大きさのほかに、腫瘍の浸潤深度を考慮すべきである。[ 72 ]
成人および小児のいくつかのシリーズは、大きな腫瘍または浸潤性腫瘍の患者は、小さな非浸潤性腫瘍の患者より、明らかに予後不良であることを示している。小児および青年の軟部肉腫に関するレトロスペクティブ研究の結果は、軟部肉腫の成人患者に適用している5cmというカットオフ値が小児、特に乳幼児には適さないことを示唆している。この研究では、腫瘍直径と体表面積の間に相関関係があることが確認された。[ 73 ]このような関連性については、その観察所見の治療上の意義を判断するために今後も研究を重ねる必要がある。
一部の小児非横紋筋肉腫性軟部肉腫は比較的良好な転帰と関連している。例えば、乳児および5歳未満の小児にみられる乳児型線維肉腫は、患児のかなりの数が手術のみで治癒が得られ、また化学療法に対する腫瘍の感受性が高いことから、予後が優れている。[ 3 ]
年長の小児および青年の軟部肉腫は、成人の軟部肉腫とほぼ同じ挙動を示すことが多い。[ 3 ][ 74 ]小児腫瘍学グループの大規模プロスペクティブ多国籍研究(ARST0332[NCT00346164])では新たに診断された30歳未満の患者が登録された。患者はリスクグループに基づく治療に割り付けられた(転移の存在、腫瘍の切除可能性および切除断端、腫瘍のサイズと悪性度により決められた;図4を参照のこと)。[ 75 ][証拠レベル:2A]
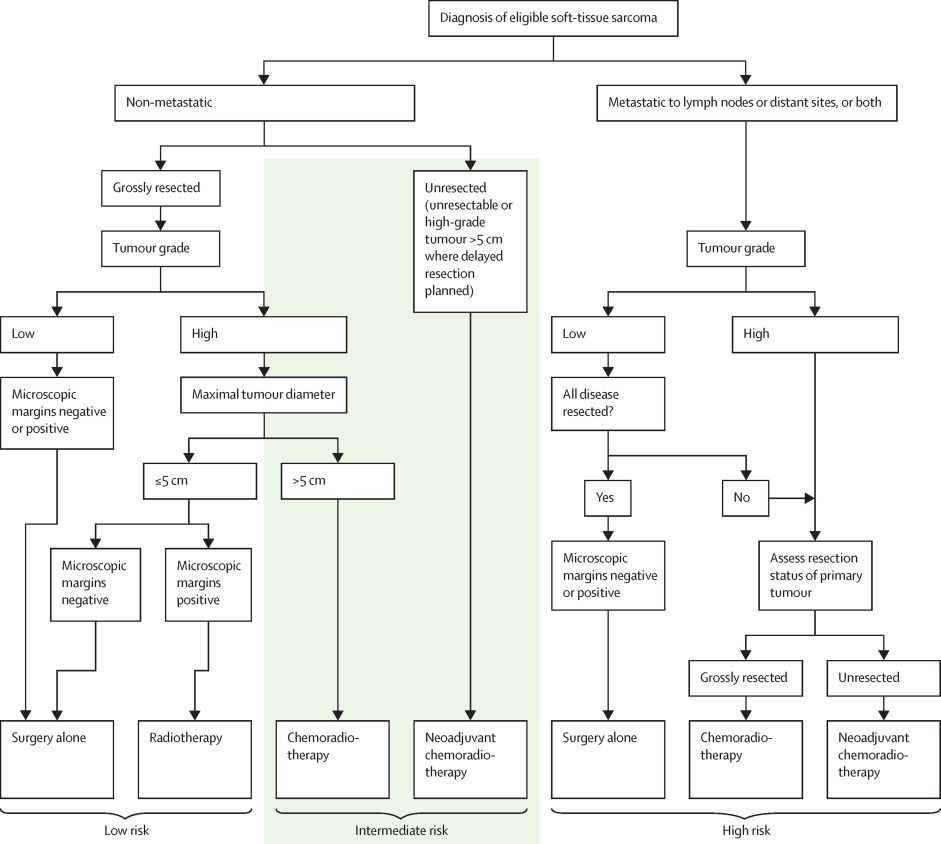
図4.小児腫瘍学グループのARST0332試験でのリスクグループおよび治療割り付け。Elsevierから許諾を得て転載:The Lancet Oncology, Volume 21 (Issue 1), Spunt SL, Million L, Chi YY, et al., A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study, Pages 145-161, Copyright © 2020. 各患者は、3つのリスクグループの1つおよび4つの治療グループの1つに割り付けられた。リスクグループは以下の通りであった:[ 75 ]
- 低リスク:非転移性のR0(切除が顕微鏡的な切除断端陰性で完全であった)またはR1(顕微鏡的な切除断端陽性)の低悪性度腫瘍、あるいは5cm以下のR1高悪性度腫瘍。
- 中リスク:非転移性のR0またはR1の5cmを超える高悪性度腫瘍、あるいは大きさまたは悪性度に関係なく切除されていない腫瘍。
- 高リスク:転移性腫瘍。
治療グループは以下の通りであった:
- 手術単独。
- 放射線療法(55.8Gy)。
- 化学放射線療法(化学療法および55.8Gyの放射線療法)。
- 術前化学放射線療法(化学療法および45Gyの放射線療法、続いて手術および切除断端に基づく放射線療法のブースト照射と化学療法の継続)。
化学療法には、6サイクルのイホスファミド(1回当たり3g/m2)、3週間ごとの1~3日目に静脈内投与および5サイクルのドキソルビシン(1回当たり37.5mg/m2)、3週間ごとの1~2日目に静脈内投与が含まれ、順序は手術または放射線療法のタイミングに応じて調整された。
登録された550人の患者について、解析には評価可能な529人の患者が含められた;生存の結果は表3に示されている。
表3.小児腫瘍学グループのARST0332試験に対する生存の結果 5年イベントフリー生存率 5年全生存率 リスクグループ イベント/患者 推定値(%) イベント/患者 推定値(%) 低リスク 26/222 88.9 (84.0–93.8) 10/222 96.2 (93.2–99.2) 中リスク 84/227 65.0 (58.2–71.8) 55/227 79.2 (73.4–85.0) 高リスク 63/80 21.2 (11.4–31.1) 52/80 35.5 (23.6–47.4) 切除不能な限局性非横紋筋肉腫性軟部肉腫の小児患者は転帰不良である。集学的治療を受けた患者で、無病生存を維持するのは約3分の1のみである。[ 69 ][ 76 ];[ 77 ][ 78 ][証拠レベル:3iiiA]内臓の非横紋筋肉腫性軟部肉腫の患者30人を対象にした1件のイタリアのレビューでは、5年経過時に生存していた患者は10人のみであった。予後不良因子は、完全切除を達成できないこと、比較的大きな腫瘍サイズ、腫瘍浸潤、組織学的サブタイプ、および肺-胸膜部位であった。[ 79 ][証拠レベル:3iiB]
米国および欧州の小児センターからのプール解析では、腫瘍摘除術が完全であったと考えられる患者の方が、不完全であった患者より転帰が優れていた。放射線療法を受けた患者の方が受けなかった患者より転帰が優れていた。[ 77 ][証拠レベル:3iiiA]
無病生存を最大化する一方で長期的な関連合併症を最小化する必要があるため、治療を開始する前に、このような予後因子を利用してそれぞれの患者に対する理想的な治療を綿密かつ個別に決定する必要がある。[ 28 ][ 80 ][ 81 ][ 82 ][ 83 ][ 84 ]
関連する要約
他の種類の肉腫に関する情報については、以下のPDQ要約を参照のこと。
参考文献- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed January 31, 2020.[PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.[PUBMED Abstract]
- Weiss SW, Goldblum JR: General considerations. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1-14.[PUBMED Abstract]
- Pappo AS, Pratt CB: Soft tissue sarcomas in children. Cancer Treat Res 91: 205-22, 1997.[PUBMED Abstract]
- Surveillance, Epidemiology, and End Results (SEER) Program: SEER*Stat Database: Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2017 Sub (1973-2015 varying) - Linked To County Attributes - Total U.S., 1969-2016 Counties [Database]. National Cancer Institute, DCCPS, Surveillance Research Program, released April 2018, based on the November 2017 submission. Available online. Last accessed February 06, 2020.[PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011.[PUBMED Abstract]
- Chang F, Syrjänen S, Syrjänen K: Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol 13 (4): 1009-22, 1995.[PUBMED Abstract]
- Plon SE, Malkin D: Childhood cancer and hereditary. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 13-31.[PUBMED Abstract]
- Groen EJ, Roos A, Muntinghe FL, et al.: Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15 (9): 2439-50, 2008.[PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007.[PUBMED Abstract]
- Wong JR, Morton LM, Tucker MA, et al.: Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol 32 (29): 3284-90, 2014.[PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.[PUBMED Abstract]
- Weiss SW, Goldblum JR: Benign tumors of peripheral nerves. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 825-901.[PUBMED Abstract]
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995.[PUBMED Abstract]
- Stark AM, Buhl R, Hugo HH, et al.: Malignant peripheral nerve sheath tumours--report of 8 cases and review of the literature. Acta Neurochir (Wien) 143 (4): 357-63; discussion 363-4, 2001.[PUBMED Abstract]
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.[PUBMED Abstract]
- Fricke BL, Donnelly LF, Casper KA, et al.: Frequency and imaging appearance of hepatic angiomyolipomas in pediatric and adult patients with tuberous sclerosis. AJR Am J Roentgenol 182 (4): 1027-30, 2004.[PUBMED Abstract]
- Adriaensen ME, Schaefer-Prokop CM, Duyndam DA, et al.: Radiological evidence of lymphangioleiomyomatosis in female and male patients with tuberous sclerosis complex. Clin Radiol 66 (7): 625-8, 2011.[PUBMED Abstract]
- Hornick JL, Fletcher CD: PEComa: what do we know so far? Histopathology 48 (1): 75-82, 2006.[PUBMED Abstract]
- Kesserwan C, Sokolic R, Cowen EW, et al.: Multicentric dermatofibrosarcoma protuberans in patients with adenosine deaminase-deficient severe combined immune deficiency. J Allergy Clin Immunol 129 (3): 762-769.e1, 2012.[PUBMED Abstract]
- Weiss SW, Goldblum JR: Malignant fibrous histiocytoma (pleomorphic undifferentiated sarcoma). In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 403-27.[PUBMED Abstract]
- Tukenova M, Guibout C, Hawkins M, et al.: Radiation therapy and late mortality from second sarcoma, carcinoma, and hematological malignancies after a solid cancer in childhood. Int J Radiat Oncol Biol Phys 80 (2): 339-46, 2011.[PUBMED Abstract]
- Bartkowiak D, Humble N, Suhr P, et al.: Second cancer after radiotherapy, 1981-2007. Radiother Oncol 105 (1): 122-6, 2012.[PUBMED Abstract]
- Casey DL, Friedman DN, Moskowitz CS, et al.: Second cancer risk in childhood cancer survivors treated with intensity-modulated radiation therapy (IMRT). Pediatr Blood Cancer 62 (2): 311-316, 2015.[PUBMED Abstract]
- McClain KL, Leach CT, Jenson HB, et al.: Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 332 (1): 12-8, 1995.[PUBMED Abstract]
- Dillon P, Maurer H, Jenkins J, et al.: A prospective study of nonrhabdomyosarcoma soft tissue sarcomas in the pediatric age group. J Pediatr Surg 27 (2): 241-4; discussion 244-5, 1992.[PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec.[PUBMED Abstract]
- Zeytoonjian T, Mankin HJ, Gebhardt MC, et al.: Distal lower extremity sarcomas: frequency of occurrence and patient survival rate. Foot Ankle Int 25 (5): 325-30, 2004.[PUBMED Abstract]
- Benesch M, von Bueren AO, Dantonello T, et al.: Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European CWS and HIT study groups. J Neurooncol 111 (3): 337-45, 2013.[PUBMED Abstract]
- Weiss SW, Goldblum JR: Miscellaneous tumors of intermediate malignancy. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1093-1160.[PUBMED Abstract]
- Federico SM, Spunt SL, Krasin MJ, et al.: Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer 60 (7): 1128-34, 2013.[PUBMED Abstract]
- Mody RJ, Bui C, Hutchinson RJ, et al.: FDG PET imaging of childhood sarcomas. Pediatr Blood Cancer 54 (2): 222-7, 2010.[PUBMED Abstract]
- Tateishi U, Hosono A, Makimoto A, et al.: Accuracy of 18F fluorodeoxyglucose positron emission tomography/computed tomography in staging of pediatric sarcomas. J Pediatr Hematol Oncol 29 (9): 608-12, 2007.[PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015.[PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008.[PUBMED Abstract]
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998.[PUBMED Abstract]
- Strauss DC, Qureshi YA, Hayes AJ, et al.: The role of core needle biopsy in the diagnosis of suspected soft tissue tumours. J Surg Oncol 102 (5): 523-9, 2010.[PUBMED Abstract]
- Chowdhury T, Barnacle A, Haque S, et al.: Ultrasound-guided core needle biopsy for the diagnosis of rhabdomyosarcoma in childhood. Pediatr Blood Cancer 53 (3): 356-60, 2009.[PUBMED Abstract]
- Tuttle R, Kane JM: Biopsy techniques for soft tissue and bowel sarcomas. J Surg Oncol 111 (5): 504-12, 2015.[PUBMED Abstract]
- Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997.[PUBMED Abstract]
- Smith LM, Watterson J, Scott SM: Medical and surgical management of pediatric soft tissue tumors. In: Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997, pp 360-71.[PUBMED Abstract]
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000.[PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000.[PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008.[PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014.[PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012.[PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013.[PUBMED Abstract]
- Wagner LM, Kremer N, Gelfand MJ, et al.: Detection of lymph node metastases in pediatric and adolescent/young adult sarcoma: Sentinel lymph node biopsy versus fludeoxyglucose positron emission tomography imaging-A prospective trial. Cancer 123 (1): 155-160, 2017.[PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002.[PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001.[PUBMED Abstract]
- Qureshi YA, Huddy JR, Miller JD, et al.: Unplanned excision of soft tissue sarcoma results in increased rates of local recurrence despite full further oncological treatment. Ann Surg Oncol 19 (3): 871-7, 2012.[PUBMED Abstract]
- Sandberg AA: Translocations in malignant tumors. Am J Pathol 159 (6): 1979-80, 2001.[PUBMED Abstract]
- Slater O, Shipley J: Clinical relevance of molecular genetics to paediatric sarcomas. J Clin Pathol 60 (11): 1187-94, 2007.[PUBMED Abstract]
- Mertens F, Antonescu CR, Hohenberger P, et al.: Translocation-related sarcomas. Semin Oncol 36 (4): 312-23, 2009.[PUBMED Abstract]
- Romeo S, Dei Tos AP: Clinical application of molecular pathology in sarcomas. Curr Opin Oncol 23 (4): 379-84, 2011.[PUBMED Abstract]
- Schaefer IM, Cote GM, Hornick JL: Contemporary Sarcoma Diagnosis, Genetics, and Genomics. J Clin Oncol 36 (2): 101-110, 2018.[PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001.[PUBMED Abstract]
- Ladanyi M: The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 4 (3): 162-73, 1995.[PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011.[PUBMED Abstract]
- Antonescu CR, Dal Cin P, Nafa K, et al.: EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46 (12): 1051-60, 2007.[PUBMED Abstract]
- Hisaoka M, Ishida T, Kuo TT, et al.: Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 32 (3): 452-60, 2008.[PUBMED Abstract]
- Barnoud R, Sabourin JC, Pasquier D, et al.: Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 24 (6): 830-6, 2000.[PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007.[PUBMED Abstract]
- Errani C, Zhang L, Sung YS, et al.: A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50 (8): 644-53, 2011.[PUBMED Abstract]
- Haller F, Knopf J, Ackermann A, et al.: Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol 238 (5): 700-10, 2016.[PUBMED Abstract]
- Jain S, Xu R, Prieto VG, et al.: Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 3 (4): 416-28, 2010.[PUBMED Abstract]
- Agaimy A, Bieg M, Michal M, et al.: Recurrent Somatic PDGFRB Mutations in Sporadic Infantile/Solitary Adult Myofibromas But Not in Angioleiomyomas and Myopericytomas. Am J Surg Pathol 41 (2): 195-203, 2017.[PUBMED Abstract]
- Spunt SL, Hill DA, Motosue AM, et al.: Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol 20 (15): 3225-35, 2002.[PUBMED Abstract]
- Spunt SL, Poquette CA, Hurt YS, et al.: Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3697-705, 1999.[PUBMED Abstract]
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005.[PUBMED Abstract]
- Brooks AD, Heslin MJ, Leung DH, et al.: Superficial extremity soft tissue sarcoma: an analysis of prognostic factors. Ann Surg Oncol 5 (1): 41-7, 1998 Jan-Feb.[PUBMED Abstract]
- Ferrari A, Miceli R, Meazza C, et al.: Soft tissue sarcomas of childhood and adolescence: the prognostic role of tumor size in relation to patient body size. J Clin Oncol 27 (3): 371-6, 2009.[PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.[PUBMED Abstract]
- Spunt SL, Million L, Chi YY, et al.: A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study. Lancet Oncol 21 (1): 145-161, 2020.[PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002.[PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011.[PUBMED Abstract]
- Smith KB, Indelicato DJ, Knapik JA, et al.: Definitive radiotherapy for unresectable pediatric and young adult nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 57 (2): 247-51, 2011.[PUBMED Abstract]
- Ferrari A, Magni C, Bergamaschi L, et al.: Pediatric nonrhabdomyosarcoma soft tissue sarcomas arising at visceral sites. Pediatr Blood Cancer 64 (9): , 2017.[PUBMED Abstract]
- Dillon PW, Whalen TV, Azizkhan RG, et al.: Neonatal soft tissue sarcomas: the influence of pathology on treatment and survival. Children's Cancer Group Surgical Committee. J Pediatr Surg 30 (7): 1038-41, 1995.[PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994.[PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997.[PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999.[PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998.[PUBMED Abstract]
- 小児軟部肉腫の病理組織学的分類
-
軟部肉腫の世界保健機関(WHO)分類
WHOのがん分類システムは、世界共通のがん命名法である。米国では、WHOのがん分類システムは肉腫の病期分類については米国がん合同委員会(AJCC)により、骨肉腫および軟部肉腫についてはCollege of American Pathologists(CAP)のがんプロトコルにより採用されている。軟部肉腫および骨肉腫の第4版WHO分類は2013年2月に発表された。[ 1 ]
軟部肉腫の悪性度分類は常に議論の的になる問題となっている。WHOでは悪性度分類において優先傾向を厳密に表明していないが、WHO分類に対して実施された主要な変更の1つとして、生物学的潜在力に関して悪性度が中等度の2つの異なるタイプ-局所侵攻性および転移はまれが指定された。[ 1 ]
WHOは、悪性線維性組織球腫(未分化多形肉腫としても知られる)および血管周皮腫(現在は孤立性線維性腫瘍の範囲に含まれると考えられる)について性質の定義が不十分であることを認めた。[ 1 ]
分子的研究および遺伝学研究における現在の進歩から、以前は分化不明の腫瘍として分類されていた血管腫様悪性線維性組織球腫および骨外性粘液型軟骨肉腫など、あるサブセットの腫瘍が新たなセクションに移動されている。複数の疾患実体が新たに認識され、皮膚腫瘍に属する2~3の疾患実体もまた、WHO分類に追加された。他の腫瘍の形態学的異型である可能性が非常に高いと明らかにされた2~3の腫瘍は現在の分類から削除されたか、または他のセクションに組み込まれた。[ 1 ]
-
脂肪細胞腫瘍。
- 良性。
- 中悪性度(局所侵攻性)。
- 悪性。
- 骨・軟骨部腫瘍。
-
線維芽細胞性/筋線維芽細胞性腫瘍。
- 良性。
- 中悪性度(局所侵攻性)。
- 中悪性度(転移はまれ)。
- 悪性。
- 骨格筋腫瘍。
-
平滑筋腫瘍。
- 良性。
- 悪性。
血管平滑筋腫は血管周囲腫瘍に再分類された。
-
いわゆる線維組織球性腫瘍。
- 良性。
- 中悪性度(転移はまれ)。
かつては悪性線維性組織球腫およびそのサブタイプとして知られていたいわゆる線維組織球性腫瘍の悪性相当物は未分化肉腫に改名されたが、以前は未分化/分類不能肉腫のセクションに分類されていた。
-
神経鞘腫瘍。
- 良性。
- 悪性。
- 周皮細胞(血管周囲)腫瘍。
-
分化不明の腫瘍。
- 良性。
- 中悪性度(局所侵攻性)。
- 中悪性度(転移はまれ)。
- 悪性。
-
未分化/分類不能肉腫。
このファミリー内で遺伝学的サブグループが出現しており、以下のような取り組みが進行中である:
-
脈管腫瘍。
- 良性。
- 中悪性度(局所侵攻性)。
- 中悪性度(転移はまれ)。
- 悪性。
参考文献- Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC Press, 2013.[PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008.[PUBMED Abstract]
- Steelman C, Katzenstein H, Parham D, et al.: Unusual presentation of congenital infantile fibrosarcoma in seven infants with molecular-genetic analysis. Fetal Pediatr Pathol 30 (5): 329-37, 2011.[PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011.[PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun.[PUBMED Abstract]
- Le Guellec S, Chibon F, Ouali M, et al.: Are peripheral purely undifferentiated pleomorphic sarcomas with MDM2 amplification dedifferentiated liposarcomas? Am J Surg Pathol 38 (3): 293-304, 2014.[PUBMED Abstract]
-
脂肪細胞腫瘍。
- 小児軟部肉腫の病期分類および悪性度判定システム
-
臨床的病期診断は、臨床転帰を予測し、小児軟部肉腫にとって最も効果的な治療法を決定するのに重要な役割を果たしている。現時点では、広く普及している病期分類システムで、あらゆる小児肉腫に適用できるものは存在しない。成人に用いられる米国がん合同委員会(AJCC)の病期分類システムは、小児を対象とした研究では検証されていない。
小児非横紋筋肉腫性軟部肉腫について標準化された病期分類システムは存在しないが、小児非横紋筋肉腫性軟部肉腫の病期分類には現在以下の2つのシステムが用いられている:[ 1 ]
Intergroup Rhabdomyosarcoma Studyの病期分類システム
非転移性疾患
転移性疾患
再発/進行性疾患
TNM病期分類システム
AJCCがん病期分類マニュアル第8版は、腫瘍の大きさ、リンパ節の状態、組織学的悪性度、および転移の4つの基準および解剖学的原発腫瘍部位(頭頸部;体幹および四肢;腹部および胸部の内臓;後腹膜;およびまれな組織型と部位)による病期判定を指定している(表4、5、6、および7を参照のこと)。[ 3 ][ 4 ][ 5 ][ 6 ][ 7 ]まれな組織型と部位に関する情報については、AJCCがん病期分類マニュアルを参照のこと。[ 7 ]
表4.体幹、四肢、および後腹膜;頭頸部;腹部および胸部の内臓の軟部肉腫について、原発腫瘍(T)の定義a T分類 体幹、四肢、および後腹膜の軟部肉腫 頭頸部の軟部肉腫 腹部および胸部の内臓の軟部肉腫 a出典:O'Sullivan et al.,[ 3 ] Yoon et al.,[ 4 ] Raut et al.,[ 5 ] and Pollock et al.[ 6 ] TX 原発腫瘍の評価が不可能。 原発腫瘍の評価が不可能。 原発腫瘍の評価が不可能。 T0 原発腫瘍を認めない。 T1 腫瘍の最大径が5cm以下。 腫瘍が2cm以下。 臓器に限局する。 T2 腫瘍の最大径が5cmを超えるが、10cm以下。 腫瘍が2cm超~4cm以下。 臓器を越えて組織への腫瘍進展。 T2a 漿膜または臓側腹膜に浸潤している。 T2b 漿膜(腸間膜)を越えた進展。 T3 腫瘍の最大径が10cmを超えるが、15cm以下。 腫瘍が4cmを超える。 別の臓器に浸潤している。 T4 腫瘍の最大径が15cmを超える。 隣接する構造への浸潤を伴う腫瘍。 多病巣性病変。 T4a 眼窩浸潤、頭蓋底/硬膜浸潤、中央区画の内臓への浸潤、顔面頭蓋への転移、または翼突筋浸潤を伴う腫瘍。 多病巣性(2部位)。 T4b 脳実質浸潤、頸動脈を狭窄させる浸潤、椎前筋浸潤、または神経周囲への浸潤を介した中枢神経系への転移を伴う腫瘍。 多病巣性(3~5部位)。 T4c 多病巣性(5部位を超える)。 表5.頭頸部;体幹および四肢;腹部および胸部の内臓;および後腹膜の軟部肉腫について、所属リンパ節(N)の定義a a出典:O'Sullivan et al.,[ 3 ] Yoon et al.,[ 4 ] Raut et al.,[ 5 ] and Pollock et al.[ 6 ] b腹部および胸部の内臓の軟部肉腫について、N0 = リンパ節転移を認めないまたはリンパ節の状態が不明、N1 = リンパ節転移を認める。 N0 所属リンパ節に転移を認めないまたはリンパ節の状態が不明。b N1 所属リンパ節に転移を認める。b 表6.頭頸部;体幹および四肢;腹部および胸部の内臓;および後腹膜の軟部肉腫について、遠隔転移(M)の定義a a出典:O'Sullivan et al.,[ 3 ] Yoon et al.,[ 4 ] Raut et al.,[ 5 ] and Pollock et al.[ 6 ] b腹部および胸部の内臓の軟部肉腫について、M0 = 遠隔転移を認めないおよびM1 = 遠隔転移を認める。 M0 遠隔転移を認めない。b M1 遠隔転移を認める。b 表7.体幹、四肢、および後腹膜の軟部肉腫に対するAJCC予後的病期グループa 病期 T N M 悪性度 T = 原発腫瘍;N = 所属リンパ節;M = 遠隔転移。 a出典:Yoon et al.[ 4 ] and Pollock et al.[ 6 ] b後腹膜の軟部肉腫についてはIIIB期;体幹および四肢の軟部肉腫についてはIV期。 IA T1 N0 M0 G1、GX IB T2、T3、T4 N0 M0 G1、GX II T1 N0 M0 G2、G3 IIIA T2 N0 M0 G2、G3 IIIB T3、T4 N0 M0 G2、G3 IIIB/IVb すべてのT N1 M0 すべてのG IV すべてのT すべてのN M1 すべてのG 軟部肉腫の腫瘍病理悪性度判定システム
ほとんどの症例では、軟部肉腫の正確な病理組織学的分類だけでは、臨床的挙動に関する最適な情報を得ることはできない。このため、悪性度判定過程では、以下のものを含め、いくつかの組織学的パラメータの評価が行われる:
この過程は、組織学的所見と臨床転帰との間の相関性を改善するために行うものである。[ 9 ]小児における軟部肉腫の悪性度判定は、4歳未満の小児において予後が良好な乳児型線維肉腫および血管周皮腫、また切除が不完全であれば局所再発の可能性があるが、通常は転移しない血管腫様線維性組織球腫および隆起性皮膚線維肉腫など、良好な予後を示す特定の腫瘍があるため誤りやすい。
このような腫瘍はまれであることから、小児集団で悪性度判定システムの妥当性を検証することは困難である。1986年3月に、Pediatric Oncology Group(POG)が横紋筋肉腫以外の小児軟部肉腫を対象としたプロスペクティブ研究を実施し、POG悪性度判定システムを考案した。横紋筋肉腫以外の限局性軟部肉腫患者を対象とした転帰解析から、腫瘍が悪性度3の患者は、悪性度1または悪性度2の患者より有意に不良な経過をたどることが明らかになった。この知見は、このシステムにより非横紋筋肉腫性軟部肉腫の臨床的挙動を正確に予測しうることを示唆している。[ 9 ][ 10 ][ 11 ]
POGおよびFrench Federation of Comprehensive Cancer Centers(Fédéation Nationale des Centres de Lutte Contre Le Cancer [FNCLCC])のSarcoma Groupによって策定された悪性度判定システムを以下に示している。これらの悪性度判定システムは、COG-ARST0332研究で中央病理検査担当者によって比較されている。この研究は中止され、結果は未発表である。
POG悪性度判定システム
POG悪性度判定システムを以下に示す。[ 9 ]これは歴史的価値をもつ旧世代の悪性度判定システムであり、現在では治療には使われていない。
悪性度I
悪性度Iの病変は、組織型、細胞組織学的特徴が高分化型、および/または患者の年齢を基にしている。
悪性度II
悪性度IIの病変は、組織学的診断により悪性度IまたはIIIに含まれない軟部肉腫である(有糸分裂像が10高倍率視野当たり5未満、または壊死が15%未満):
悪性度III
悪性度IIIの病変は、悪性度IIの病変に類似しており、組織学的診断(有糸分裂像が10高倍率視野当たり4を超える、または壊死が15%を超える)および悪性度I以外の腫瘍であるという理由で臨床的に侵攻性であることが知られている以下の特定腫瘍を含む:
FNCLCC悪性度判定システム
FNCLCC組織学的悪性度判定システムは、軟部肉腫の成人用に開発された。この悪性度判定システムの目的は、どの患者が転移を来し、その後の術後化学療法が有益となるかを予測することである。[ 12 ][ 13 ]この悪性度判定システムを表8および表9に示す。
表8.FNCLCC組織学的悪性度判定システム FNCLCC = Fédération Nationale des Centres de Lutte Contre Le Cancer;HPF = 高倍率視野。 腫瘍分化度 スコア1 正常な成人の間葉組織に酷似している肉腫(例えば、高分化型脂肪肉腫) スコア2 組織型が確定している腫瘍(例えば、粘液型脂肪肉腫) スコア3 胎児型および未分化型肉腫、組織型が疑わしい肉腫、滑膜肉腫 有糸分裂像の数 スコア1 有糸分裂像が10HPF当たり0~9 スコア2 有糸分裂像が10HPF当たり10~19 スコア3 有糸分裂像が10HPF当たり20以上 腫瘍壊死 スコア0 壊死なし スコア1 腫瘍壊死が50%未満 スコア2 腫瘍壊死が50%以上 表9.総スコアにより判定した組織学的悪性度 総スコア 組織学的悪性度 2–3 悪性度I 4–5 悪性度II 6–8 悪性度III 腫瘍悪性度判定の予後的意義
POGおよびFNCLCCの悪性度判定システムは、小児および成人の非横紋筋肉腫性軟部肉腫において予後的価値があることが証明されている。[ 14 ][ 15 ][ 16 ][ 17 ][ 18 ]3件のプロスペクティブ臨床試験に登録された非横紋筋肉腫性軟部肉腫の小児および青年から採取した130の腫瘍を調べた研究では、POG指定の悪性度とFNCLCC指定の悪性度との間に相関性が認められた。しかしながら、すべての症例で悪性度指定が相関していたわけではなかった;腫瘍の悪性度指定が一致しなかった患者44人(POGで悪性度3、FNCLCCで悪性度1または2)は、一致した悪性度3、および悪性度1および2の間の転帰を示した。有糸分裂指数が10以上という指標が、重要な予後因子として浮上した。[ 19 ]
完了したCOG-ARST0332試験では、データを解析し、POGとFNCLCCの病理学的悪性度判定システムを比較することで、いずれのシステムが臨床転帰とより良好な相関性を示すかが判定される。募集が締め切られているCOG試験(ARST1321[NCT02180867])では、組織学的悪性度の割り付けにFNCLCCシステムが用いられた。
参考文献- American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. New York, NY: Springer, 2002.[PUBMED Abstract]
- Maurer HM, Beltangady M, Gehan EA, et al.: The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 61 (2): 209-20, 1988.[PUBMED Abstract]
- O'Sullivan B, Maki RG, Agulnik M, et al.: Soft tissue sarcoma of the head and neck. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 499-505.[PUBMED Abstract]
- Yoon SS, Maki RG, Asare EA, et al.: Soft tissue sarcoma of the trunk and extremities. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 507-15.[PUBMED Abstract]
- Raut CP, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the abdomen and thoracic visceral organs. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 517-21.[PUBMED Abstract]
- Pollock RE, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the retroperitoneum. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 531-7.[PUBMED Abstract]
- Maki RG, Folpe AL, Guadagnolo BA, et al.: Soft tissue sarcoma - unusual histologies and sites. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 539-45.[PUBMED Abstract]
- Waxweiler TV, Rusthoven CG, Proper MS, et al.: Non-Rhabdomyosarcoma Soft Tissue Sarcomas in Children: A Surveillance, Epidemiology, and End Results Analysis Validating COG Risk Stratifications. Int J Radiat Oncol Biol Phys 92 (2): 339-48, 2015.[PUBMED Abstract]
- Parham DM, Webber BL, Jenkins JJ, et al.: Nonrhabdomyosarcomatous soft tissue sarcomas of childhood: formulation of a simplified system for grading. Mod Pathol 8 (7): 705-10, 1995.[PUBMED Abstract]
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998.[PUBMED Abstract]
- Skytting B, Meis-Kindblom JM, Larsson O, et al.: Synovial sarcoma--identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand 70 (6): 543-54, 1999.[PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001.[PUBMED Abstract]
- Guillou L, Coindre JM, Bonichon F, et al.: Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 15 (1): 350-62, 1997.[PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec.[PUBMED Abstract]
- Pisters PW, Leung DH, Woodruff J, et al.: Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 14 (5): 1679-89, 1996.[PUBMED Abstract]
- Coindre JM, Terrier P, Bui NB, et al.: Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol 14 (3): 869-77, 1996.[PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994.[PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998.[PUBMED Abstract]
- Khoury JD, Coffin CM, Spunt SL, et al.: Grading of nonrhabdomyosarcoma soft tissue sarcoma in children and adolescents: a comparison of parameters used for the Fédération Nationale des Centers de Lutte Contre le Cancer and Pediatric Oncology Group Systems. Cancer 116 (9): 2266-74, 2010.[PUBMED Abstract]
- 小児軟部肉腫に対する治療法選択肢の概要
-
小児非横紋筋肉腫性軟部肉腫はまれなことから、この種の腫瘍を来したすべての小児、青年、および若年成人については、腫瘍専門医(小児または内科)、病理医、外科医、および放射線腫瘍医からなる集学的チームによる治療の連係を考慮すべきである。さらに、この種の腫瘍の自然経過および治療に対する反応をさらに明らかにするために、まれな腫瘍に罹患している小児については、国または施設の治療プロトコルへの登録を考慮すべきである。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
小児腫瘍学グループ(COG)により、軟部肉腫患者を対象にした1件のプロスペクティブ非ランダム化試験が実施された。[ 1 ]
原発腫瘍の外科的切除は、以下のように分類された:
患者は、以下の3つのリスクグループのいずれかに割り付けられた:
- 低リスク:非転移性のR0またはR1の低悪性度腫瘍、あるいは5cm以下のR1の高悪性度腫瘍。
- 中リスク:非転移性のR0またはR1の5cmを超える高悪性度腫瘍、あるいは大きさまたは悪性度に関係なく切除されていない腫瘍。
- 高リスク:転移性腫瘍。
治療グループは以下の通りであった:
- 手術単独。
- 放射線療法(55.8Gy)。
- 化学放射線療法(化学療法および55.8Gyの放射線療法)。
- 術前化学放射線療法(化学療法および45Gyの放射線療法、続いて手術および切除断端に基づく放射線療法のブースト照射と化学療法の継続)。
化学療法には、6サイクルのイホスファミド(1回当たり3g/m2)、3週間ごとの1~3日目に静脈内投与および5サイクルのドキソルビシン(1回当たり37.5mg/m2)、3週間ごとの1~2日目に静脈内投与が含まれ、順序は手術または放射線療法のタイミングに応じて調整された。
529人の評価可能な患者が解析に含まれた:低リスク(n = 222)、中リスク(n = 227)、および高リスク(n = 80);手術単独(n = 205)、放射線療法(n = 17)、化学放射線療法(n = 111)、および術前化学放射線療法(n = 196)。
追跡期間中央値6.5年(四分位範囲[IQR]、4.9-7.9)で、5年イベントフリー生存(EFS)率および全生存(OS)率はリスクグループ別に、以下の通りであった:
著者らにより、治療失敗リスクを有効に決定し、非横紋筋肉腫性軟部肉腫の若年患者をリスク調整治療に層別化するために治療前の臨床的特徴が利用可能であると結論付けられた。ほとんどの低リスク患者は補助療法なしに治癒可能であり、治療による既知の長期合併症が回避される。中リスクおよび高リスク患者に対する生存は依然として最適以下であり、これらの患者に対しては新たな治療法が必要である。
手術
適切な生検および病理学的診断の後、原発腫瘍を可能な限り断端陰性で局所切除することを目指し、その施行前または施行後に化学療法および/または放射線療法を行う。軟部肉腫の切除に関し専門の技術・経験を有する外科医が加わることが強く望まれる。
手術実施時期の決定に際しては、手術の実施可能性と合併症の査定が必要となる。初回手術で切除組織の病理学的な断端陰性が得られなかった場合、またはがんの存在を知らずに初回手術が行われた場合は、断端陰性が得られるように、だが不必要に広範囲とすることなく、病変領域の再切除を施行する。[ 2 ][ 3 ][ 4 ][ 5 ]この手術方針は、たとえ初回手術後の磁気共鳴画像法(MRI)で腫瘤が検出されない場合であっても変わらない。[ 6 ];[ 7 ][証拠レベル:3iiA]
診断時の所属リンパ節転移はまれであり、類上皮肉腫および明細胞肉腫の患者に最も多くみられる。[ 8 ][ 9 ]さまざまな施設のシリーズで、軟部肉腫の患児における病期分類検査としてのセンチネルリンパ節生検の実施可能性および有効性が実証されている。[ 10 ][ 11 ][ 12 ][ 13 ][ 14 ][ 15 ]
放射線療法
放射線療法の検討は、手術単独または手術と化学療法の併用により、重要臓器を喪失したり、重大な機能的、審美的、または心理的な障害を生じたりすることなく局所制御を得られる可能性に基づいて行う。これは、以下により異なる:
放射線療法は術前または術後に施行することができる。放射線療法はまた、外科的切除が実施されないまれな状況では根治治療として実施できる。[ 16 ]照射野の大きさや線量は、患者と腫瘍変数および外科的処置を基にして決定する。[ 17 ]放射線療法は術前または術後に実施した場合、手術単独と比較してOSが改善した。[ 18 ]
術前放射線療法に伴って、優れた局所制御率が得られている。[ 19 ][ 20 ][ 21 ]このアプローチの利点は、術後に腫瘍床を治療する必要がなく、より少ない腫瘍量を治療すること、および手術による脈管系の破壊および瘢痕化からもたらされる相対的な低酸素症がみられないため、照射線量がいくぶん少なくなることである。成人では、術前放射線療法に伴って、主に下肢腫瘍における創傷合併症の発生率が高くなっている;しかしながら、この合併症発生率の程度は疑わしい。[ 22 ]逆に、術前放射線療法では、術後アプローチよりもおそらく治療腫瘍量が少なく線量が低いため、線維症が少なくなる可能性がある。[ 23 ]放射線技術は正常組織の温存に影響を与える可能性がある;三次元原体照射療法と比較して、強度変調放射線療法では、皮膚および骨端(四肢肉腫に照射する場合)への放射線量を低下できる可能性がある。[ 24 ]
後腹膜肉腫は、腸が放射線感受性により損傷しやすいことから、術後放射線療法があまり望ましくないという点で独特である。[ 25 ][ 26 ]放射線の照射線量にかかわらず、術後癒着および腸の不動により損傷リスクが高まることがある。これは、腫瘍により腸がしばしば照射野外に移動し、曝露される腸があっても可動性が高いため、特定の腸区画に対する曝露量が低減される術前アプローチとは対照的である。
放射線療法を術後に施行することもできる。一般的に、外科的切除断端が不十分な患者および腫瘍が大きく悪性度も高い患者には、放射線療法が適応となる。[ 27 ][ 28 ]腫瘍切除断端が1cm未満の高悪性度腫瘍では、これが特に重要となる。[ 29 ][ 30 ];[ 31 ][証拠レベル:3iiDiv]R0(切除断端陰性)の手術と放射線療法を併用すると、四肢肉腫患者の約90%、後腹膜肉腫患者の70~75%、および患者全体では80%で原発腫瘍の局所制御を達成できる。[ 32 ][ 33 ][ 34 ][ 35 ][ 36 ]
特定の状況下においては、密封小線源治療や術中照射を適用できる場合もある。[ 33 ][ 37 ][ 38 ];[ 39 ][証拠レベル:3iiiDii]
放射線の照射容積および線量は、前述の患者因子、腫瘍因子、および外科的因子とともに、以下の事項によって変化させる:
放射線の線量は、一般に術前で45~50Gyであり、切除断端が顕微鏡的または肉眼的に陽性の場合は10~20Gyの術後追加照射、または切除が完全ではないと予想される場合に計画される密封小線源治療の検討も含める。しかしながら、術後追加照射の効力を証明するデータは不足している。[ 40 ]術後放射線の線量は、R0の切除で55Gy~60Gy、R1(顕微鏡的な切除断端陽性)の切除で最大65Gyで、切除不能な肉眼的残存病変が存在する状況では全般的な治療の目標(例、根治的局所制御 vs 症状緩和)に応じて、これより高い線量が照射される。
化学療法
術後化学療法の役割は、依然として不明である。[ 43 ]
証拠(術後化学療法に関する明瞭さの欠如):
- 軟部肉腫の成人を対象にしたすべてのランダム化試験データのメタアナリシスにより、以下が観察された:[ 44 ]
- 欧州の1件の試験において、軟部肉腫を完全切除された成人患者が経過観察またはイホスファミドとドキソルビシンによる術後化学療法にランダムに割り付けられた。[ 45 ][証拠レベル:1iiA]
- 小児を対象とした最大規模のプロスペクティブ試験では、ビンクリスチン、ダクチノマイシン、シクロホスファミド、およびドキソルビシンによる術後化学療法についていかなる有益性も実証されなかった。[ 32 ]
- ドキソルビシンおよびイホスファミドはリスクに基づくCOG ARST0332(NCT00346164)試験で使用された。[ 1 ][証拠レベル:3iiiA]
標的療法
血管新生抑制薬および哺乳類ラパマイシン標的蛋白(mTOR)阻害薬の使用が成人軟部肉腫の治療において研究されているが、小児科では研究されていない。
証拠(軟部肉腫の成人における標的療法):
小児軟部肉腫の治療に関する特別な考慮事項
小児および青年におけるがんはまれであるが、小児がんの全発生率は、1975年以降徐々に増加している。[ 49 ]小児および青年のがん患者は、小児期および青年期に発生するがんの治療経験を有する専門家から構成される集学的チームのある医療機関に紹介されるべきである。この集学的チームのアプローチとは、至適な生存期間および生活の質を得られるような治療、支持療法およびリハビリテーションを小児が確実に受けられるようにするため、以下の医療専門家らの技能を集結させたものである。
(小児および青年のがんの支持療法に関する具体的な情報については、PDQの支持療法と緩和ケアの要約を参照のこと。)
米国小児科学会は、小児がん施設とそれらが小児がん患者の治療において担う役割に関するガイドラインを概説している。[ 50 ]このような小児がん施設では、小児および青年に発症するほとんどの種類のがんに関する臨床試験が行われており、大半の患者/家族に参加する機会が与えられている。これらの患児が最適な臨床転帰を確実に得られるようにするため、外科的専門知識および放射線療法の専門技能を有する小児がん施設で集学的に評価することがきわめて重要である。かなりの割合の患者で放射線療法を併用するまたは併用しない手術で治癒が得られるが、化学療法を追加することで一部のがん患者に利益がもたらされる可能性がある;したがって、臨床試験への登録が勧められる。小児および青年のがんに関する臨床試験は一般に、現在標準とされている治療法と、それより効果的であると思われる治療法とを比較するようデザインされている。小児がんの治癒を目指した治療法の進歩の大部分は、このような臨床試験によって達成されたものである。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
軟部腫瘍を有する小児および青年に対する治療戦略の多くは、成人患者に対するものと同様であるが、重要な差がある。例えば、小児患者におけるこの種の腫瘍の生物学的特徴は、成人におけるそれとは劇的に異なる場合がある。さらに小児患者では、患肢温存療法の試行がより困難となる。また放射線療法による罹病率が、成人で観察されるよりもはるかに高い場合があり、特に乳児と幼児で顕著である。[ 51 ]
集学的治療により軟部肉腫の成人および小児患者の治療成績は過去20年間で劇的に改善されたが、小児例については(特に小児の余命が成人のそれより長いことを考慮した場合)この治療法による長期的副作用に対する懸念が高まってきている。このことから、腫瘍を最大限に制御し、長期的な合併症を最小限にするために、非横紋筋肉腫性軟部肉腫に罹患している小児および青年の治療は個別化する必要がある。このような患者は、起こりうる合併症を正確に評価するためのプロスペクティブ研究に登録すべきである。[ 52 ]
参考文献- Spunt SL, Million L, Chi YY, et al.: A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study. Lancet Oncol 21 (1): 145-161, 2020.[PUBMED Abstract]
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002.[PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001.[PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002.[PUBMED Abstract]
- Paulino AC, Ritchie J, Wen BC: The value of postoperative radiotherapy in childhood nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 43 (5): 587-93, 2004.[PUBMED Abstract]
- Kaste SC, Hill A, Conley L, et al.: Magnetic resonance imaging after incomplete resection of soft tissue sarcoma. Clin Orthop (397): 204-11, 2002.[PUBMED Abstract]
- Chandrasekar CR, Wafa H, Grimer RJ, et al.: The effect of an unplanned excision of a soft-tissue sarcoma on prognosis. J Bone Joint Surg Br 90 (2): 203-8, 2008.[PUBMED Abstract]
- Daigeler A, Kuhnen C, Moritz R, et al.: Lymph node metastases in soft tissue sarcomas: a single center analysis of 1,597 patients. Langenbecks Arch Surg 394 (2): 321-9, 2009.[PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987.[PUBMED Abstract]
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000.[PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000.[PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008.[PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014.[PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012.[PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013.[PUBMED Abstract]
- Haas RL, Gronchi A, van de Sande MAJ, et al.: Perioperative Management of Extremity Soft Tissue Sarcomas. J Clin Oncol 36 (2): 118-124, 2018.[PUBMED Abstract]
- Crompton JG, Ogura K, Bernthal NM, et al.: Local Control of Soft Tissue and Bone Sarcomas. J Clin Oncol 36 (2): 111-117, 2018.[PUBMED Abstract]
- Nussbaum DP, Rushing CN, Lane WO, et al.: Preoperative or postoperative radiotherapy versus surgery alone for retroperitoneal sarcoma: a case-control, propensity score-matched analysis of a nationwide clinical oncology database. Lancet Oncol 17 (7): 966-975, 2016.[PUBMED Abstract]
- Virkus WW, Mollabashy A, Reith JD, et al.: Preoperative radiotherapy in the treatment of soft tissue sarcomas. Clin Orthop (397): 177-89, 2002.[PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Preoperative vs. postoperative radiation therapy for soft tissue sarcoma: a retrospective comparative evaluation of disease outcome. Int J Radiat Oncol Biol Phys 56 (2): 482-8, 2003.[PUBMED Abstract]
- Dickie C, Parent A, Griffin AM, et al.: The value of adaptive preoperative radiotherapy in management of soft tissue sarcoma. Radiother Oncol 122 (3): 458-463, 2017.[PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002.[PUBMED Abstract]
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005.[PUBMED Abstract]
- Rao AD, Chen Q, Million L, et al.: Preoperative Intensity Modulated Radiation Therapy Compared to Three-Dimensional Conformal Radiation Therapy for High-Grade Extremity Sarcomas in Children: Analysis of the Children's Oncology Group Study ARST0332. Int J Radiat Oncol Biol Phys 103 (1): 38-44, 2019.[PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015.[PUBMED Abstract]
- Bishop AJ, Zagars GK, Torres KE, et al.: Combined Modality Management of Retroperitoneal Sarcomas: A Single-Institution Series of 121 Patients. Int J Radiat Oncol Biol Phys 93 (1): 158-65, 2015.[PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997.[PUBMED Abstract]
- Delaney TF, Kepka L, Goldberg SI, et al.: Radiation therapy for control of soft-tissue sarcomas resected with positive margins. Int J Radiat Oncol Biol Phys 67 (5): 1460-9, 2007.[PUBMED Abstract]
- Blakely ML, Spurbeck WW, Pappo AS, et al.: The impact of margin of resection on outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Pediatr Surg 34 (5): 672-5, 1999.[PUBMED Abstract]
- Skytting B: Synovial sarcoma. A Scandinavian Sarcoma Group project. Acta Orthop Scand Suppl 291: 1-28, 2000.[PUBMED Abstract]
- Hua C, Gray JM, Merchant TE, et al.: Treatment planning and delivery of external beam radiotherapy for pediatric sarcoma: the St. Jude Children's Research Hospital experience. Int J Radiat Oncol Biol Phys 70 (5): 1598-606, 2008.[PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999.[PUBMED Abstract]
- Merchant TE, Parsh N, del Valle PL, et al.: Brachytherapy for pediatric soft-tissue sarcoma. Int J Radiat Oncol Biol Phys 46 (2): 427-32, 2000.[PUBMED Abstract]
- Karakousis CP, Driscoll DL: Treatment and local control of primary extremity soft tissue sarcomas. J Surg Oncol 71 (3): 155-61, 1999.[PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Prognostic factors for disease-specific survival after first relapse of soft-tissue sarcoma: analysis of 402 patients with disease relapse after initial conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys 57 (3): 739-47, 2003.[PUBMED Abstract]
- Raut CP, Miceli R, Strauss DC, et al.: External validation of a multi-institutional retroperitoneal sarcoma nomogram. Cancer 122 (9): 1417-24, 2016.[PUBMED Abstract]
- Schomberg PJ, Gunderson LL, Moir CR, et al.: Intraoperative electron irradiation in the management of pediatric malignancies. Cancer 79 (11): 2251-6, 1997.[PUBMED Abstract]
- Nag S, Shasha D, Janjan N, et al.: The American Brachytherapy Society recommendations for brachytherapy of soft tissue sarcomas. Int J Radiat Oncol Biol Phys 49 (4): 1033-43, 2001.[PUBMED Abstract]
- Viani GA, Novaes PE, Jacinto AA, et al.: High-dose-rate brachytherapy for soft tissue sarcoma in children: a single institution experience. Radiat Oncol 3: 9, 2008.[PUBMED Abstract]
- Al Yami A, Griffin AM, Ferguson PC, et al.: Positive surgical margins in soft tissue sarcoma treated with preoperative radiation: is a postoperative boost necessary? Int J Radiat Oncol Biol Phys 77 (4): 1191-7, 2010.[PUBMED Abstract]
- Wang D, Bosch W, Kirsch DG, et al.: Variation in the gross tumor volume and clinical target volume for preoperative radiotherapy of primary large high-grade soft tissue sarcoma of the extremity among RTOG sarcoma radiation oncologists. Int J Radiat Oncol Biol Phys 81 (5): e775-80, 2011.[PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013.[PUBMED Abstract]
- Ferrari A: Role of chemotherapy in pediatric nonrhabdomyosarcoma soft-tissue sarcomas. Expert Rev Anticancer Ther 8 (6): 929-38, 2008.[PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997.[PUBMED Abstract]
- Woll PJ, Reichardt P, Le Cesne A, et al.: Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncol 13 (10): 1045-54, 2012.[PUBMED Abstract]
- Demetri GD, Chawla SP, Ray-Coquard I, et al.: Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol 31 (19): 2485-92, 2013.[PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012.[PUBMED Abstract]
- Mir O, Brodowicz T, Italiano A, et al.: Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17 (12): 1732-1742, 2016.[PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.[PUBMED Abstract]
- Suit H, Spiro I: Radiation as a therapeutic modality in sarcomas of the soft tissue. Hematol Oncol Clin North Am 9 (4): 733-46, 1995.[PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.[PUBMED Abstract]
- 新規診断小児軟部肉腫の治療
-
脂肪細胞腫瘍
脂肪肉腫
脂肪肉腫は20歳未満の患者における軟部肉腫の3%を占める(表1を参照のこと)。
脂肪肉腫は、小児集団ではまれである。成人型肉腫の患児182人を対象としたレビューでは、脂肪肉腫と診断されたのはわずか14人であった。[ 1 ]あるレトロスペクティブ研究で、1960年から2011年に22歳未満の患者34人が特定された。[ 2 ]男性と女性の患者数はほぼ等しく、年齢中央値は18歳であった。国際的な臨床病理学的レビューで、小児脂肪肉腫82症例の特徴が報告された。[ 3 ]中央年齢は15.5歳で、女性の方が多く罹患していた。両報告で大多数の患者が粘液型脂肪肉腫であった。[ 2 ][ 3 ]
臨床像
小児および青年の年齢層における脂肪肉腫のほとんどが低悪性度であり、皮下に位置している。リンパ節への転移はまれで、大多数の転移部位は肺である。末梢に発生した腫瘍は、低悪性度で粘液型となる傾向が高い。中枢に発生した腫瘍は、高悪性度で多形型、さらに転移がみられるか、転移を伴って再発する傾向が高い。
予後
高悪性度または中枢の腫瘍は、有意に高い死亡リスクと関連している。国際的なレトロスペクティブ・レビューで、中枢腫瘍の5年生存率は42%であった。多形性粘液性脂肪肉腫の患者10人中7人がこの疾患で死亡した。[ 3 ]14人の患者を対象としたレトロスペクティブ研究では、5年生存率は78%であり、腫瘍の悪性度、組織学的サブタイプ、および原発部位が生存率と相関していた。[ 2 ]
治療
脂肪肉腫に対する治療法の選択肢には以下のものがある:
- 手術。腫瘍が完全に切除されていない場合または局所再発した場合は、2回目の手術を施行してもよい。[ 6 ][ 7 ][ 8 ]
- 化学療法およびその後の手術。
- 放射線療法およびその後の手術、または手術およびその後の放射線療法(成人での研究に基づく証拠)。[ 9 ][ 10 ]
脂肪肉腫に対して最も重要な治療法は手術である。高分化型または粘液性脂肪肉腫の外科的完全切除後におけるイベントフリー生存(EFS)率および全生存(OS)率は、約90%である。[ 11 ]初回手術が不完全な場合は、再切除を実施して、広い切除断端を得るべきである。局所再発が確認されているが、特に低悪性度脂肪肉腫は腫瘍の二次切除により制御されている。放射線療法もまた、追加の手術および放射線療法の審美的/機能的結果に応じて術前または術後に検討される。[ 12 ][ 13 ]
特に中枢の腫瘍で完全切除を容易にするために、手術の前に化学療法を用いて脂肪肉腫のサイズを小さくした報告がある。[ 14 ][ 15 ]脂肪肉腫に対する術後化学療法の役割は明確に定義されていない。完全切除された粘液型脂肪肉腫に対しては、いかなる術後療法の必要性もないと考えられている。術後化学療法を使用しても、多形型脂肪肉腫の場合の生存率は依然として不良である。[ 16 ]
進行粘液型脂肪肉腫の成人患者において、トラベクテジンが有望な反応をもたらしている。[ 17 ]ある研究において、再発脂肪肉腫および平滑筋肉腫の成人患者がトラベクテジンまたはダカルバジンによる治療にランダムに割り付けられた。トラベクテジンによる治療を受けた患者では疾患増悪の45%の低減が得られた。[ 18 ][証拠レベル:1iiDiii]小児患者においてトラベクテジンの使用を支持するデータは非常に少ない。[ 19 ]
非タキサン系微小管ダイナミクス阻害剤のエリブリンを用いた治療はダカルバジンと比較して、再発脂肪肉腫の成人患者における生存を有意に改善し、OS期間中央値はそれぞれ、15.6ヵ月 vs 8.4ヵ月であった。脱分化型および多形型脂肪肉腫の患者では、生存の差がより顕著であった。エリブリンは、高悪性度または中悪性度腫瘍を有する患者の生存期間を延長させる上で有効であった。[ 20 ][証拠レベル:1iiA]エリブリンに関する小児の第I相試験には、脂肪肉腫の患者が登録されなかった。[ 21 ]
骨・軟骨部腫瘍
骨・軟骨部腫瘍として、以下のサブタイプがある:
骨外性間葉性軟骨肉腫
骨性および軟骨性新生物は20歳未満の軟部肉腫患者の0.8%を占める(表1を参照のこと)。
病理組織学および分子的特徴
間葉性軟骨肉腫は小円形細胞および硝子軟骨を特徴とするまれな腫瘍であり、若年成人に生じることが多く、頭頸部領域に好発する。
間葉性軟骨肉腫は一貫性のある染色体再構成と関連付けられている。間葉性軟骨肉腫症例についてのレトロスペクティブ解析で、検査された15件の標本中10件にHEY1-NCOA2融合が確認された。[ 22 ]この遺伝子融合は核型分析により発見可能な染色体変化と関連していなかった。一例として、ある症例の間葉性軟骨肉腫で転座t(1;5)(q42;q32)が同定され、新たなIRF2BP-CDX1融合遺伝子と関連していることが示されている。[ 23 ]
予後
欧州の施設のレトロスペクティブな調査により、間葉性軟骨肉腫の小児および成人患者113人が同定された。良好な転帰と関連する因子には以下のものがあった:[ 24 ][証拠レベル:3iiiA]
1973年から2011年までのSurveillance, Epidemiology, and End Results(SEER)データのレトロスペクティブ解析で、間葉性軟骨肉腫の患者205人が確認された;82人の患者が骨性原発腫瘍を有し、123人の患者が骨外性原発腫瘍を有した。[ 25 ]骨性および骨外性原発腫瘍の転帰は同じであった。転帰に関連する因子には、以下のものが挙げられた:
単一施設での1件のレトロスペクティブ・レビューで、1979年から2010年までに間葉性軟骨肉腫の症例43人が確認された。[ 26 ]限局性病変が認められた30人の患者が評価された。診断時平均年齢は33歳(範囲、11~65歳)であった。5年OS率は51%で、10年OS率は37%であった。比較的年齢が若いこと(30歳未満)および男性であることが、不良なOSおよび無病生存(DFS)に関連した。補助放射線療法を受けなかった患者では、局所再発を来す可能性が高かった。
治療
骨外性間葉性軟骨肉腫に対する治療法の選択肢には以下のものがある:
- 手術。腫瘍が完全に切除されない場合は放射線療法も施行することができる。
- 放射線療法とその後の手術または手術とその後の放射線療法。[ 9 ][ 10 ]
- 化学療法とその後の手術および追加の化学療法。放射線療法を施行することもできる。
German Cooperative Soft Tissue Sarcoma Study Group(軟部組織病変を有する11人)およびGerman-Austrian-Swiss Cooperative Osteosarcoma Study Group(原発骨病変を有する4人)によるプロトコルから、26歳未満の患者15人を対象としたレビューによると、局所制御のためには、完全な外科的切除、または不完全切除後の放射線療法が必要であることが示唆される。[ 27 ][証拠レベル:3iiA]
単一施設での1件のレトロスペクティブ・レビューで間葉性軟骨肉腫の小児患者12人が同定された。[ 28 ]これらの患児において、腫瘍内のNCOA2再構成の存在が報告されている。治癒を得るためには外科的切除が必要であることも確認された。11人の患児が限局性病変を、1人が肺結節を呈した。全患児が化学療法を受けた - 6人は外科的切除の前後に、6人は切除後にのみ受けた。全患児が放射線療法(線量中央値、59.4Gy)を併用したまたは併用しない術後化学療法(イホスファミド/ドキソルビシンが最も多かった)を受けた。追跡期間中央値4.8年で、5年DFS率は68.2%(95%信頼区間[CI]、39.8%-96.6%)およびOS率は88.9%(95%CI、66.9%-100%)であった。
骨外性粘液型軟骨肉腫および間葉性軟骨肉腫患者を対象にした日本の1件の研究では、患者がトラベクテジンまたは最適な支持療法のいずれかによる治療にランダムに割り付けられた。[ 29 ]患者の年齢中央値は38歳(範囲、21~77歳)であった。トラベクテジンを受ける群に割り付けられた患者のOSは、最適な支持療法を受ける群に割り付けられた患者のOSよりも優れていた。
骨外性骨肉腫
骨性および軟骨性新生物は20歳未満の患者における軟部肉腫の0.8%を占める(表1を参照のこと)。
小児および青年の年齢層における骨外性骨肉腫はきわめてまれである。SEERデータの解析で、1973年から2009年までの高悪性度骨肉腫患者4,173人のうち骨外性骨肉腫を有する256人の患者(6%)が確認された。骨性骨肉腫と比較して、骨外性骨肉腫患者は年齢が高く、女性であり、体軸の原発腫瘍を有し、所属リンパ節転移を来している可能性が高かった。予後不良な特徴として、転移性病変の存在、比較的大きな腫瘍サイズ、年齢が高いこと、および体軸の原発腫瘍部位が挙げられた。[ 30 ]
分子的特徴
骨外性骨肉腫の成人患者32人のレビューでは、一貫していくつかの変化が明らかにされた。[ 31 ]頻繁なゲノム変化として、コピー数の減少がCDKN2A(70%)、TP53(56%)、およびRB1(49%)においてみられた。メチル化/脱メチル反応(40%)、クロマチン再構成(27%)、およびWNT/SHH経路(27%)に影響する変異が同定された。同時にTP53とRB1の両アレル性コピー数の減少がみられた症例では、DFSおよびOSが不良であった。
予後
骨外性骨肉腫は、局所再発および肺転移のリスクの高さと関連している。[ 32 ]1件の単一施設のレトロスペクティブ・レビューで骨外性骨肉腫患者43人が確認された;37人の患者が限局性病変を有し、6人の患者が転移性病変を有した。[ 33 ]年齢中央値は55歳(範囲、7~81歳)であった。無増悪生存(PFS)期間中央値は21ヵ月であった;OS期間中央値は50ヵ月であった。75%の患者が化学療法を受けた。化学療法を受けた患者では生存が比較的良好な傾向がみられ、シスプラチンを含む化学療法を受けた患者では、生存における統計的に有意な改善が得られた。
診断時年齢中央値57歳(範囲、12~91歳)の患者274人を対象にしたレビューにおいて、5年DFS率およびOS率は化学療法を受けた患者で有意に良好であり、骨肉腫向けのレジメンの使用は奏効率の改善に関連した。[ 34 ][証拠レベル:3iiiA]
European Musculoskeletal Oncology Societyにより、1981年から2014年に治療された骨外性骨肉腫の適格患者266人のレトロスペクティブ解析が実施された。[ 34 ]50人の患者(19%)が転移性病変を有した。原発腫瘍の外科的切除後に完全寛解を達成した211人の患者を解析したところ、51%の5年OS率および43%の5年DFS率が示された。通常は骨性骨肉腫患者に用いられる化学療法で治療された患者で生存が良好な傾向がみられた。多変量解析では、より良好な予後に関連する因子として、比較的年齢が低いこと(40歳未満)、小さい腫瘍、および化学療法の使用が挙げられた。
線維芽細胞性/筋線維芽細胞性腫瘍
線維芽細胞性/筋線維芽細胞性腫瘍として、以下のサブタイプがある:
- 線維芽細胞性/筋線維芽細胞性腫瘍。
- 中悪性度(局所侵攻性)。
- 中悪性度(転移はまれ)。
- 悪性。
デスモイド型線維腫症
デスモイド型線維腫症は以前はデスモイド腫瘍または侵襲性線維腫症と呼ばれていた。
危険因子
少数のデスモイド型線維腫症が、(腸管ポリープおよび結腸がんの高い発生率を伴う)APC遺伝子における変異に関連して発生することがある。デスモイド型線維腫症と診断された10歳以上の患者519人を対象にした研究では、39人の患者(7.5%、過小評価の可能性がある)が家族性大腸腺腫症(FAP)を有することが判明した。[ 35 ]FAPおよびデスモイド型線維腫症を有する患者は、FAPを伴わないデスモイド型線維腫症の患者と比べて若年で、男性の頻度が高く、腹壁腫瘍または腸間膜腫瘍が多くみられた。
結腸がんの家族歴、網膜色素上皮の先天性過形成が認められる[ 36 ][ 37 ]、またはデスモイド型線維腫症が腹部または腹壁に位置している場合[ 35 ]は遺伝カウンセラーに紹介すべきである。現在のところ、デスモイド型線維腫症の小児における遺伝子検査について一般的な推奨事項はない。腫瘍の病理学および分子的特徴はスクリーニングの指針を提供するに過ぎない。もし腫瘍にCTNNB1体細胞突然変異がみられる場合、この状況でのAPC遺伝子の突然変異は記載されていないため、スクリーニングは必要ない。もしCTNNB1変異が同定されないなら、APC突然変異を調べるスクリーニングが必要となりうる。[ 38 ][ 39 ](詳しい情報については、大腸がんの遺伝学に関するPDQ要約の家族性大腸腺腫症[FAP]のセクションを参照のこと。)
予後
デスモイド型線維腫症は転移の可能性がきわめて低い。この腫瘍は局所的に浸潤するため、正常構造の温存の必要性から外科的制御が困難となることがある。
デスモイド型線維腫症は局所再発の可能性が高い。これらの腫瘍は自然史が非常に多様であり、十分に証明された自然退縮の症例もみられる。[ 40 ]デスモイド型線維腫症の80%超にCTNNB1遺伝子のエクソン3における突然変異がみられ、45Fの突然変異では再発リスクが高いという関連性が指摘されている。[ 41 ]再度の外科的切除により、ときに再発病変を制御下に置くことが可能となる場合もある。[ 42 ]
治療
デスモイド型線維腫症の自然経過は非常に多様で、患者の最大20%で部分退縮が示されるため、その治療のための介入の有益性評価はきわめて困難となっている。[ 43 ]現在では、大規模な複数の成人患者シリーズと比較的小規模な複数の小児患者シリーズから、腫瘍の長期の安定、さらには全身療法なしでの腫瘍退縮すらも報告されている。[ 42 ][ 44 ];[ 45 ][証拠レベル:3iiiDi]例えば、デスモイド腫瘍の成人患者におけるソラフェニブに関する1件の大規模プラセボ対照試験において、治療を受けなかった(観察/プラセボ)患者は20%の部分退縮率を示し、プラセボ群患者の46%では1年経過時に進行が認められなかった。[ 43 ]
デスモイド型線維腫症に対する治療法の選択肢には以下のものがある:
- 経過観察。上述のように、デスモイド腫瘍の自然史は多様であるため、ときに経過観察が実行可能な選択肢である。これは特に無症状の病変、重要臓器に対し危険をもたらさない病変、および切除が不完全であった腫瘍に当てはまる。[ 42 ][ 46 ][ 47 ][ 48 ][ 49 ][ 50 ][ 51 ][ 52 ]
- 切除不能腫瘍または再発腫瘍に対しては化学療法。
- チロシンキナーゼ阻害薬。
- NSAID。デスモイド型線維腫症に対してスリンダクのようなNSAIDが単一症例群で用いられている;確認された奏効は通常、病勢の安定化であった。[ 62 ]
- 抗エストロゲン療法。通常はタモキシフェンを用いた抗エストロゲン療法にスリンダクを加えることでも病勢の安定化が得られている。[ 63 ]タモキシフェンとスリンダクの併用に関する1件のプロスペクティブ試験では副作用はほとんど報告されなかったが、女児では無症状の卵巣嚢胞が一般的である。この併用について奏効率およびPFS率で測定したところ、活性は相対的にほとんど示されなかった。[ 64 ][証拠レベル:2Diii]
- 手術。手術が選択される場合、その目的は切除断端陰性の達成である。しかしながら、St. Jude Children's Research Hospital(SJCRH)でデスモイド型線維腫症の手術を受けた小児を対象としたレトロスペクティブ・レビューでは、外科的切除断端と再発リスク間には相関性がないことが報告された。[ 52 ]
- 放射線療法。
- NOTCH経路阻害剤。
隆起性皮膚線維肉腫
皮膚線維肉腫はすべての年齢層で発生しうるまれな腫瘍であるが、報告された症例の多くは小児で発生したものである。[ 68 ][ 69 ][ 70 ]SEERデータベースにおける20歳未満の小児の症例451例のレビューにより、発生率は100万人当たり1例で、15~19歳の黒人患者で最も高かった。最も一般的な部位は体幹および四肢であったが、これは成人にみられるものと類似している。95%の患者が手術を受けた。OS率は5年で100%、15年で98%、30年で97%であった。男性の生存率は女性よりも低かった(P < 0.05)。[ 71 ][証拠レベル:3iA]
分子的特徴
この腫瘍では、COL1A1遺伝子とPDGFRB遺伝子の融合を引き起こす染色体転座t(17;22)(q22;q13)が一貫して認められる。
治療
隆起性皮膚線維肉腫に対する治療法の選択肢には以下のものがある:
- 手術。
- 放射線療法とその後の手術または手術とその後の放射線療法。
- 切除不能腫瘍または再発腫瘍に対する放射線療法およびイマチニブ療法。
皮膚線維肉腫のほとんどの腫瘍は、外科的な完全切除により治癒が可能である。切除断端陰性の広範囲切除またはモース手術/修正モース手術により、ほとんどの腫瘍で再発を回避できる。[ 72 ]腫瘍の振る舞いが局所侵攻性であるにもかかわらず、リンパ節または内臓への転移はまれである。
複数のレトロスペクティブ・レビューでは、不完全切除後の術後放射線療法により再発可能性が低下した可能性がある。[ 73 ][ 74 ]
外科的切除を遂行できない場合や腫瘍が再発した場合でも、イマチニブによる治療が有効とされている。[ 75 ][ 76 ][ 77 ]複数回の再発後は転移性疾患の可能性が高いため、外科的に管理できない再発患者では放射線や他の補助療法を検討すべきである。[ 69 ][ 71 ]
隆起性皮膚線維肉腫に対する精密検査および管理のためのガイドラインが発表されている。[ 78 ]
炎症性筋線維芽細胞性腫瘍
炎症性筋線維芽細胞性腫瘍はまれな間葉系腫瘍であり、小児および青年に好発する。[ 79 ][ 80 ][ 81 ]
臨床像
炎症性筋線維芽細胞性腫瘍はまれな腫瘍であり、小児および若年成人の軟部組織および内臓に生じる。[ 82 ]転移はまれだが、局所浸潤性となることが多い。病変が生じる一般的な解剖学的部位には軟部組織、肺、脾臓、結腸、および乳房がある。[ 79 ]小児における膀胱の炎症性筋線維芽細胞性腫瘍42症例のレビューが2015年に発表された。[ 83 ]
分子的特徴
炎症性筋線維芽細胞性腫瘍の約半数は、染色体2p23における未分化リンパ腫キナーゼ(ALK)受容体チロシンキナーゼ遺伝子を活性化するクローン変異を示す。[ 84 ]免疫組織化学検査でALK陰性であった症例11人中8人(73%)で、ROS1およびPDGFRBキナーゼ融合が同定されている。[ 85 ][証拠レベル:3iiiDiv]
治療
炎症性筋線維芽細胞性腫瘍に対する治療法の選択肢には以下のものがある:
- 手術。
- 化学療法。
- ステロイド療法。
- NSAIDによる治療。
- 標的療法(ALK阻害薬)。
実施可能な場合は、外科的完全切除が治療の中心である。[ 86 ]9人の患者を対象にしたシリーズにおいて、完全切除後に4人の患者が持続的寛解を達成し、残存病変が認められた3人の患者は再発したが、後に持続的寛解を達成し、転移性病変が認められた1人の患者が多剤併用化学療法への反応を示した。[ 87 ][証拠レベル:3iiA]化学療法の有益性が症例報告で認められている。[ 88 ]ドイツの研究のレビューで、炎症性筋線維芽細胞性腫瘍を有する21歳未満の患者37人が確認された。[ 89 ][証拠レベル:3iiA]全体の5年EFS率は75%で、OS率は91%であった。20人の患者のうち、17人が完全切除を受け、再発は認められなかった。他の患者は全員、手術とさまざまな化学療法レジメンの併用で治療された。外科的切除は、形状および機能を維持する手技に限定できる。
ステロイドまたはNSAIDのいずれかに対して奏効が認められた症例報告がいくつかある。[ 90 ][ 91 ]18歳以下の患者32人のシリーズで、完全切除が治療の中心であるが、一部の患者はステロイドまたは細胞毒性化学療法で治療されたことが明らかにされた。OS率は94%であった;3人の患者が再燃し、このうち2人がこの疾患により死亡した。ステロイドなど、他の治療を併用するまたは併用しない完全切除を実施した場合に、この疾患を有する患者に対して高い生存率が得られた。[ 92 ][証拠レベル:3iiA]
炎症性筋線維芽細胞性腫瘍は、以下のようにALK阻害薬療法に反応する:
乳児型線維肉腫
小児および青年における線維肉腫には、明らかに異なる種類として次の2つがある:乳児型線維肉腫(先天性線維肉腫とも呼ばれる)、および成人にみられる線維肉腫と区別できない線維肉腫。両者は病理学的診断が明らかに異なり、別の治療法が必要である。成人型線維肉腫について以下で取り扱う。
臨床像
乳児型線維肉腫は、通常は急速に増殖する腫瘤を示し、しばしば出生時に認められ、出生前の超音波検査で確認されることもある。発症時に腫瘍がきわめて大きくなっていることが多い。[ 99 ]新生児においてこの疾患の初診時の特徴として、副甲状腺ホルモン関連蛋白の濃度上昇に続発する高カルシウム血症が報告されている。[ 100 ]
分子的特徴
通常、腫瘍には細胞遺伝学的に特徴的なt(12;15)(ETV-NTRK3)の転座が認められる。乳児型線維肉腫にもこの転座が認められ、組織学的所見が間葉芽腎腫と実質的に一致している。
乳児型線維肉腫は通常、1歳未満の乳児に発生する。ときには4歳までの小児に発生することもある。より年齢の高い小児において形態学的に類似した腫瘍が同定されている;これらの年齢の高い小児における腫瘍には、より年齢の低い患者で特徴的なt(12;15)(ETV-NTRK3)転座は認められない。[ 101 ]乳児型線維肉腫の症例においてBRAF遺伝子内欠失が報告されており、NTRK3融合が同時発生している。[ 102 ]
予後
このような腫瘍では、診断時に転移が認められる可能性は低い。
治療
乳児型線維肉腫に対する治療法の選択肢には以下のものがある:
- 手術およびその後の経過観察。
- 手術およびその後の化学療法。
- 化学療法およびその後の手術。
- 標的療法。
乳児型線維肉腫患者のほとんどが完全切除により治癒可能である。しかしながら、病変のサイズが大きいと、重大な機能的後遺症をもたらすことなしに切除することが不可能になる頻度が高い。例えば、四肢の腫瘍では完全切除に切断術がしばしば必要になる。欧州の小児科グループは、手術後のグループII病変の患者では経過観察も選択肢となりうることを報告している。[ 103 ]グループII病変の患者12人は追加治療を受けず、2人が再燃した。1人は化学療法後に完全寛解を得た。より悪性度の高い病変群の患者および増悪を来した患者に対し術後化学療法が施行された。その後の研究では、グループII病変の患者7人のうち、経過観察中に増悪を来した患者は1人のみであった;その患者は化学療法により完全寛解を達成した。[ 104 ][証拠レベル:3iiA]
術前化学療法により、さらに保存的な外科アプローチが可能となる;この設定で活性のある薬剤には、ビンクリスチン、ダクチノマイシン、シクロホスファミド、およびイホスファミドがある。[ 105 ][ 106 ];[ 104 ][ 107 ][証拠レベル:3iiA];[ 108 ][証拠レベル:3iiB]乳児型線維肉腫の患者を対象とした3件の研究から、アルキル化剤を含まないレジメンが有効であり、肉眼的病変を有する患児において第一選択治療法として使用すべきであることが示唆されている。[ 103 ][ 104 ][ 109 ]
LMNA-NTRK1融合の変異型を示す2例がクリゾチニブに反応した。[ 110 ][ 111 ]
TRK A、B、およびCの経口のATP競合的阻害剤であるラロトレクチニブに関する第I/II相試験では、NTRK融合が認められた再発乳児型線維肉腫の患者8人全員に持続的な客観的奏効が示された。ラロトレクチニブの術前補助療法後に部分奏効を達成した5人の患者のうち3人が切除断端陰性での外科的完全切除を受け、非常に優れた病理学的奏効(98%を超える治療効果)を達成し、手術後7~15ヵ月経過時に無病状態を維持していた。[ 112 ][ 113 ];[ 114 ][証拠レベル:3iiD]この試験でETV6-NTRK3再構成がみられた乳児型線維肉腫患者8人中1人では、ラロトレクチニブ療法の8ヵ月後に疾患が進行し、G623Rにおいて耐性を獲得した変異を有することが明らかにされた。この患者は、反復性のキナーゼドメイン変異の媒介によって獲得された耐性を克服するようにデザインされた選択的TRK阻害剤のLOXO-195で治療され、一時的な部分奏効を経験した。[ 115 ]1件の第I相小児試験において、ラロトレクチニブの小児に特異的な薬物動態および毒性が記述された。[ 116 ]
乳児型線維肉腫を有する生後2ヵ月の患者は、最初に化学療法で治療された。疾患進行時には、パゾパニブで奏効が示された。[ 117 ]
治療なしに自然退縮を生じたまれな症例が1例報告されている。[ 118 ][証拠レベル:3iiiDiv]
臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
成人型線維肉腫
これらの腫瘍では、乳児型線維肉腫にみられる転座がみられない。これらの腫瘍は、ほとんどの非横紋筋肉腫と類似しており、管理アプローチも同様である。
粘液線維肉腫
粘液線維肉腫はまれな病変であり、特に小児ではまれである。一般には外科的完全切除による治療が実施される。
低悪性度線維粘液性肉腫
低悪性度線維粘液性肉腫は、若年成人および中年期の成人が最も多く罹患する組織学的見かけがあてにならない軟部新生物であり、四肢の深部にみられることが多く、FUS-CREB3L3の転座(およびまれに、FUS-CREB3L1やEWSR1-CREB3L1といった別の転座)を特徴とする。[ 4 ][ 119 ][ 120 ][ 121 ][ 122 ]
予後
低悪性度線維粘液性肉腫患者33人(3人は18歳未満であった)を対象としたレビューで、33人中21人が診断から最長15年(中央値で3.5年)経過後に局所再発を来した;15人の患者が診断から最長45年(中央値で5年)経過後に主に肺および胸膜に転移を来したことから、これらの患者を継続的に追跡する必要があることが浮き彫りとなっている。[ 119 ]転移した後でも、疾患の経過が緩慢な場合がある。[ 123 ]
他の報告では、患者73人中14人が18歳未満であった。追跡期間が比較的短い(中央値24ヵ月)このシリーズでは、適切な追跡を受けた患者54人中8人のみが局所(9%)または遠隔(6%)再発を生じた。この報告は、この腫瘍の挙動がかつて報告されていたものより顕著に良好である可能性を示唆するものである。[ 124 ]しかしながら、晩期に転移が発生するため、これらの患者については注意深いモニタリングが必要となる。
最新の小児腫瘍学グループ(COG)試験(ARST0332[NCT00346164])でこの腫瘍の患者11人が登録された。診断時の年齢中央値は13歳で、男児の方が多く罹患していた。最好発部位は下肢および上肢(n = 9)で、中央値2.7年間の追跡後に局所または遠隔転移を生じていた患児はいなかった。[ 125 ]
治療
低悪性度線維粘液性肉腫に対する治療法の選択肢には以下のものがある:
- 手術。
低悪性度線維粘液性肉腫についての限られた治療情報は、この腫瘍にあまり化学療法感受性がないことから、手術が選択すべき治療法であることを示唆している。[ 123 ]German Cooperative Weichteilsarkom Studiengruppe(CWS)により、21歳未満の低悪性度線維粘液性肉腫患者31人に対する研究結果が報告された。[ 121 ][証拠レベル:3iiDi]5年EFS率は71%(95%CI、±18.6%)、5年局所無再燃生存率は76%(95%CI、±17.6%)、および5年OS率は100%であった。R0切除を受けた患者24人中、5人(21%)の患者が再燃を起こした(3人が局所、1人が転移性、および1人が複合)。R1切除を受けた患者7人中、3人(43%)の患者が局所再燃を起こした。
この腫瘍に対する化学療法および/または放射線療法の使用に関するデータはほとんどない。1件の報告から、低悪性度線維粘液性肉腫の治療においてトラベクテジンが有効となる可能性が示唆されている。[ 126 ]
骨格筋腫瘍
横紋筋肉腫
詳しい情報については、小児横紋筋肉腫の治療に関するPDQ要約を参照のこと。
平滑筋腫瘍
平滑筋肉腫
平滑筋肉腫は20歳未満の患者における軟部肉腫の2%を占める(表1を参照のこと)。
危険因子
腫瘍の発生をみたHIV/AIDSの小児43人のうち、8人にエプスタイン-バーウイルス関連平滑筋肉腫の発生がみられた。[ 130 ]遺伝性網膜芽細胞腫の生存者は、平滑筋肉腫の発症リスクが統計的に有意に高く、これらの患者の78%が網膜芽細胞腫の最初の診断から30年以上経過後に診断された。[ 131 ]
治療
平滑筋肉腫に対する治療法の選択肢には以下のものがある:
- 化学療法(トラベクテジン)。
トラベクテジンは平滑筋肉腫の成人で研究されている。研究の結果は、以下の通りである:
小児患者においてトラベクテジンの使用を支持するデータは存在しない。
いわゆる線維組織球性腫瘍
いわゆる線維組織球性腫瘍として、以下のサブタイプがある:
叢状線維組織球性腫瘍
叢状線維組織球性腫瘍はまれで、小児および若年成人が最も多く罹患する低悪性度~中悪性度の腫瘍である。シリーズにもよるが、初診時の年齢中央値は8~14.5歳の範囲である;しかしながら、生後3ヵ月という若い患児でも報告がある。[ 133 ][ 134 ]
臨床像
腫瘍は、皮膚または皮下組織に痛みを伴わない腫瘤として多く発生し、指、手、手首などの上肢に現れることが最も多い。[ 135 ][ 136 ][ 137 ]所属リンパ節または肺へ転移したという腫瘍の報告はまれである。[ 133 ][ 137 ][ 138 ]
分子的特徴
一貫性のある染色体異常は検出されていないが、t(4;15)(q21;q15)転座が報告されている。[ 139 ]
予後
叢状線維組織球性腫瘍は中悪性度の腫瘍であり、転移はまれである。
神経鞘腫瘍
悪性末梢神経鞘腫瘍
悪性末梢神経鞘腫瘍は、20歳未満の患者における軟部肉腫の5%を占める(表1を参照のこと)。
危険因子
悪性末梢神経鞘腫瘍は散発性に、また神経線維腫症1型(NF1)の小児に発生することがある。[ 141 ]NF1患者における悪性末梢神経鞘腫瘍の家族歴は、早期発症型悪性末梢神経鞘腫瘍の発症リスク増加に関連している。[ 142 ]
神経線維腫症2型(NF2)および良性の神経線維腫が明らかにされた小児のまれな例では、5度の再発がみられた;この間に、病変では次第にマーカー(S-100など)が検出できなくなり、この疾患におけるNOTCH2の最初の例を含めて複数のマーカーで示された悪性末梢神経鞘腫瘍への悪性転換の明確な徴候を獲得した。[ 143 ]
分子的特徴
悪性末梢神経鞘腫瘍の分子的特徴には以下のものがある:
予後
良好な予後に関連する特徴として、以下のものがある:[ 141 ][ 147 ][ 148 ][ 149 ]
不良な予後に関連する特徴として、以下のものがある:[ 151 ]
MD Anderson Cancer Centerの研究における限局性病変の患者では、NF1を合併した患者と合併していない患者において転帰に有意差は認められなかった。[ 148 ]他の諸研究において、NF1を合併していないことが良好な予後因子であるかどうかについては、良好[ 147 ]および不良[ 141 ][ 147 ][ 149 ]な転帰の両方で関連性が指摘されているため明らかではなかった。French Sarcoma Group研究において、NF1は他の予後不良な特徴に関連していたが、不良な転帰の独立した予測因子ではなかった。[ 151 ]オランダのがん登録データのレトロスペクティブ解析で、悪性末梢神経鞘腫瘍の患者784人が確認された;患者のうち70人は18歳以下であった。[ 154 ][証拠レベル:3iA]NF1の小児では、大きな腫瘍サイズが一般的であった(5cm超、92.3% vs 59.1%)。全体として、限局性悪性末梢神経鞘腫瘍とNF1を合併している患者の推定5年生存率は52.4%(標準誤差[SE]、10.1%)であったのに対し、NF1が認められない患者では75.8%(SE、7.1%)であった。
Italian Sarcoma Groupにより、悪性末梢神経鞘腫瘍の小児および青年73人における再発後の転帰が報告された。[ 155 ][証拠レベル:3iiiA]初回再燃後のOS期間中央値は11ヵ月で、1年生存率は39.2%で、5年生存率は15.8%であった。再燃したこれらの患者に対するより良好な予後に関連する因子は、最初の腫瘍の浸潤性が比較的低いこと、再燃までの期間が長いこと、および第二完全寛解の達成(根治的手術の実施可能性に関係した)であった。
治療
悪性末梢神経鞘腫瘍に対する治療法の選択肢には以下のものがある:
腫瘍の外科的な完全切除が実施可能な場合は、常に治療の柱となる。
放射線療法の役割については評価が困難であるが、術後の顕微鏡的な残存腫瘍が既知の場合、放射線療法後に局所制御が持続するかどうかは確実ではない。
- 小児悪性末梢神経鞘腫瘍では、化学療法により客観的奏効が得られている。ドイツおよびイタリアにおける悪性末梢神経鞘腫瘍の経験に関する大規模なレトロスペクティブ解析では、測定可能な腫瘍の65%がイホスファミドを含む化学療法レジメンに対して客観的奏効を示したが、この解析では化学療法による生存率の改善について、決定的な実証はなされなかった。[ 141 ]このレトロスペクティブ解析では、術後放射線療法により転帰が改善する傾向もみられた。[ 141 ]
- 悪性末梢神経鞘腫瘍とNF1を合併している若年患者37人を対象としたシリーズでは、ほとんどの患者が化学療法に対する反応の乏しい大きな浸潤性腫瘍を有していることが示された;PFS率は19%、5年OS率は28%であった。[ 156 ]
- EpSSGにより、悪性末梢神経鞘腫瘍を有する21歳以下の患者を対象としてプロスペクティブ研究が実施された。[
157
]原発腫瘍の外科的切除は、切除が顕微鏡的な切除断端陰性で完全であった場合のR0、切除断端が顕微鏡的に陽性の場合のR1、および切除しても肉眼的残存腫瘍が残った場合のR2として分類された。患者は、以下の4つの治療グループのいずれかに非ランダムに割り付けられた:
化学療法を受けた患者について、治療は4コースのイホスファミド/ドキソルビシンおよび2コースのイホスファミドと放射線療法(50.4-54Gy)の同時併用で構成された。病変が測定可能な患者における化学療法に対する奏効率(部分奏効 + 完全奏効)は46%であった。NF1の存在(51%の患者)はOSおよびEFSに対する独立した不良な予後因子であった。
再発悪性末梢神経鞘腫瘍
1979年から2004年のイタリアの小児プロトコルに登録された患者120人を解析したところ、悪性末梢神経鞘腫瘍が再燃した21歳未満の患者73人が同定された。初回診断から再燃までの期間は1ヵ月から204ヵ月に及び、再燃までの期間中央値は7ヵ月であった。初回再燃からのOS期間中央値は11ヵ月で、1年OS率は39%、5年OS率は16%であった。再燃後のより高い生存の確率に関連した因子は、初発時の低い腫瘍浸潤性、再燃までの期間が長いこと、および再燃時の腫瘍の外科的完全切除であった。[ 155 ]
臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
悪性トリトン腫瘍
悪性トリトン腫瘍は悪性末梢神経鞘腫瘍の1亜型である。この腫瘍はNF1患者に最も好発し、神経原性および横紋筋芽細胞性成分からなる。悪性トリトン腫瘍は高悪性度の腫瘍である。この腫瘍は通常は35歳までに生じ、小児ではきわめてまれである(症例報告のみ)。[ 158 ]
治療
悪性トリトン腫瘍は通常は化学療法および放射線療法に反応しないが、横紋筋肉腫に対して行われる治療法による治療が行われている。[ 158 ][証拠レベル:3iiiA](詳しい情報については、小児横紋筋肉腫の治療に関するPDQ要約を参照のこと。)
外胚葉性間葉腫
外胚葉性間葉腫はまれな神経鞘腫瘍で、主に小児に生じる。この腫瘍は間葉性成分および外胚葉性成分の両方を示す二重表現型軟部肉腫である。
周皮性(血管周囲性)腫瘍
筋周皮腫
筋周皮腫の亜型の1つである乳児型血管周皮腫は、原発不明で高度な血管新生を示す腫瘍である。
血管周皮腫の1歳未満の乳児は、血管周皮腫の1歳以上の小児より予後が良好と考えられる。[ 162 ][ 163 ][ 164 ]
組織型
組織学的に、血管周皮腫は複雑な脈管構造の周りに密に配列した円形細胞または紡錘状細胞から構成され、多くの分岐様構造を形成する。硝子化がよくみられる。乳児型血管周皮腫は組織学的に類似しており、多くの場合、腫瘍の腫瘤の外側に脈管構造を伴う多葉性となっている。[ 165 ]
治療および転帰
乳児型血管周皮腫に対する治療法の選択肢には以下のものがある:
- 化学療法。
小児17人のシリーズで、成人と乳児の血管周皮腫では転移の可能性および治療に対する反応に差があることが明確に実証された。[ 166 ]11人の小児が1歳を過ぎていた。これらの患者の数人にリンパ節または肺の病変が認められた。II期またはIII期の患者6人が進行して死亡した。I期の患者3人が生存していたが、1人の患者では肺に再発が認められた。6人の患者が乳児型血管周皮腫を有し、そのうち5人がI期を超えていた。6人の患者全員が生存していたが、3人はビンクリスチン、アクチノマイシン、およびシクロホスファミドに対して良好な反応を示した。
数件の研究で、乳児筋線維腫症(本要約の乳児筋線維腫症のセクションを参照のこと)または血管周皮腫に比較的類似した小児における腫瘍について報告されている。[ 111 ][ 167 ]先天性乳児型線維肉腫にみられるETV6-NTRK3融合蛋白ではなく、LMNA-NTRK1融合蛋白が同定された。[ 168 ]この融合を保有した1人の患者がクリゾチニブに反応した。
分化不明の腫瘍
分化不明の腫瘍として、以下のサブタイプがある:
滑膜肉腫、NOS
滑膜肉腫は20歳未満の患者における軟部肉腫の9%を占める(表1を参照のこと)。
滑膜肉腫は、小児および青年において最もよくみられる非横紋筋肉腫性軟部肉腫の1つである。1973年から2005年までのSEERのレビューでは、滑膜肉腫患者1,268人が特定された。これらの患者の約17%が小児および青年で、診断時年齢の中央値は34歳であった。[ 178 ]
組織学的分類
滑膜肉腫は、以下のタイプに下位分類可能である:
臨床像
最もよくみられる腫瘍部位は四肢で、体幹および頭頸部が続く。[ 178 ]滑膜肉腫はまれに心臓または心膜に発生することがある。[ 179 ]
転移好発部位は肺である。[ 180 ][ 181 ]転移リスクは腫瘍の大きさによって大きく影響を受ける;腫瘍の大きさが5cmを超える患者は、それ以外の患者と比べて転移するリスクが32倍高いと推定される。
診断的評価および分子的特徴
滑膜肉腫の診断は、免疫組織化学的分析、超微細構造所見、および特異的なt(X;18)(p11.2;q11.2)の染色体転座の立証によって確定される。この異常は滑膜肉腫に特有のものであり、すべての形態学的亜型にみられるものである。滑膜肉腫では、第18番染色体上のSYT遺伝子とX染色体上のSSX遺伝子の亜型(1、2または4)の1つとの間で再構成が起こる。[ 182 ][ 183 ]SYT/SSX18の転写産物は、重要な腫瘍抑制遺伝子のエピジェネティックサイレンシングを促進すると考えられている。[ 184 ]
1件の報告において、免疫組織化学的染色でINI1核内反応の低下は49例の滑膜肉腫でみられ、このパターンが滑膜肉腫と他の組織型を見分けるのに役立つ可能性があることが示唆されている。[ 185 ]
予後
10歳未満の患者は、これより年齢が高い患者よりも良好な転帰および臨床的特徴-四肢原発、小さい腫瘍、および限局性腫瘍-を有する。[ 178 ][ 186 ][ 187 ]メタアナリシスで、化学療法に対する奏効が生存延長に関係していることも示唆された。[ 188 ]
以下の諸研究から転帰不良と関連する複数の因子が報告されている:
治療
滑膜肉腫に対する治療法の選択肢には以下のものがある:
COGおよびEuropean Pediatric Soft Tissue Sarcoma Study Groupにより、21歳未満の限局性滑膜肉腫患者60人が補助放射線療法または化学療法を併用しない手術にプロスペクティブに割り付けられた併合解析について報告された。[ 198 ]登録は、すべての腫瘍サイズの悪性度2の腫瘍または5cm以下のグレード3の腫瘍を有し、組織学的に切除断端陰性の初回完全切除が実施された患者に限定された。3年EFS率は90%であった(追跡期間中央値、5.2年;範囲、1.9-9.1)。8つすべてのイベントが腫瘍の局所再発であった;転移性再発はみられなかった。疾患が再発した患者はすべて第二選択療法により有効に治療され、OSは100%であった。
滑膜肉腫は、他の多くの軟部肉腫よりも化学療法に対する感受性が高いようであり、滑膜肉腫の小児は滑膜肉腫の成人よりも予後が良好と考えられる。[ 15 ][ 181 ][ 193 ][ 199 ][ 200 ][ 201 ][ 202 ][ 203 ]滑膜肉腫の治療に最も一般的に用いられるレジメンには、イホスファミドおよびドキソルビシンが組み入れられている。[ 188 ][ 202 ][ 204 ]イホスファミドおよびドキソルビシンのレジメンに対する奏効率は、他の非横紋筋肉腫性軟部肉腫より高い。[ 205 ]
諸研究から以下の化学療法関連の治療所見が報告されている:
再発滑膜肉腫、NOS
再燃後の生存率は不良である(5年で30~40%)。再燃後の転帰に関連する因子としては、初回寛解の持続期間(18ヵ月を超えるまたは18ヵ月以下)および二回目の寛解なしが挙げられる。[ 211 ][ 212 ]ドイツの経験では、転移巣の外科的切除は二回目の完全寛解を達成するための最も一般的な方法であった。[ 212 ]経口トロホスファミド、イダルビシン、およびエトポシドまたは経口シクロホスファミドおよび静脈内ビンブラスチンによる維持化学療法が個別に実施された。
選択された肺転移巣を標的にするために放射線療法(定位放射線療法)が使用可能である。放射線療法は通常、転移病変を確認するため少なくとも1回の切除後に検討される。放射線療法は、気管支に隣接する位置のために空気交換を脅かすか、胸壁に浸潤することで疼痛の原因となる病変を有する患者に特に適している。[ 213 ]
臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
類上皮肉腫
類上皮肉腫は、組織発生が不明で多系統への分化を示すまれな間葉系腫瘍である。[ 214 ]
臨床像
類上皮肉腫は、深部の軟部組織を基盤とした増殖の遅い硬結節として多くみられる;近位型は主に成人にみられ、体軸骨格および近位部位に生じる。高度に侵攻性の腫瘍であり、リンパ節転移を来す可能性もある。
分子的特徴
類上皮肉腫はSMARCB1遺伝子の不活化を特徴とし、これは類上皮肉腫の通常型および近位型の両方において認められる。[ 215 ]この異常により、EZH2への依存度の増加および腫瘍形成が生じる。[ 216 ]
治療
類上皮肉腫に対する治療法の選択肢には以下のものがある:
- 化学療法。
- 手術。
- 放射線療法とその後の手術または手術とその後の放射線療法。
リンパ節転移の有無について患者を注意深く評価すべきである;疑わしいリンパ節については生検が行われる。原発腫瘍および再発腫瘍の外科的切除が最も有効な治療法である。[ 217 ][証拠レベル:3iiiA]この腫瘍はリンパ節に潜在転移を来す傾向があるため、四肢または殿部の類上皮肉腫について(画像検査または身体診察により)臨床で腫大したリンパ節が認められない場合に、センチネルリンパ節生検が推奨される。[ 218 ]
類上皮肉腫の小児患者30人(発症時年齢の中央値は12歳)を対象としたレビューによると、肉腫向けをベースにしたレジメンを用いた患者の40%に化学療法に対する奏効が報告され、初回診断から5年後の時点で60%の患者が生存していた。[ 219 ]小児および成人を含む20人の患者(年齢中央値、27.3歳)を対象とした単一施設のレトロスペクティブ・レビューでは、化学療法を受けた患者と受けなかった患者とで再発確率に差が認められなかったことから、放射線療法が有用となる可能性が示唆された。[ 217 ]
類上皮肉腫の小児、青年、および若年成人67人(年齢中央値、14歳)を対象にしたGerman Cooperative Weichteilsarkom Studiengruppeのレトロスペクティブ解析において、53人の患者が限局性病変を呈し、14人の患者が転移病変を呈した。[ 220 ][証拠レベル:3iiA]患者67人中58人が一次切除で治療された。35人の患者で顕微鏡的完全切除が実施され、12人の患者で顕微鏡的不完全切除が実施され、20人の患者で肉眼的不完全切除が実施された。49人の患者が化学療法を受け、33人の患者が放射線療法を受けた。限局性病変を呈した53人の患者中45人(85%)で、完全寛解が達成された。中央値で0.9年(範囲、0.1~2.3年)後に27人の患者が再燃した。限局性病変を呈した患者の5年EFS率は35%(95%CI、±12%)およびOS率は48%(95%CI、±14%)であった。転移病変を呈した患者の5年EFS率は7%(95%CI、±14%)およびOS率は9%(95%CI、±16%)であった。限局性病変を呈した患者において、小さい腫瘍のサイズ、低いIRSグループ、低い腫瘍浸潤性、リンパ節転移陰性状態、および顕微鏡的完全切除が予後良好と相関した。
1件のレトロスペクティブ解析で、類上皮肉腫を有する30歳未満の患者が登録されたCOGおよびEpSSGのプロスペクティブ臨床試験がレビューされた。[ 221 ][証拠レベル:2A]この解析で、2005年7月から2015年11月までに治療された63人の患者が確認された。患者は臨床的特徴と受けた治療を組み合わせて、3つのリスクグループに層別化された。低リスク患者は放射線療法を併用するまたは併用しない手術を受け、主として非転移性の5cm以下の腫瘍が広範囲にまたはわずかに切除された患者が含められた。中リスク群には、非転移性で高悪性度の5cm超の腫瘍または切除不能な腫瘍を有する患者が含められた。リンパ節または遠隔転移病変を有する患者は、腫瘍の悪性度または大きさにかかわらず高リスクであった。術前補助療法を受けた患者22人中11人(50%)で部分奏効が観察された。イベントは局所再発(n = 10)および遠隔再発(n = 15)であった;推定5年OS率は低リスク患者で86.4%、中リスク患者で63.5%、および高リスク患者で0%であった。局所領域リンパ節転移、浸潤性腫瘍、高悪性度、および切除の程度が比較的少ないことが、転移が認められない患者における不良なEFSを予測した。
EZH2阻害薬のタゼメトスタットの第I相試験では、INI1陰性の類上皮肉腫患者2人で治療開始後20ヵ月以上の長期の疾患安定が得られた。[ 222 ]
臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
胞巣状軟部肉腫
胞巣状軟部肉腫は20歳未満の患者における軟部肉腫の1.4%を占める(表1を参照のこと)。
臨床像
発症時の年齢中央値は25歳であり、胞巣状軟部肉腫は四肢に最も好発するが、口腔および顎顔面領域にも発生しうる。[ 223 ][ 224 ][ 225 ]小児の胞巣状軟部肉腫は転移病変の証拠を呈することがある。[ 226 ]脳および肺への後発転移はまれである。[ 223 ]
連続した4件のCWS試験およびSoTiSaR登録において治療された胞巣状軟部肉腫患者61人のシリーズでは、46人の患者が限局性病変を呈し、15人の患者が診断時に転移の証拠を示した。[ 227 ]1980年から2014年に4つの大規模施設で治療された30歳未満の胞巣状軟部肉腫の9人の小児について、診断時年齢中央値は17歳で、患者の64%が女性であった。最も好発する病変部位は下肢で、26人の患者にASSPL-TFE3転座がみられた。Intergroup Rhabdomyosarcoma Study(IRS)グループ別の分布は次の通りであった:19人の患者がIRS Iの腫瘍を有し、7人の患者がIRS IIの腫瘍を有し、5人の患者がIRS IIIの腫瘍を有し、38人の患者がIRS IVの腫瘍を有した。[ 228 ]
予後
小児の胞巣状軟部肉腫は緩徐な経過をたどる場合がある。[ 226 ]胞巣状軟部肉腫の患者では、長期間にわたる見かけ上の寛解を経て、数年後に再燃がみられることがある。[ 227 ][ 231 ]このような腫瘍はまれであるため、胞巣状軟部肉腫の小児はすべてプロスペクティブ臨床試験への登録を検討すべきである。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
治療を受けた患者19人についての1件の症例集積研究において、報告された5年OS率は80%、局所疾患患者のOS率は91%、腫瘍が5cm以下の患者のOS率は100%、腫瘍が5cmより大きい患者のOS率は31%であった。[ 232 ]33人の患者を対象とした別のシリーズにおけるOSは、診断から5年で68%、診断から10年で53%であった。腫瘍が小さい(5cm以下)ほど、さらに完全に腫瘍が切除された場合ほど、生存は良好であった。[ 233 ][証拠レベル:3iiA]
4つの施設の小児および30歳未満の若年成人(年齢中央値、17歳;範囲、1.5~30歳)を対象にした1件のレトロスペクティブ・レビューで、1980年から2014年の間に最初に手術で治療された69人の患者が確認された。[ 228 ][証拠レベル:3iiA]ASPL-TFE3転座が検査された26人の患者全員で認められた。IRSの術後の病期分類で19人の患者がグループIの腫瘍(28%)を有し、7人の患者がIRSグループIIの腫瘍(10%)を有し、5人の患者がIRSグループIIIの腫瘍(7%)を有し、38人の患者がIRSグループIVの腫瘍(55%)を有した。限局性腫瘍(手術後のIRSグループI、II、およびIII)を有する患者31人について5年EFS率は80%でOS率は87%であった。転移性腫瘍(手術後のIRSグループIV)を有する患者38人について5年EFS率は7%でOS率は61%であった。
胞巣状軟部肉腫患者では、転移の存在が一般的であり、しばしば緩慢な臨床経過が長期に及ぶ。ドイツの連続した研究で治療された患者のシリーズにおいて、患者61人中15人(25%)が転移を来し、しばしばそれらの性質は粟粒性であった。化学療法への反応がみられなかったにもかかわらず、5年OS率は61%で、EFS率は20%であった。[ 227 ]
治療
胞巣状軟部肉腫に対する治療法の選択肢には以下のものがある:
標準的アプローチは原発病変の完全切除である。[ 232 ]完全切除が不可能な場合は放射線療法が施行される。中国の1件の研究により、口腔および顎顔面領域の胞巣状軟部肉腫の患者18人について報告された;15人の患者が30歳未満であった。[ 225 ][証拠レベル:3iiDii]切除断端陰性での外科的切除が初回治療であった。すべての患者が生存し、1人の患者においてのみ転移性病変の再発がみられた。
胞巣状軟部肉腫を有する0~21歳の小児患者51人のシリーズでは、10年OS率が78%およびEFS率は約63%であった。限局性疾患の患者(n = 37)に対する10年OS率は87%で、診断時に転移が認められた14人の患者の10年OS率は44%であったが、これは一部の患者で原発腫瘍と肺転移巣の外科的切除を行えた結果であろう。測定可能な病変を有する患者18人中、従来の肉腫に対する化学療法で反応が得られたのは3人のみ(17%)であったが、スニチニブで治療された患者4人中2人で部分奏効が得られた。[ 223 ][証拠レベル:3iiiA]
ドイツの連続した研究で治療された患者のシリーズにおいて、発症時に転移のない患者のPFSは原発腫瘍の完全切除により改善するようであった;5年EFS率は腫瘍が完全切除された患者で100%であったのに対し、顕微鏡的または肉眼的残存病変が認められた患者では50%であった。[ 227 ]
インターフェロンアルファおよびベバシズマブに客観的奏効が得られたとする報告が散見される。[ 223 ][ 235 ][ 236 ]
チロシンキナーゼ阻害薬に関する研究で以下が観察されている:
胞巣状軟部肉腫に対して臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
軟部明細胞肉腫
明細胞肉腫(以前は軟部悪性黒色腫という不適切な名称で呼ばれていた)はまれな軟部肉腫であり、典型的には四肢の深部の軟部組織に生じる。この腫瘍は腱および腱膜の明細胞肉腫とも呼ばれる。この腫瘍は青年および若年成人に好発する。
分裂速度が低く組織学的悪性度が中等度の小さな限局腫瘍の患者は最良の経過をとる。[ 244 ]
臨床像
この腫瘍は下肢、特に足、踵、足関節部に最も好発する。[ 245 ][ 246 ]この腫瘍はリンパ節播種、特に所属リンパ節への転移を生じる傾向が強い(12~43%)。[ 246 ][ 247 ]この腫瘍は典型的には緩徐な臨床経過をたどる。
治療
軟部明細胞肉腫に対する治療法の選択肢には以下のものがある:
Italian and German Soft Tissue Cooperative Studiesにより報告された小児患者28人を対象としたシリーズでは、診断時年齢の中央値は14歳で、原発部位として最も多かったのは下肢であった(50%)。放射線療法を併用するまたは併用しない手術が選択すべき治療法であり、治癒が得られる確率が最も高い。このシリーズでは、完全切除を受けた患者13人中12人が治癒した。より進行した患者では転帰は不良であり、化学療法が奏効することはまれである。[ 250 ];[ 251 ][証拠レベル:3iiDii]European Organization for Research and Treatment of Cancerによる1件の研究において、転移病変を有し、EWSR1再構成が実証された明細胞肉腫患者26人がクリゾチニブで治療された。[ 252 ]1人の患者が部分奏効を達成し、17人の患者で疾患の安定が得られた。
骨外性粘液型軟骨肉腫
骨外性粘液型軟骨肉腫は、軟部肉腫の中では比較的まれで、すべての軟部肉腫に占める割合はわずか2.3%である。[ 253 ]また、小児および青年において報告されている。[ 254 ]
分子的特徴
骨外性粘液型軟骨肉腫は多結節性の新生物である。この円形細胞は、コンドロイチン硫酸による粘液性の基礎環境では索状に配列し糸状となる。細胞遺伝学的にいくつかの異常が特定されており(表2を参照のこと)、EWSR1-NR4A3遺伝子に関わるt(9;22)(q22;q12)の転座が最も高頻度に認められる。[ 255 ]
予後
この腫瘍は従来低悪性度の可能性があるとみなされている。[ 256 ]しかしながら、大規模施設からの最近の報告によると、骨外性粘液型軟骨肉腫は、特に長期にわたって患者を監視すると、悪性度が有意に高いことが示された。[ 257 ][ 258 ]患者は長期にわたり緩徐な経過を示す傾向がある。リンパ節転移については、多くの報告がなされている。局所再発(57%)および肺への転移(26%)が報告されている。[ 258 ]
治療
骨外性粘液型軟骨肉腫に対する治療法の選択肢には以下のものがある:
- 手術。
- 放射線療法。
積極的な局所制御および転移病変の切除により、5年OS率が87%、10年OS率が63%となった。この腫瘍は放射線療法に対し比較的抵抗性が高かった。[ 257 ]治療上の化学療法の有益性については、まだ確立されていない。
小分子に対する遺伝的標的薬の可能性は考えられるが、これらは臨床試験の一環として研究すべきである。成人を対象とした1件の研究で、スニチニブ投与を受けた患者10人中6人が部分奏効を得た。[ 259 ]
臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
骨外性ユーイング肉腫
(詳しい情報については、ユーイング肉腫の治療に関するPDQ要約を参照のこと。)
線維形成性小円形細胞腫瘍
線維形成性小円形細胞腫瘍はまれな原始的肉腫である。
臨床像
線維形成性小円形細胞腫瘍は、腹部腹膜、骨盤、および/または陰嚢腹膜(peritoneum into the scrotal sac)に最も好発するが、腎臓や他の固形臓器に発生することもある。[ 260 ][ 261 ][ 262 ][ 263 ][ 264 ]数ダースから何百個にも及ぶ腹腔内播種がしばしば発見される。この腫瘍は男性に発生し(85%)、肺およびその他の部位に転移することがある。[ 264 ][ 265 ]
65人の患者を対象にした単一施設の大規模シリーズでは、ほとんどの患者(n = 54)で実施されたコンピュータ断層撮影(CT)スキャンとポジトロン放射断層撮影(PET)-CTキャン(n = 11)が比較された。PET-CTキャンでは偽陰性の結果が非常に少なく、従来のCTキャンで見逃されていた転移部位が発見された。[ 265 ]
分子的特徴
この腫瘍の細胞遺伝学的検査では、WT1遺伝子とEWSR1遺伝子の融合として特徴付けられている反復性転座t(11;22)(p13;q12)が実証されている。[ 263 ][ 266 ]WT1-EWSR1融合により線維形成性小円形細胞腫瘍の診断が確定する。
予後
線維形成性小円形細胞腫瘍の全般的な予後は依然としてきわめて不良であり、報告されている死亡率は90%である。発症時または術前化学療法後のいずれかで切除された腫瘍が90%を超えることが、OSについての良好な予後因子となる可能性がある。[ 267 ][ 268 ];[ 269 ][証拠レベル:3iiiA]術前補助化学療法に対する反応および完全切除(100%近い)は治療成績の改善と関連している。[ 264 ][ 270 ]
治療
線維形成性小円形細胞腫瘍の治療に標準的アプローチは存在しない。
線維形成性小円形細胞腫瘍に対する治療法の選択肢には以下のものがある:
- 手術。
- 化学療法およびその後の手術。
- 放射線療法。
外科的完全切除はまれで、通常は専門性の高い施設で実施されるが、生存の改善に不可欠である。成功を収めている治療法として、ユーイング肉腫向けの術前補助化学療法に続いて、広範な腹腔内腫瘍の外科的完全切除とその後の全腹部放射線療法が挙げられる。この集学的治療法により、5年経過時に30~40%の患者で生存が達成可能である。[ 260 ][ 261 ][ 267 ][ 271 ][ 272 ][ 273 ][ 274 ]
外科的完全切除(腫瘍減量手術)への腹腔内温熱化学療法(HIPEC)の追加は、1件の第I相臨床試験において2006年に初めて小児に適用された新たな技術である。線維形成性小円形細胞腫瘍に対する腫瘍減量手術およびHIPECは集学的アプローチの一部であり、高度に専門化された施設でのみ実施されている。手術は12時間に及ぶことがあり、この特有の腫瘍切除の技術的側面を考慮すべきである。HIPECは腹腔内の顕微鏡的病変の制御を改善しうる局所治療法である。その理論は、外科的切除後(手術時)に腹腔に注入される化学療法に熱を加えることで、腹部内に残存する顕微鏡的細胞に対して相乗的な細胞傷害活性が得られるということである。[ 275 ]
1件の単一施設の第II相研究により、HIPECは外科的完全切除に対する潜在的に有望な追加治療であることが示された。線維形成性小円形細胞腫瘍患者14人および別の肉腫の患者5人が登録された。これらの高度に選択された患者は腹腔に限局した腫瘍を有し、ユーイング肉腫向けの術前補助化学療法に対して部分奏効を示し、外科的完全切除とシスプラチンを用いるHIPECを受け、全腹部補助放射線療法に続いて補助化学療法を受けた。この標準化されたアプローチにより、線維形成性小円形細胞腫瘍患者のOS率は30ヵ月経過時に80%、50ヵ月経過時に40%であった。肝転移が認められない線維形成性小円形細胞腫瘍患者では腹腔内再発が認められなかったのに対し、肝転移または門脈に病変が認められた患者では87%が再発した。[ 275 ]
他の施設でも、線維形成性小円形細胞腫瘍患者において腫瘍減量手術およびHIPECを併用するこのアプローチが用いられている。フランスの施設の線維形成性小円形細胞腫瘍患者を対象にしたレトロスペクティブ研究において、患者が腫瘍減量手術およびHIPECで治療された。22人の患者が選択され、診断時年齢中央値は14.8歳(範囲、4.2~17.6歳)であった。7人の患者が腹膜中皮腫を有し、7人の患者が線維形成性小円形細胞腫瘍を有し、8人の患者が他の組織型を有した。肉眼的完全切除(CC-0、CCは腫瘍減量の完全性[completeness of cytoreduction]を示す)が16例(73%)で達成された。16人の患者(72%)が中央値にして9.6ヵ月(範囲、1.4~86.4ヵ月)後に再燃し、9人の患者(41%)が中央値にして5.3ヵ月(範囲、0.1~36.1ヵ月)後に疾患再燃により死亡した。線維形成性小円形細胞腫瘍を有する7人の患者全員が完全切除を受けたわけではなかった。[ 276 ][証拠レベル:3iii]
フランスの別の研究により、線維形成性小円形細胞腫瘍を有し、病変が腹部に限局していた患者の治療法として腫瘍減量手術およびHIPECの使用がレビューされた。線維形成性小円形細胞腫瘍を有する患者107人について、48人は腹膜外への転移を来しておらず、腫瘍減量手術を受けた。48人の患者のうち、38人の患者(79%)は術前および/または術後化学療法を受け、23人の患者(48%)が腹部骨盤領域全体に術後放射線療法を受けた。腹腔内化学療法が11人の患者(23%)に実施された;2人の患者が術後早期腹腔内化学療法(EPIC)を受け、9人の患者がHIPECを受けた。追跡期間中央値30ヵ月後、コホート全体のOS期間中央値は42ヵ月であった。2年OS率は72%で、5年OS率は19%であった。2年DFS率は30%で、5年DFS率は12%であった。腹部骨盤領域全体の放射線療法は、腫瘍減量手術後の長期の腹膜無再燃生存およびDFSに関連する唯一の変数であった。腹腔内化学療法(HIPECまたはEPIC)を受けた11人の患者では、6つの異なる化学療法レジメンが用いられた。この集団の生存または転帰はこの文書で報告されなかった。HIPEC/EPICがOSおよびDFSに及ぼす影響は統計的に有意ではなかったが、すべての患者で標準化されたレジメンが用いられたわけではなかったため、結果の判定は困難となった。[ 277 ]
Center for International Blood and Marrow Transplant Researchが、そのレジストリーの、大量化学療法および自家幹細胞を用いた再構築による地固めを受けた線維形成性小円形細胞腫瘍患者を分析した。[ 278 ]このレトロスペクティブ・レジストリー分析でこのアプローチについて若干の有益性が示唆された一方で、過度の毒性および効力の欠如のためにこのアプローチを放棄した研究者もいる。[ 267 ]
単一施設研究の報告では、線維形成性小円形細胞腫瘍が再発した患者5人中5人が、ビノレルビン、シクロホスファミド、およびテムシロリムスを併用する治療に部分奏効を示した。[ 279 ]
腎外性(頭蓋外)ラブドイド腫瘍
悪性ラブドイド腫瘍は1981年に腎腫瘍の小児において初めて報告され(詳しい情報については、ウィルムス腫瘍とその他の小児腎腫瘍の治療に関するPDQ要約の腎ラブドイド腫瘍のセクションを参照のこと)、後に腎外のさまざまな部位で発見された。これらの腫瘍はまれで悪性度が高く、特に2歳未満の小児でその傾向が強い。
腎外性(頭蓋外)ラブドイド腫瘍は20歳未満の患者における軟部肉腫の2%を占める(表1を参照のこと)。
分子的特徴
軟部組織の腎外性頭蓋外悪性ラブドイド腫瘍の小児患者26人を対象にした最初の大規模シリーズは、病理学資料のレビュー中にIntergroup Rhabdomyosarcoma Studies I~IIIに登録した患者からのものである。無病状態で生存していた患者はわずか5人(19%)であった。[ 280 ]後に、脳の非定型奇形腫様/ラブドイド腫瘍の小児のほか、腎および腎外性の悪性ラブドイド腫瘍の小児が調査され、検査された29の腫瘍すべてでSMARCB1遺伝子の生殖細胞変異および後天性変異が明らかにされた。[ 281 ]ラブドイド腫瘍はSMARCB1遺伝子の生殖細胞変異に関連していると考えられ、見たところ罹患していない一方の親から遺伝している可能性がある。[ 282 ]この観察は、診断時平均年齢が12ヵ月の患者のすべての部位にある32の悪性ラブドイド腫瘍に拡大された。[ 283 ]
予後
腎臓、中枢神経系(CNS)、および腎外性の悪性ラブドイド腫瘍の患者229人を対象にしたSEER研究では、2~18歳の患者の年齢、腫瘍の範囲が限られていること、放射線療法の実施が他の患者と比較して治療成績に良好に作用することが示された(それぞれの比較についてP < 0.002)。原発腫瘍部位は予後的に重要ではなかった。5年OS率は33%であった。[ 284 ]
治療
腎外性(頭蓋外)ラブドイド腫瘍に対する治療法の選択肢には、以下のものがある:[ 285 ][証拠レベル:3iA];[ 286 ][ 287 ][証拠レベル:3iiiB]
- 可能な場合の外科的切除。
- 軟部肉腫に用いられている化学療法(ただし、現在最善のものとして認められている単一のレジメンはない)。
- 放射線療法。
アリセルチブに対する反応がCNS非定型奇形腫様/ラブドイド腫瘍の患者4人において報告されている。[ 288 ](CNS非定型奇形腫様/ラブドイド腫瘍に関する詳しい情報については、小児中枢神経系非定型奇形腫様/ラブドイド腫瘍の治療に関するPDQ要約を参照のこと。)
臨床評価段階にある治療法の選択肢
NCIが支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
血管周囲類上皮細胞への分化を伴う腫瘍(PEComa)
危険因子および分子的特徴
良性のPEComaは、腎細胞がんおよび脳腫瘍の素因もある常染色体優性遺伝症候群の結節硬化症に多くみられる。結節硬化症は、TSC1(9q34)またはTSC2(16p13.3)のいずれかの生殖細胞系不活化により引き起こされ、散発性PEComaではこの同じ腫瘍抑制遺伝子が体細胞で不活化されている。[ 289 ]いずれかの遺伝子の不活化により、mTOR経路が刺激されることから、手術によらずmTOR阻害薬により治癒可能な同様の遺伝子の不活化が認められる腫瘍(リンパ管平滑筋腫症および血管筋脂肪腫)に対する治療の基礎が得られる。[ 290 ][ 291 ]ごく一部のPEComaはSFPQ/PSFおよびRAD51Bを含むさまざまな遺伝子が関わる融合を伴うTFE3再構成を有する。[ 292 ]
臨床像
PEComaは、まれに消化管、肺、女性器、尿生殖器などのさまざまな部位に発生する。軟部組織、内臓、および女性器のPEComaは、中年女性患者により多くみられ、通常結節硬化症とは無関係である。[ 293 ]疾患の経過は緩慢な場合がある。
予後
ほとんどのPEComaは、良性の臨床経過を示すが、悪性の挙動が報告されており、腫瘍サイズ、分裂速度、および壊死の有無を基に予想可能である。[ 294 ]
未分化/分類不能肉腫
1972年から2006年まで未分化軟部肉腫の患者は、Intergroup Rhabdomyosarcoma Study GroupおよびCOGの調整で実施された横紋筋肉腫の臨床試験への参加に適格とされてきた。その根拠は、未分化軟部肉腫を有する患者は病巣部位と治療成績が胞巣型横紋筋肉腫の患者に類似しているという観察結果にあった。軟部肉腫の成人に対する治療試験には未分化肉腫および他の病歴の患者も含まれ、イホスファミドとドキソルビシンに加え、ときには他の化学療法薬、手術および放射線療法を使用して同様の治療を行っている。
COGのARST0332(NCT00346164)試験では、高悪性度未分化肉腫患者がイホスファミドおよびドキソルビシンをベースとしたレジメンによる治療を受け、また過去のIntergroup Rhabdomyosarcoma Study Groupの諸研究で用いられた横紋筋肉腫を対象とする治療法を受け、非転移患者での推定5年生存率は72%であった。[ 297 ][証拠レベル:3iiA]
ARST0332(NCT00346164)試験に登録された未分化軟部肉腫患者32人の報告において、登録時の年齢中央値は13.6歳で、患者の3分の2が男性であった。最も一般的な原発部位は傍脊髄部および四肢であった。5人の患者が転移性疾患を呈した。[ 298 ]
未分化多形肉腫/悪性線維性組織球腫(高悪性度)
悪性線維組織球腫は、かつては成人の軟部肉腫の中で単一で最も頻度の高い組織型とされていた。悪性線維組織球腫は、1960年代初期に初めて認知されて以降、その組織発生および臨床病理学的疾患としての妥当性の両面において絶えず議論の的となってきた。最新のWHO分類は、現在では悪性線維組織球腫を別個の診断カテゴリーとして含めておらず、むしろ未分化多形肉腫の亜型の1つとしている。[ 4 ][ 299 ]
この腫瘍はすべての小児軟部肉腫の2~6%を占める。[ 300 ]
分子的特徴
未分化多形肉腫は、以前はほとんどの場合、悪性線維性組織球腫と呼ばれていた。歴史的に、この疾患実体は診断基準が移行しているため、評価困難となっている。特定の型を示さない悪性線維性組織球症、花むしろ状の配列を有する(storiform)または多形悪性線維性組織球腫、多形肉腫、または未分化多形肉腫と診断された症例70例を解析したところ、特定の反復性の異常を伴わない高度に複雑な核型が示された。[ 301 ]
MDM2およびCDK4を含む12q13-15の増幅を伴う未分化肉腫は、脱分化型脂肪肉腫として分類するのが最良である[ 301 ];この腫瘍と紡錘細胞形態学を示す未分化/未分類腫瘍ファミリーとの関係はあまり明らかになっていないままである。
危険因子
これらの腫瘍は、過去に放射線が照射された部位に、または網膜芽細胞腫患者における二次悪性腫瘍として現れることがある。[ 302 ]
臨床像および治療
また、主に10歳代に発生する。患者10人のシリーズにおける年齢中央値は10歳で、腫瘍は主に四肢に局在する場合が最も多かった。このシリーズでは、すべての腫瘍が限局性で、(追跡可能であった)9人中5人が生存しており、初回寛解状態にあった。[ 300 ]悪性線維組織球腫の小児患者17人を対象とした別のシリーズでは、診断時年齢の中央値は5歳で、四肢に病変がみられたのは8例であった。[ 303 ]転移したすべての患者が死亡し、ドキソルビシンをベースとしたレジメンに対する臨床的奏効が2人の患者に認められた。
(骨悪性線維性組織球腫の治療に関する詳しい情報については、骨肉腫および骨悪性線維性組織球腫の治療に関するPDQ要約を参照のこと。)
BCOR-CCNB3再構成を伴う未分化円形細胞肉腫
(詳しい情報については、ユーイング肉腫の治療に関するPDQ要約のBCOR-CCNB3再構成を伴う未分化円形細胞肉腫およびユーイング肉腫のゲノム情報のセクションを参照のこと。)
CIC-DUX4再構成を伴う未分化円形細胞肉腫
(詳しい情報については、ユーイング肉腫の治療に関するPDQ要約のCIC-DUX4再構成を伴う未分化円形細胞肉腫およびユーイング肉腫のゲノム情報のセクションを参照のこと。)
CIC-NUTM1再構成を伴う未分化円形細胞肉腫
(詳しい情報については、ユーイング肉腫の治療に関するPDQ要約のCIC-NUTM1再構成を伴う未分化円形細胞肉腫のセクションを参照のこと。)
脈管腫瘍
脈管腫瘍は、常に良性と考えられる血管腫から悪性度の高い血管肉腫まで多様である。[ 304 ]悪性脈管腫瘍として、以下のサブタイプがある:
類上皮血管内皮腫
発生率および転帰
この腫瘍は1982年にWeissおよびEnzingerにより軟部組織において最初に記述された。類上皮血管内皮腫は若年で発生することがあるが、発生率のピークは30~40歳代である。腫瘍は緩慢な経過をたどる場合も、非常に侵攻性の経過をたどる場合もあり、5年全生存率は73%である。非常に侵攻性の経過をたどる患者がいるのに対して、未治療の多発性病変を有しながら非常に良性の経過をたどる患者の症例報告もある。病理医はリスクを評価し治療を調整するために患者を層別化しようと試みているが、さらなる研究が必要である。[ 305 ][ 306 ][ 307 ][ 308 ][ 309 ][ 310 ][ 311 ]
1件の多施設ケースシリーズで、2~26歳の類上皮血管内皮腫患者24人について報告された。[ 312 ][証拠レベル:3iiiDii]ほとんどの患者が多臓器病変を呈した。63%の患者で進行がみられ、進行までの平均期間は18.4ヵ月(範囲、0~72ヵ月)であった。
滲出液の存在、3cmを超える腫瘍サイズ、および高い分裂指数(有糸分裂像が50高倍率視野当たり3を超える)が不良な転帰と関連している。[ 307 ]
病理組織学および分子的特徴
患者の大部分でWWTR1-CAMTA1遺伝子融合が認められている;頻度は少ないが、YAP1-TFE3遺伝子融合が報告されている。[ 305 ]これらの融合は現在の薬物を用いた直接の標的として治療できていない。多発性肝病変で単クローン性が記述されており、転移の過程が示唆されている。
組織学的に、これらの病変は血管腔をほとんど認めない巣状、糸状、および索状パターンで配列した類上皮病変を特徴としている。侵攻性の臨床的挙動に関連しうる特徴としては、細胞異型、10高倍率視野当たり1つ以上の有糸分裂像、高い割合の紡錘形細胞、病巣壊死、および化生性骨形成が挙げられる。[ 307 ]
文献で報告されている小児患者の数は限られている。
臨床像および診断的評価
一般的な病変の部位は、肝単独(21%)、肝 + 肺(18%)、肺単独(12%)、および骨単独(14%)である。[ 307 ][ 313 ][ 314 ]臨床像は以下に示すように、病変の部位によって異なる:
類上皮血管内皮腫の治療
類上皮血管内皮腫に対する治療法の選択肢には以下のものがある:
- 経過観察。
- 手術。
- 免疫療法。
- 標的療法。
- 化学療法。
- 放射線療法。
経過が緩慢な症例には、観察が妥当である。経過がより侵攻性の症例には、インターフェロン、サリドマイド、ソラフェニブ、パゾパニブ、シロリムスなど複数の医薬品が使用されている。[ 315 ]経過が最も侵攻性の症例は、血管肉腫と同様の化学療法で治療される。切除が可能な場合には手術が実施される。侵攻性の肝病変では、転移の有無にかかわらず、肝移植が用いられている。[ 307 ][ 316 ][ 317 ][ 318 ][ 319 ]
1件の多施設ケースシリーズで、2~26歳の類上皮血管内皮腫患者24人について報告された。[ 312 ][証拠レベル:3iiiDii]シロリムスで治療された3人の患者が、2.5年間以上、疾患の安定または部分奏効を達成した。
臨床的に顕著な活性を示す標準的な薬剤がないため、疾患に向けた追加の治療を希望する患者または家族は新たな治療アプローチの試験への参加を考慮すべきである。
進行時に疾患に向けた治療を行う決定がなされたかどうかに関係なく、緩和ケアは依然として管理の中心となっている。これにより、QOLを最大化しながら、末期疾患に関連する症状とストレスを緩和する試みが確保される。
類上皮血管内皮腫に対して臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
- NCT03148275(転移性、局所進行性、または手術で切除不可能な類上皮血管内皮腫患者の治療におけるトラメチニブ):これは、患者報告による転帰を2番目の目的としてトラメチニブの効力を評価する第II相試験である。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
軟部血管肉腫
発生率
血管肉腫はまれな(肉腫の2%を占める)侵攻性の脈管腫瘍で身体のあらゆる部位に生じうるが、軟部組織においてより一般的である。血管肉腫の推定発生率は100万人当たり2例である;米国では、年間約600人(典型的には60~70歳)が罹患する。[ 320 ]
血管肉腫は小児ではきわめてまれであり、小児集団におけるこの腫瘍の病態生理学が異なるものであるかどうかは不明である。新生児および歩き始めの幼児において多発性の皮膚病変および肝病変(これらの一部はGLUT1陽性である)の発症を伴う症例が報告されている。[ 321 ][ 322 ][ 323 ][ 324 ]ほとんどの血管肉腫は皮膚および表在性軟部組織に病変がみられるが、肝臓、脾臓、および肺にみられることもある;骨に病変がみられることはまれである。
危険因子
確立された危険因子には以下のものがある:[ 325 ]
病理組織学および分子的特徴
血管肉腫は一般に異数体腫瘍である。血管腫などの良性病変から発生する血管肉腫のまれな症例は、研究が必要な異なる経路を有している。MYC増幅は放射線誘発性血管肉腫においてみられる。KDR-VEGFR2の突然変異およびFLT4-VEGFR3の増幅がみられる頻度は50%未満である。[ 325 ]
病理組織学的診断は、多様な異型の範囲が存在する可能性があるため非常に困難である。一般的な特徴は、真皮コラーゲン束に沿って切断したようなパターンの不規則なチャンネルのネットワークである。多様な細胞の形状、サイズ、有糸分裂、内皮多層構造、および乳頭状構造がみられる。類上皮細胞がみられることもある。壊死および出血が一般的である。腫瘍は第VIII因子、CD31、およびCD34に染色される。一部の肝病変は乳児血管腫に酷似し、病巣のGLUT1が陽性である。こうした肝病変の命名法は難しく、1971年に提案された旧式の専門用語(例、I型血管内皮腫:乳児血管腫;II型血管内皮腫:低悪性度血管肉腫;III型血管内皮腫:高悪性度血管肉腫)の使用により混乱させるものとなっている。[ 322 ]
軟部血管肉腫の治療
軟部血管肉腫に対する治療法の選択肢には以下のものがある:
- 手術(限局性腫瘍)。
- 放射線療法(成人における限局性皮膚病変)。
- 手術、化学療法、および放射線療法(転移性腫瘍)。
限局性疾患は積極的な手術により治癒可能である。血管肉腫およびリンパ管肉腫患者が長期生存するには、局所療法または全身療法で治療された一部の患者で腫瘍の縮小を示した証拠が存在するにもかかわらず、外科的完全切除がきわめて重要なようである。[ 323 ][ 326 ][ 327 ][ 328 ]患者222人(年齢中央値62歳;範囲15~90歳)を対象としたレビューでは、全疾患特異的生存率(DSS)が5年で38%であったことを示した。5年DSS率は、限局性腫瘍を切除した患者138人では44%であったが、診断時に転移が認められた患者43人ではわずか16%であった。[ 328 ]限局性血管肉腫に対する肝移植のデータは限られている。[ 329 ][証拠レベル:3iiA]
限局性腫瘍、特に皮膚血管肉腫は放射線療法で治療できる。これらの報告されている症例のほとんどは成人である。[ 330 ]
転移性腫瘍に対しては、手術、全身化学療法、および放射線療法による集学的治療が用いられるが、治癒が得られることはまれである。[ 331 ][ 332 ]転移性血管肉腫では疾患の制御が目標であり、発表されている無増悪生存期間は3~7ヵ月[ 333 ]および全生存(OS)期間中央値は14~18ヵ月[ 334 ]である。成人と小児の両方で、20~35%の5年OS率が報告されている。[ 323 ][ 324 ][ 335 ]
乳児血管腫からの悪性転換に続発する血管肉腫と診断された1人の小児では、血管内皮増殖因子に対するモノクローナル抗体のベバシズマブと全身化学療法とを併用する治療での反応が報告されている。[ 321 ][ 331 ]小児における肝血管肉腫8症例の報告により、血管内皮腫という用語の誤用とこれらの腫瘍の早期診断および治療の重要性が強調された。[ 336 ]
血管形成を阻害する生物学的製剤が血管肉腫の成人で活性を示している。[ 322 ][ 335 ]
臨床的に顕著な活性を示す標準的な薬剤がないため、疾患に向けた追加の治療を希望する患者または家族は新たな治療アプローチの試験への参加を考慮すべきである。
進行時に疾患に向けた治療を行う決定がなされたかどうかに関係なく、緩和ケアは依然として管理の中心となっている。これにより、QOLを最大化しながら、末期疾患に関連する症状とストレスを緩和する試みが確保される。
軟部血管肉腫に対して臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
- NCT02834013(まれな腫瘍を有する患者の治療におけるニボルマブおよびイピリムマブ):これは、まれな腫瘍を有する患者を治療するためのニボルマブおよびイピリムマブに関する第II相研究である。ニボルマブおよびイピリムマブといったモノクローナル抗体による免疫療法は、身体の免疫システムががんを攻撃する助けとなり、腫瘍細胞が増殖し転移する能力を阻害しうる。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005.[PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Sidebotham EL, et al.: Prognostic factors and survival in pediatric and adolescent liposarcoma. Sarcoma 2012: 870910, 2012.[PUBMED Abstract]
- Alaggio R, Coffin CM, Weiss SW, et al.: Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 33 (5): 645-58, 2009.[PUBMED Abstract]
- Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC Press, 2013.[PUBMED Abstract]
- Sreekantaiah C, Karakousis CP, Leong SP, et al.: Cytogenetic findings in liposarcoma correlate with histopathologic subtypes. Cancer 69 (10): 2484-95, 1992.[PUBMED Abstract]
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002.[PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001.[PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002.[PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013.[PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015.[PUBMED Abstract]
- La Quaglia MP, Spiro SA, Ghavimi F, et al.: Liposarcoma in patients younger than or equal to 22 years of age. Cancer 72 (10): 3114-9, 1993.[PUBMED Abstract]
- Lee ATJ, Thway K, Huang PH, et al.: Clinical and Molecular Spectrum of Liposarcoma. J Clin Oncol 36 (2): 151-159, 2018.[PUBMED Abstract]
- Beane JD, Yang JC, White D, et al.: Efficacy of adjuvant radiation therapy in the treatment of soft tissue sarcoma of the extremity: 20-year follow-up of a randomized prospective trial. Ann Surg Oncol 21 (8): 2484-9, 2014.[PUBMED Abstract]
- Ferrari A, Casanova M, Spreafico F, et al.: Childhood liposarcoma: a single-institutional twenty-year experience. Pediatr Hematol Oncol 16 (5): 415-21, 1999 Sep-Oct.[PUBMED Abstract]
- Cecchetto G, Alaggio R, Dall'Igna P, et al.: Localized unresectable non-rhabdo soft tissue sarcomas of the extremities in pediatric age: results from the Italian studies. Cancer 104 (9): 2006-12, 2005.[PUBMED Abstract]
- Huh WW, Yuen C, Munsell M, et al.: Liposarcoma in children and young adults: a multi-institutional experience. Pediatr Blood Cancer 57 (7): 1142-6, 2011.[PUBMED Abstract]
- Gronchi A, Bui BN, Bonvalot S, et al.: Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol 23 (3): 771-6, 2012.[PUBMED Abstract]
- Demetri GD, von Mehren M, Jones RL, et al.: Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol 34 (8): 786-93, 2016.[PUBMED Abstract]
- Baruchel S, Pappo A, Krailo M, et al.: A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children's Oncology Group. Eur J Cancer 48 (4): 579-85, 2012.[PUBMED Abstract]
- Demetri GD, Schöffski P, Grignani G, et al.: Activity of Eribulin in Patients With Advanced Liposarcoma Demonstrated in a Subgroup Analysis From a Randomized Phase III Study of Eribulin Versus Dacarbazine. J Clin Oncol 35 (30): 3433-3439, 2017.[PUBMED Abstract]
- Schafer ES, Rau RE, Berg S, et al.: A phase 1 study of eribulin mesylate (E7389), a novel microtubule-targeting chemotherapeutic agent, in children with refractory or recurrent solid tumors: A Children's Oncology Group Phase 1 Consortium study (ADVL1314). Pediatr Blood Cancer 65 (8): e27066, 2018.[PUBMED Abstract]
- Wang L, Motoi T, Khanin R, et al.: Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 51 (2): 127-39, 2012.[PUBMED Abstract]
- Nyquist KB, Panagopoulos I, Thorsen J, et al.: Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One 7 (11): e49705, 2012.[PUBMED Abstract]
- Frezza AM, Cesari M, Baumhoer D, et al.: Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur J Cancer 51 (3): 374-81, 2015.[PUBMED Abstract]
- Schneiderman BA, Kliethermes SA, Nystrom LM: Survival in Mesenchymal Chondrosarcoma Varies Based on Age and Tumor Location: A Survival Analysis of the SEER Database. Clin Orthop Relat Res 475 (3): 799-805, 2017.[PUBMED Abstract]
- Kawaguchi S, Weiss I, Lin PP, et al.: Radiation therapy is associated with fewer recurrences in mesenchymal chondrosarcoma. Clin Orthop Relat Res 472 (3): 856-64, 2014.[PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008.[PUBMED Abstract]
- Bishop MW, Somerville JM, Bahrami A, et al.: Mesenchymal Chondrosarcoma in Children and Young Adults: A Single Institution Retrospective Review. Sarcoma 2015: 608279, 2015.[PUBMED Abstract]
- Morioka H, Takahashi S, Araki N, et al.: Results of sub-analysis of a phase 2 study on trabectedin treatment for extraskeletal myxoid chondrosarcoma and mesenchymal chondrosarcoma. BMC Cancer 16: 479, 2016.[PUBMED Abstract]
- Thampi S, Matthay KK, Boscardin WJ, et al.: Clinical Features and Outcomes Differ between Skeletal and Extraskeletal Osteosarcoma. Sarcoma 2014: 902620, 2014.[PUBMED Abstract]
- Jour G, Wang L, Middha S, et al.: The molecular landscape of extraskeletal osteosarcoma: A clinicopathological and molecular biomarker study. J Pathol Clin Res 2 (1): 9-20, 2016.[PUBMED Abstract]
- Sordillo PP, Hajdu SI, Magill GB, et al.: Extraosseous osteogenic sarcoma. A review of 48 patients. Cancer 51 (4): 727-34, 1983.[PUBMED Abstract]
- Paludo J, Fritchie K, Haddox CL, et al.: Extraskeletal Osteosarcoma: Outcomes and the Role of Chemotherapy. Am J Clin Oncol 41 (9): 832-837, 2018.[PUBMED Abstract]
- Longhi A, Bielack SS, Grimer R, et al.: Extraskeletal osteosarcoma: A European Musculoskeletal Oncology Society study on 266 patients. Eur J Cancer 74: 9-16, 2017.[PUBMED Abstract]
- Nieuwenhuis MH, Casparie M, Mathus-Vliegen LM, et al.: A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 129 (1): 256-61, 2011.[PUBMED Abstract]
- Rossato M, Rigotti M, Grazia M, et al.: Congenital hypertrophy of the retinal pigment epithelium (CHRPE) and familial adenomatous polyposis (FAP). Acta Ophthalmol Scand 74 (4): 338-42, 1996.[PUBMED Abstract]
- Baker RH, Heinemann MH, Miller HH, et al.: Hyperpigmented lesions of the retinal pigment epithelium in familial adenomatous polyposis. Am J Med Genet 31 (2): 427-35, 1988.[PUBMED Abstract]
- Kattentidt Mouravieva AA, Geurts-Giele IR, de Krijger RR, et al.: Identification of Familial Adenomatous Polyposis carriers among children with desmoid tumours. Eur J Cancer 48 (12): 1867-74, 2012.[PUBMED Abstract]
- Wang WL, Nero C, Pappo A, et al.: CTNNB1 genotyping and APC screening in pediatric desmoid tumors: a proposed algorithm. Pediatr Dev Pathol 15 (5): 361-7, 2012 Sep-Oct.[PUBMED Abstract]
- Lewis JJ, Boland PJ, Leung DH, et al.: The enigma of desmoid tumors. Ann Surg 229 (6): 866-72; discussion 872-3, 1999.[PUBMED Abstract]
- Lazar AJ, Tuvin D, Hajibashi S, et al.: Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol 173 (5): 1518-27, 2008.[PUBMED Abstract]
- Faulkner LB, Hajdu SI, Kher U, et al.: Pediatric desmoid tumor: retrospective analysis of 63 cases. J Clin Oncol 13 (11): 2813-8, 1995.[PUBMED Abstract]
- Gounder MM, Mahoney MR, Van Tine BA, et al.: Sorafenib for Advanced and Refractory Desmoid Tumors. N Engl J Med 379 (25): 2417-2428, 2018.[PUBMED Abstract]
- Merchant NB, Lewis JJ, Woodruff JM, et al.: Extremity and trunk desmoid tumors: a multifactorial analysis of outcome. Cancer 86 (10): 2045-52, 1999.[PUBMED Abstract]
- Honeyman JN, Theilen TM, Knowles MA, et al.: Desmoid fibromatosis in children and adolescents: a conservative approach to management. J Pediatr Surg 48 (1): 62-6, 2013.[PUBMED Abstract]
- Bonvalot S, Ternès N, Fiore M, et al.: Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol 20 (13): 4096-102, 2013.[PUBMED Abstract]
- Bonvalot S, Eldweny H, Haddad V, et al.: Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 34 (4): 462-8, 2008.[PUBMED Abstract]
- Merchant TE, Nguyen D, Walter AW, et al.: Long-term results with radiation therapy for pediatric desmoid tumors. Int J Radiat Oncol Biol Phys 47 (5): 1267-71, 2000.[PUBMED Abstract]
- Zelefsky MJ, Harrison LB, Shiu MH, et al.: Combined surgical resection and iridium 192 implantation for locally advanced and recurrent desmoid tumors. Cancer 67 (2): 380-4, 1991.[PUBMED Abstract]
- Weiss AJ, Lackman RD: Low-dose chemotherapy of desmoid tumors. Cancer 64 (6): 1192-4, 1989.[PUBMED Abstract]
- Klein WA, Miller HH, Anderson M, et al.: The use of indomethacin, sulindac, and tamoxifen for the treatment of desmoid tumors associated with familial polyposis. Cancer 60 (12): 2863-8, 1987.[PUBMED Abstract]
- Soto-Miranda MA, Sandoval JA, Rao B, et al.: Surgical treatment of pediatric desmoid tumors. A 12-year, single-center experience. Ann Surg Oncol 20 (11): 3384-90, 2013.[PUBMED Abstract]
- Orbach D, Brennan B, Bisogno G, et al.: The EpSSG NRSTS 2005 treatment protocol for desmoid-type fibromatosis in children: an international prospective case series. Lancet Child Adolesc Health 1 (4): 284-292, 2017.[PUBMED Abstract]
- Gladdy RA, Gupta AA: If Active Surveillance is the Standard of Care for Desmoid Patients, When Should Intervention be Considered? Ann Surg Oncol 26 (13): 4185-4187, 2019.[PUBMED Abstract]
- Skapek SX, Ferguson WS, Granowetter L, et al.: Vinblastine and methotrexate for desmoid fibromatosis in children: results of a Pediatric Oncology Group Phase II Trial. J Clin Oncol 25 (5): 501-6, 2007.[PUBMED Abstract]
- Gega M, Yanagi H, Yoshikawa R, et al.: Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol 24 (1): 102-5, 2006.[PUBMED Abstract]
- Constantinidou A, Jones RL, Scurr M, et al.: Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer 45 (17): 2930-4, 2009.[PUBMED Abstract]
- Ananth P, Werger A, Voss S, et al.: Liposomal doxorubicin: Effective treatment for pediatric desmoid fibromatosis. Pediatr Blood Cancer 64 (7): , 2017.[PUBMED Abstract]
- Ferrari A, Orbach D, Affinita MC, et al.: Evidence of hydroxyurea activity in children with pretreated desmoid-type fibromatosis: A new option in the armamentarium of systemic therapies. Pediatr Blood Cancer 66 (1): e27472, 2019.[PUBMED Abstract]
- Agresta L, Kim H, Turpin BK, et al.: Pazopanib therapy for desmoid tumors in adolescent and young adult patients. Pediatr Blood Cancer 65 (6): e26968, 2018.[PUBMED Abstract]
- Toulmonde M, Pulido M, Ray-Coquard I, et al.: Pazopanib or methotrexate-vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): a non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet Oncol 20 (9): 1263-1272, 2019.[PUBMED Abstract]
- Meazza C, Bisogno G, Gronchi A, et al.: Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 116 (1): 233-40, 2010.[PUBMED Abstract]
- Hansmann A, Adolph C, Vogel T, et al.: High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 100 (3): 612-20, 2004.[PUBMED Abstract]
- Skapek SX, Anderson JR, Hill DA, et al.: Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a Children's Oncology Group (COG) phase II study. Pediatr Blood Cancer 60 (7): 1108-12, 2013.[PUBMED Abstract]
- Rutenberg MS, Indelicato DJ, Knapik JA, et al.: External-beam radiotherapy for pediatric and young adult desmoid tumors. Pediatr Blood Cancer 57 (3): 435-42, 2011.[PUBMED Abstract]
- Shang H, Braggio D, Lee YJ, et al.: Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer 121 (22): 4088-96, 2015.[PUBMED Abstract]
- Messersmith WA, Shapiro GI, Cleary JM, et al.: A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res 21 (1): 60-7, 2015.[PUBMED Abstract]
- Buckley PG, Mantripragada KK, Benetkiewicz M, et al.: A full-coverage, high-resolution human chromosome 22 genomic microarray for clinical and research applications. Hum Mol Genet 11 (25): 3221-9, 2002.[PUBMED Abstract]
- Valdivielso-Ramos M, Torrelo A, Campos M, et al.: Pediatric dermatofibrosarcoma protuberans in Madrid, Spain: multi-institutional outcomes. Pediatr Dermatol 31 (6): 676-82, 2014 Nov-Dec.[PUBMED Abstract]
- Gooskens SL, Oranje AP, van Adrichem LN, et al.: Imatinib mesylate for children with dermatofibrosarcoma protuberans (DFSP). Pediatr Blood Cancer 55 (2): 369-73, 2010.[PUBMED Abstract]
- Rubio GA, Alvarado A, Gerth DJ, et al.: Incidence and Outcomes of Dermatofibrosarcoma Protuberans in the US Pediatric Population. J Craniofac Surg 28 (1): 182-184, 2017.[PUBMED Abstract]
- Meguerditchian AN, Wang J, Lema B, et al.: Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. Am J Clin Oncol 33 (3): 300-3, 2010.[PUBMED Abstract]
- Dagan R, Morris CG, Zlotecki RA, et al.: Radiotherapy in the treatment of dermatofibrosarcoma protuberans. Am J Clin Oncol 28 (6): 537-9, 2005.[PUBMED Abstract]
- Sun LM, Wang CJ, Huang CC, et al.: Dermatofibrosarcoma protuberans: treatment results of 35 cases. Radiother Oncol 57 (2): 175-81, 2000.[PUBMED Abstract]
- Price VE, Fletcher JA, Zielenska M, et al.: Imatinib mesylate: an attractive alternative in young children with large, surgically challenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 44 (5): 511-5, 2005.[PUBMED Abstract]
- McArthur GA, Demetri GD, van Oosterom A, et al.: Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol 23 (4): 866-73, 2005.[PUBMED Abstract]
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al.: Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 28 (10): 1772-9, 2010.[PUBMED Abstract]
- Miller SJ, Alam M, Andersen JS, et al.: Dermatofibrosarcoma protuberans. J Natl Compr Canc Netw 10 (3): 312-8, 2012.[PUBMED Abstract]
- Kovach SJ, Fischer AC, Katzman PJ, et al.: Inflammatory myofibroblastic tumors. J Surg Oncol 94 (5): 385-91, 2006.[PUBMED Abstract]
- Brodlie M, Barwick SC, Wood KM, et al.: Inflammatory myofibroblastic tumours of the respiratory tract: paediatric case series with varying clinical presentations. J Laryngol Otol 125 (8): 865-8, 2011.[PUBMED Abstract]
- Xiao Y, Zhou S, Ma C, et al.: Radiological and histopathological features of hepatic inflammatory myofibroblastic tumour: analysis of 10 cases. Clin Radiol 68 (11): 1114-20, 2013.[PUBMED Abstract]
- Karnak I, Senocak ME, Ciftci AO, et al.: Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 36 (6): 908-12, 2001.[PUBMED Abstract]
- Collin M, Charles A, Barker A, et al.: Inflammatory myofibroblastic tumour of the bladder in children: a review. J Pediatr Urol 11 (5): 239-45, 2015.[PUBMED Abstract]
- Coffin CM, Hornick JL, Fletcher CD: Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 31 (4): 509-20, 2007.[PUBMED Abstract]
- Lovly CM, Gupta A, Lipson D, et al.: Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 4 (8): 889-95, 2014.[PUBMED Abstract]
- Devaney KO, Lafeir DJ, Triantafyllou A, et al.: Inflammatory myofibroblastic tumors of the head and neck: evaluation of clinicopathologic and prognostic features. Eur Arch Otorhinolaryngol 269 (12): 2461-5, 2012.[PUBMED Abstract]
- Mehta B, Mascarenhas L, Zhou S, et al.: Inflammatory myofibroblastic tumors in childhood. Pediatr Hematol Oncol 30 (7): 640-5, 2013.[PUBMED Abstract]
- Favini F, Resti AG, Collini P, et al.: Inflammatory myofibroblastic tumor of the conjunctiva: response to chemotherapy with low-dose methotrexate and vinorelbine. Pediatr Blood Cancer 54 (3): 483-5, 2010.[PUBMED Abstract]
- Kube S, Vokuhl C, Dantonello T, et al.: Inflammatory myofibroblastic tumors-A retrospective analysis of the Cooperative Weichteilsarkom Studiengruppe. Pediatr Blood Cancer 65 (6): e27012, 2018.[PUBMED Abstract]
- Doski JJ, Priebe CJ, Driessnack M, et al.: Corticosteroids in the management of unresected plasma cell granuloma (inflammatory pseudotumor) of the lung. J Pediatr Surg 26 (9): 1064-6, 1991.[PUBMED Abstract]
- Diop B, Konate I, Ka S, et al.: Mesenteric myofibroblastic tumor: NSAID therapy after incomplete resection. J Visc Surg 148 (4): e311-4, 2011.[PUBMED Abstract]
- Dalton BG, Thomas PG, Sharp NE, et al.: Inflammatory myofibroblastic tumors in children. J Pediatr Surg 51 (4): 541-4, 2016.[PUBMED Abstract]
- Butrynski JE, D'Adamo DR, Hornick JL, et al.: Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 363 (18): 1727-33, 2010.[PUBMED Abstract]
- Mossé YP, Lim MS, Voss SD, et al.: Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 14 (6): 472-80, 2013.[PUBMED Abstract]
- Gaudichon J, Jeanne-Pasquier C, Deparis M, et al.: Complete and Repeated Response of a Metastatic ALK-rearranged Inflammatory Myofibroblastic Tumor to Crizotinib in a Teenage Girl. J Pediatr Hematol Oncol 38 (4): 308-11, 2016.[PUBMED Abstract]
- Mossé YP, Voss SD, Lim MS, et al.: Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study. J Clin Oncol 35 (28): 3215-3221, 2017.[PUBMED Abstract]
- Nishio M, Murakami H, Horiike A, et al.: Phase I Study of Ceritinib (LDK378) in Japanese Patients with Advanced, Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer or Other Tumors. J Thorac Oncol 10 (7): 1058-66, 2015.[PUBMED Abstract]
- Brivio E, Zwaan CM: ALK inhibition in two emblematic cases of pediatric inflammatory myofibroblastic tumor: Efficacy and side effects. Pediatr Blood Cancer 66 (5): e27645, 2019.[PUBMED Abstract]
- Sulkowski JP, Raval MV, Browne M: Margin status and multimodal therapy in infantile fibrosarcoma. Pediatr Surg Int 29 (8): 771-6, 2013.[PUBMED Abstract]
- Hirschfeld R, Welch JJG, Harrison DJ, et al.: Two cases of humoral hypercalcemia of malignancy complicating infantile fibrosarcoma. Pediatr Blood Cancer 64 (10): , 2017.[PUBMED Abstract]
- Kao YC, Fletcher CDM, Alaggio R, et al.: Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol 42 (1): 28-38, 2018.[PUBMED Abstract]
- Wegert J, Vokuhl C, Collord G, et al.: Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nat Commun 9 (1): 2378, 2018.[PUBMED Abstract]
- Orbach D, Rey A, Cecchetto G, et al.: Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 28 (2): 318-23, 2010.[PUBMED Abstract]
- Orbach D, Brennan B, De Paoli A, et al.: Conservative strategy in infantile fibrosarcoma is possible: The European paediatric Soft tissue sarcoma Study Group experience. Eur J Cancer 57: 1-9, 2016.[PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.[PUBMED Abstract]
- Loh ML, Ahn P, Perez-Atayde AR, et al.: Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children's Hospital, Boston. J Pediatr Hematol Oncol 24 (9): 722-6, 2002.[PUBMED Abstract]
- Akyüz C, Küpeli S, Varan A, et al.: Infantile fibrosarcoma: retrospective analysis of eleven patients. Tumori 97 (2): 166-9, 2011 Mar-Apr.[PUBMED Abstract]
- Gallego S, Pericas N, Barber I, et al.: Infantile fibrosarcoma of the retroperitoneum: a site of unfavorable prognosis? Pediatr Hematol Oncol 28 (5): 451-3, 2011.[PUBMED Abstract]
- Parida L, Fernandez-Pineda I, Uffman JK, et al.: Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int 29 (7): 703-8, 2013.[PUBMED Abstract]
- Mody RJ, Wu YM, Lonigro RJ, et al.: Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 314 (9): 913-25, 2015.[PUBMED Abstract]
- Wong V, Pavlick D, Brennan T, et al.: Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib. J Natl Cancer Inst 108 (1): , 2016.[PUBMED Abstract]
- Kummar S, Lassen UN: TRK Inhibition: A New Tumor-Agnostic Treatment Strategy. Target Oncol 13 (5): 545-556, 2018.[PUBMED Abstract]
- Drilon A, Laetsch TW, Kummar S, et al.: Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 378 (8): 731-739, 2018.[PUBMED Abstract]
- DuBois SG, Laetsch TW, Federman N, et al.: The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 124 (21): 4241-4247, 2018.[PUBMED Abstract]
- Drilon A, Nagasubramanian R, Blake JF, et al.: A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov 7 (9): 963-972, 2017.[PUBMED Abstract]
- Laetsch TW, DuBois SG, Mascarenhas L, et al.: Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 19 (5): 705-714, 2018.[PUBMED Abstract]
- Yanagisawa R, Noguchi M, Fujita K, et al.: Preoperative Treatment With Pazopanib in a Case of Chemotherapy-Resistant Infantile Fibrosarcoma. Pediatr Blood Cancer 63 (2): 348-51, 2016.[PUBMED Abstract]
- Madden NP, Spicer RD, Allibone EB, et al.: Spontaneous regression of neonatal fibrosarcoma. Br J Cancer Suppl 18: S72-5, 1992.[PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011.[PUBMED Abstract]
- Guillou L, Benhattar J, Gengler C, et al.: Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol 31 (9): 1387-402, 2007.[PUBMED Abstract]
- Scheer M, Vokuhl C, Veit-Friedrich I, et al.: Low-grade fibromyxoid sarcoma: A report of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer 67 (2): e28009, 2020.[PUBMED Abstract]
- Mohamed M, Fisher C, Thway K: Low-grade fibromyxoid sarcoma: Clinical, morphologic and genetic features. Ann Diagn Pathol 28: 60-67, 2017.[PUBMED Abstract]
- O'Sullivan MJ, Sirgi KE, Dehner LP: Low-grade fibrosarcoma (hyalinizing spindle cell tumor with giant rosettes) with pulmonary metastases at presentation: case report and review of the literature. Int J Surg Pathol 10 (3): 211-6, 2002.[PUBMED Abstract]
- Folpe AL, Lane KL, Paull G, et al.: Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 24 (10): 1353-60, 2000.[PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015.[PUBMED Abstract]
- Maretty-Nielsen K, Baerentzen S, Keller J, et al.: Low-Grade Fibromyxoid Sarcoma: Incidence, Treatment Strategy of Metastases, and Clinical Significance of the FUS Gene. Sarcoma 2013: 256280, 2013.[PUBMED Abstract]
- Prieto-Granada C, Zhang L, Chen HW, et al.: A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer 54 (1): 28-38, 2015.[PUBMED Abstract]
- Chew W, Benson C, Thway K, et al.: Clinical Characteristics and efficacy of chemotherapy in sclerosing epithelioid fibrosarcoma. Med Oncol 35 (11): 138, 2018.[PUBMED Abstract]
- Arbajian E, Puls F, Antonescu CR, et al.: In-depth Genetic Analysis of Sclerosing Epithelioid Fibrosarcoma Reveals Recurrent Genomic Alterations and Potential Treatment Targets. Clin Cancer Res 23 (23): 7426-7434, 2017.[PUBMED Abstract]
- Pollock BH, Jenson HB, Leach CT, et al.: Risk factors for pediatric human immunodeficiency virus-related malignancy. JAMA 289 (18): 2393-9, 2003.[PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007.[PUBMED Abstract]
- Samuels BL, Chawla S, Patel S, et al.: Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 24 (6): 1703-9, 2013.[PUBMED Abstract]
- Enzinger FM, Zhang RY: Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am J Surg Pathol 12 (11): 818-26, 1988.[PUBMED Abstract]
- Black J, Coffin CM, Dehner LP: Fibrohistiocytic tumors and related neoplasms in children and adolescents. Pediatr Dev Pathol 15 (1 Suppl): 181-210, 2012.[PUBMED Abstract]
- Moosavi C, Jha P, Fanburg-Smith JC: An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol 11 (5): 313-9, 2007.[PUBMED Abstract]
- Billings SD, Folpe AL: Cutaneous and subcutaneous fibrohistiocytic tumors of intermediate malignancy: an update. Am J Dermatopathol 26 (2): 141-55, 2004.[PUBMED Abstract]
- Remstein ED, Arndt CA, Nascimento AG: Plexiform fibrohistiocytic tumor: clinicopathologic analysis of 22 cases. Am J Surg Pathol 23 (6): 662-70, 1999.[PUBMED Abstract]
- Salomao DR, Nascimento AG: Plexiform fibrohistiocytic tumor with systemic metastases: a case report. Am J Surg Pathol 21 (4): 469-76, 1997.[PUBMED Abstract]
- Redlich GC, Montgomery KD, Allgood GA, et al.: Plexiform fibrohistiocytic tumor with a clonal cytogenetic anomaly. Cancer Genet Cytogenet 108 (2): 141-3, 1999.[PUBMED Abstract]
- Luzar B, Calonje E: Cutaneous fibrohistiocytic tumours - an update. Histopathology 56 (1): 148-65, 2010.[PUBMED Abstract]
- Carli M, Ferrari A, Mattke A, et al.: Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol 23 (33): 8422-30, 2005.[PUBMED Abstract]
- Malbari F, Spira M, B Knight P, et al.: Malignant Peripheral Nerve Sheath Tumors in Neurofibromatosis: Impact of Family History. J Pediatr Hematol Oncol 40 (6): e359-e363, 2018.[PUBMED Abstract]
- Agresta L, Salloum R, Hummel TR, et al.: Malignant peripheral nerve sheath tumor: Transformation in a patient with neurofibromatosis type 2. Pediatr Blood Cancer 66 (2): e27520, 2019.[PUBMED Abstract]
- Zhang M, Wang Y, Jones S, et al.: Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46 (11): 1170-2, 2014.[PUBMED Abstract]
- Röhrich M, Koelsche C, Schrimpf D, et al.: Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol 131 (6): 877-87, 2016.[PUBMED Abstract]
- Kaplan HG, Rostad S, Ross JS, et al.: Genomic Profiling in Patients With Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways With Targetable Mutations. J Natl Compr Canc Netw 16 (8): 967-974, 2018.[PUBMED Abstract]
- Hagel C, Zils U, Peiper M, et al.: Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol 82 (2): 187-92, 2007.[PUBMED Abstract]
- Zou C, Smith KD, Liu J, et al.: Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249 (6): 1014-22, 2009.[PUBMED Abstract]
- Okada K, Hasegawa T, Tajino T, et al.: Clinical relevance of pathological grades of malignant peripheral nerve sheath tumor: a multi-institution TMTS study of 56 cases in Northern Japan. Ann Surg Oncol 14 (2): 597-604, 2007.[PUBMED Abstract]
- Amirian ES, Goodman JC, New P, et al.: Pediatric and adult malignant peripheral nerve sheath tumors: an analysis of data from the surveillance, epidemiology, and end results program. J Neurooncol 116 (3): 609-16, 2014.[PUBMED Abstract]
- Valentin T, Le Cesne A, Ray-Coquard I, et al.: Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer 56: 77-84, 2016.[PUBMED Abstract]
- Høland M, Kolberg M, Danielsen SA, et al.: Inferior survival for patients with malignant peripheral nerve sheath tumors defined by aberrant TP53. Mod Pathol 31 (11): 1694-1707, 2018.[PUBMED Abstract]
- Krawczyk MA, Karpinsky G, Izycka-Swieszewska E, et al.: Immunohistochemical assessment of cyclin D1 and p53 is associated with survival in childhood malignant peripheral nerve sheath tumor. Cancer Biomark 24 (3): 351-361, 2019.[PUBMED Abstract]
- Martin E, Coert JH, Flucke UE, et al.: Neurofibromatosis-associated malignant peripheral nerve sheath tumors in children have a worse prognosis: A nationwide cohort study. Pediatr Blood Cancer 67 (4): e28138, 2020.[PUBMED Abstract]
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018.[PUBMED Abstract]
- Ferrari A, Bisogno G, Macaluso A, et al.: Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 109 (7): 1406-12, 2007.[PUBMED Abstract]
- van Noesel MM, Orbach D, Brennan B, et al.: Outcome and prognostic factors in pediatric malignant peripheral nerve sheath tumors: An analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 prospective study. Pediatr Blood Cancer 66 (10): e27833, 2019.[PUBMED Abstract]
- Okur FV, Oguz A, Karadeniz C, et al.: Malignant triton tumor of the pelvis in a 2-year-old boy. J Pediatr Hematol Oncol 28 (3): 173-6, 2006.[PUBMED Abstract]
- Griffin BB, Chou PM, George D, et al.: Malignant Ectomesenchymoma: Series Analysis of a Histologically and Genetically Heterogeneous Tumor. Int J Surg Pathol 26 (3): 200-212, 2018.[PUBMED Abstract]
- Huang SC, Alaggio R, Sung YS, et al.: Frequent HRAS Mutations in Malignant Ectomesenchymoma: Overlapping Genetic Abnormalities With Embryonal Rhabdomyosarcoma. Am J Surg Pathol 40 (7): 876-85, 2016.[PUBMED Abstract]
- Dantonello TM, Leuschner I, Vokuhl C, et al.: Malignant ectomesenchymoma in children and adolescents: report from the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer 60 (2): 224-9, 2013.[PUBMED Abstract]
- Rodriguez-Galindo C, Ramsey K, Jenkins JJ, et al.: Hemangiopericytoma in children and infants. Cancer 88 (1): 198-204, 2000.[PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Hemangiopericytoma in pediatric ages: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 92 (10): 2692-8, 2001.[PUBMED Abstract]
- Bien E, Stachowicz-Stencel T, Godzinski J, et al.: Retrospective multi-institutional study on hemangiopericytoma in Polish children. Pediatr Int 51 (1): 19-24, 2009.[PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008.[PUBMED Abstract]
- Fernandez-Pineda I, Parida L, Jenkins JJ, et al.: Childhood hemangiopericytoma: review of St Jude Children's Research Hospital. J Pediatr Hematol Oncol 33 (5): 356-9, 2011.[PUBMED Abstract]
- Haller F, Knopf J, Ackermann A, et al.: Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol 238 (5): 700-10, 2016.[PUBMED Abstract]
- Doebele RC, Davis LE, Vaishnavi A, et al.: An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov 5 (10): 1049-57, 2015.[PUBMED Abstract]
- Wiswell TE, Davis J, Cunningham BE, et al.: Infantile myofibromatosis: the most common fibrous tumor of infancy. J Pediatr Surg 23 (4): 315-8, 1988.[PUBMED Abstract]
- Chung EB, Enzinger FM: Infantile myofibromatosis. Cancer 48 (8): 1807-18, 1981.[PUBMED Abstract]
- Modi N: Congenital generalised fibromatosis. Arch Dis Child 57 (11): 881-2, 1982.[PUBMED Abstract]
- Levine E, Fréneaux P, Schleiermacher G, et al.: Risk-adapted therapy for infantile myofibromatosis in children. Pediatr Blood Cancer 59 (1): 115-20, 2012.[PUBMED Abstract]
- Larralde M, Hoffner MV, Boggio P, et al.: Infantile myofibromatosis: report of nine patients. Pediatr Dermatol 27 (1): 29-33, 2010 Jan-Feb.[PUBMED Abstract]
- Cheung YH, Gayden T, Campeau PM, et al.: A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92 (6): 996-1000, 2013.[PUBMED Abstract]
- Agaimy A, Bieg M, Michal M, et al.: Recurrent Somatic PDGFRB Mutations in Sporadic Infantile/Solitary Adult Myofibromas But Not in Angioleiomyomas and Myopericytomas. Am J Surg Pathol 41 (2): 195-203, 2017.[PUBMED Abstract]
- Gopal M, Chahal G, Al-Rifai Z, et al.: Infantile myofibromatosis. Pediatr Surg Int 24 (3): 287-91, 2008.[PUBMED Abstract]
- Weaver MS, Navid F, Huppmann A, et al.: Vincristine and Dactinomycin in Infantile Myofibromatosis With a Review of Treatment Options. J Pediatr Hematol Oncol 37 (3): 237-41, 2015.[PUBMED Abstract]
- Sultan I, Rodriguez-Galindo C, Saab R, et al.: Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115 (15): 3537-47, 2009.[PUBMED Abstract]
- Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013.[PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994.[PUBMED Abstract]
- Ferrari A, De Salvo GL, Oberlin O, et al.: Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer 48 (9): 1370-5, 2012.[PUBMED Abstract]
- van de Rijn M, Barr FG, Collins MH, et al.: Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 112 (1): 43-9, 1999.[PUBMED Abstract]
- Krsková L, Sumerauer D, Stejskalová E, et al.: A novel variant of SYT-SSX1 fusion gene in a case of spindle cell synovial sarcoma. Diagn Mol Pathol 16 (3): 179-83, 2007.[PUBMED Abstract]
- Su L, Sampaio AV, Jones KB, et al.: Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21 (3): 333-47, 2012.[PUBMED Abstract]
- Arnold MA, Arnold CA, Li G, et al.: A unique pattern of INI1 immunohistochemistry distinguishes synovial sarcoma from its histologic mimics. Hum Pathol 44 (5): 881-7, 2013.[PUBMED Abstract]
- Vlenterie M, Ho VK, Kaal SE, et al.: Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113 (11): 1602-6, 2015.[PUBMED Abstract]
- Smolle MA, Parry M, Jeys L, et al.: Synovial sarcoma: Do children do better? Eur J Surg Oncol 45 (2): 254-260, 2019.[PUBMED Abstract]
- Okcu MF, Munsell M, Treuner J, et al.: Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21 (8): 1602-11, 2003.[PUBMED Abstract]
- Brecht IB, Ferrari A, Int-Veen C, et al.: Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer 46 (1): 11-7, 2006.[PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Healey JH, et al.: Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 20 (1): 73-9, 2013.[PUBMED Abstract]
- Trassard M, Le Doussal V, Hacène K, et al.: Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19 (2): 525-34, 2001.[PUBMED Abstract]
- Guillou L, Benhattar J, Bonichon F, et al.: Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22 (20): 4040-50, 2004.[PUBMED Abstract]
- Ferrari A, Gronchi A, Casanova M, et al.: Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101 (3): 627-34, 2004.[PUBMED Abstract]
- Lagarde P, Przybyl J, Brulard C, et al.: Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31 (5): 608-15, 2013.[PUBMED Abstract]
- Stegmaier S, Leuschner I, Poremba C, et al.: The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer 64 (1): 89-95, 2017.[PUBMED Abstract]
- Scheer M, Dantonello T, Hallmen E, et al.: Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer 63 (7): 1198-206, 2016.[PUBMED Abstract]
- Orbach D, Mosseri V, Pissaloux D, et al.: Genomic complexity in pediatric synovial sarcomas (Synobio study): the European pediatric soft tissue sarcoma group (EpSSG) experience. Cancer Med 7 (4): 1384-1393, 2018.[PUBMED Abstract]
- Ferrari A, Chi YY, De Salvo GL, et al.: Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 78: 1-6, 2017.[PUBMED Abstract]
- McGrory JE, Pritchard DJ, Arndt CA, et al.: Nonrhabdomyosarcoma soft tissue sarcomas in children. The Mayo Clinic experience. Clin Orthop (374): 247-58, 2000.[PUBMED Abstract]
- Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al.: Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 17 (1): 150-7, 1999.[PUBMED Abstract]
- Koscielniak E, Harms D, Henze G, et al.: Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17 (12): 3706-19, 1999.[PUBMED Abstract]
- Pappo AS, Devidas M, Jenkins J, et al.: Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 23 (18): 4031-8, 2005.[PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999.[PUBMED Abstract]
- Brennan B, Stevens M, Kelsey A, et al.: Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55 (1): 85-90, 2010.[PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011.[PUBMED Abstract]
- Raney RB: Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 27 (4): 207-11, 2005.[PUBMED Abstract]
- Orbach D, Mc Dowell H, Rey A, et al.: Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer 57 (7): 1130-6, 2011.[PUBMED Abstract]
- Ladenstein R, Treuner J, Koscielniak E, et al.: Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 71 (11): 3647-55, 1993.[PUBMED Abstract]
- Venkatramani R, Anderson JR, Million L, et al.: Risk-based treatment for synovial sarcoma in patients under 30 years of age: Children's Oncology Group study ARST0332. [Abstract] J Clin Oncol 33 (15 Suppl): A-10012, 2015. Also available online. Last accessed February 06, 2020.[PUBMED Abstract]
- Ferrari A, De Salvo GL, Brennan B, et al.: Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26 (3): 567-72, 2015.[PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012.[PUBMED Abstract]
- Scheer M, Dantonello T, Hallmen E, et al.: Synovial Sarcoma Recurrence in Children and Young Adults. Ann Surg Oncol 23 (Suppl 5): 618-626, 2016.[PUBMED Abstract]
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012.[PUBMED Abstract]
- Chbani L, Guillou L, Terrier P, et al.: Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 131 (2): 222-7, 2009.[PUBMED Abstract]
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009.[PUBMED Abstract]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.[PUBMED Abstract]
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution's experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012.[PUBMED Abstract]
- Hawkins DS, Spunt SL, Skapek SX, et al.: Children's Oncology Group's 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer 60 (6): 1001-8, 2013.[PUBMED Abstract]
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006.[PUBMED Abstract]
- Sparber-Sauer M, Koscielniak E, Vokuhl C, et al.: Epithelioid sarcoma in children, adolescents, and young adults: Localized, primary metastatic and relapsed disease. Treatment results of five Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 66 (9): e27879, 2019.[PUBMED Abstract]
- Spunt SL, Francotte N, De Salvo GL, et al.: Clinical features and outcomes of young patients with epithelioid sarcoma: an analysis from the Children's Oncology Group and the European paediatric soft tissue Sarcoma Study Group prospective clinical trials. Eur J Cancer 112: 98-106, 2019.[PUBMED Abstract]
- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.[PUBMED Abstract]
- Orbach D, Brennan B, Casanova M, et al.: Paediatric and adolescent alveolar soft part sarcoma: A joint series from European cooperative groups. Pediatr Blood Cancer 60 (11): 1826-32, 2013.[PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011.[PUBMED Abstract]
- Wang HW, Qin XJ, Yang WJ, et al.: Alveolar soft part sarcoma of the oral and maxillofacial region: clinical analysis in a series of 18 patients. Oral Surg Oral Med Oral Pathol Oral Radiol 119 (4): 396-401, 2015.[PUBMED Abstract]
- Kayton ML, Meyers P, Wexler LH, et al.: Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg 41 (1): 187-93, 2006.[PUBMED Abstract]
- Sparber-Sauer M, Seitz G, von Kalle T, et al.: Alveolar soft-part sarcoma: Primary metastatic disease and metastatic relapse occurring during long-term follow-up: Treatment results of four Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 65 (12): e27405, 2018.[PUBMED Abstract]
- Flores RJ, Harrison DJ, Federman NC, et al.: Alveolar soft part sarcoma in children and young adults: A report of 69 cases. Pediatr Blood Cancer 65 (5): e26953, 2018.[PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001.[PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011.[PUBMED Abstract]
- Lieberman PH, Brennan MF, Kimmel M, et al.: Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 63 (1): 1-13, 1989.[PUBMED Abstract]
- Casanova M, Ferrari A, Bisogno G, et al.: Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol 11 (11): 1445-9, 2000.[PUBMED Abstract]
- Pennacchioli E, Fiore M, Collini P, et al.: Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 33 patients at a single institution. Ann Surg Oncol 17 (12): 3229-33, 2010.[PUBMED Abstract]
- Wilky BA, Trucco MM, Subhawong TK, et al.: Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol 20 (6): 837-848, 2019.[PUBMED Abstract]
- Roozendaal KJ, de Valk B, ten Velden JJ, et al.: Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer 89 (2): 243-5, 2003.[PUBMED Abstract]
- Conde N, Cruz O, Albert A, et al.: Antiangiogenic treatment as a pre-operative management of alveolar soft-part sarcoma. Pediatr Blood Cancer 57 (6): 1071-3, 2011.[PUBMED Abstract]
- Stacchiotti S, Negri T, Zaffaroni N, et al.: Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22 (7): 1682-90, 2011.[PUBMED Abstract]
- Jagodzińska-Mucha P, Świtaj T, Kozak K, et al.: Long-term results of therapy with sunitinib in metastatic alveolar soft part sarcoma. Tumori 103 (3): 231-235, 2017.[PUBMED Abstract]
- Kummar S, Allen D, Monks A, et al.: Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31 (18): 2296-302, 2013.[PUBMED Abstract]
- Cohen JW, Widemann BC, Derdak J, et al.: Cediranib phase-II study in children with metastatic alveolar soft-part sarcoma (ASPS). Pediatr Blood Cancer 66 (12): e27987, 2019.[PUBMED Abstract]
- Judson I, Morden JP, Kilburn L, et al.: Cediranib in patients with alveolar soft-part sarcoma (CASPS): a double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol 20 (7): 1023-1034, 2019.[PUBMED Abstract]
- Kim M, Kim TM, Keam B, et al.: A Phase II Trial of Pazopanib in Patients with Metastatic Alveolar Soft Part Sarcoma. Oncologist 24 (1): 20-e29, 2019.[PUBMED Abstract]
- Stacchiotti S, Mir O, Le Cesne A, et al.: Activity of Pazopanib and Trabectedin in Advanced Alveolar Soft Part Sarcoma. Oncologist 23 (1): 62-70, 2018.[PUBMED Abstract]
- Coindre JM, Hostein I, Terrier P, et al.: Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 107 (5): 1055-64, 2006.[PUBMED Abstract]
- Meis-Kindblom JM: Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 13 (6): 286-92, 2006.[PUBMED Abstract]
- Dim DC, Cooley LD, Miranda RN: Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med 131 (1): 152-6, 2007.[PUBMED Abstract]
- Blazer DG, Lazar AJ, Xing Y, et al.: Clinical outcomes of molecularly confirmed clear cell sarcoma from a single institution and in comparison with data from the Surveillance, Epidemiology, and End Results registry. Cancer 115 (13): 2971-9, 2009.[PUBMED Abstract]
- Fujimura Y, Siddique H, Lee L, et al.: EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 20 (46): 6653-9, 2001.[PUBMED Abstract]
- Hisaoka M, Ishida T, Kuo TT, et al.: Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 32 (3): 452-60, 2008.[PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 94 (12): 3269-76, 2002.[PUBMED Abstract]
- Karita M, Tsuchiya H, Yamamoto N, et al.: Caffeine-potentiated chemotherapy for clear cell sarcoma: a report of five cases. Int J Clin Oncol 18 (1): 33-7, 2013.[PUBMED Abstract]
- Schöffski P, Wozniak A, Stacchiotti S, et al.: Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 'CREATE'. Ann Oncol 28 (12): 3000-3008, 2017.[PUBMED Abstract]
- Tsuneyoshi M, Enjoji M, Iwasaki H, et al.: Extraskeletal myxoid chondrosarcoma--a clinicopathologic and electron microscopic study. Acta Pathol Jpn 31 (3): 439-47, 1981.[PUBMED Abstract]
- Hachitanda Y, Tsuneyoshi M, Daimaru Y, et al.: Extraskeletal myxoid chondrosarcoma in young children. Cancer 61 (12): 2521-6, 1988.[PUBMED Abstract]
- Hisaoka M, Ishida T, Imamura T, et al.: TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40 (4): 325-8, 2004.[PUBMED Abstract]
- Enzinger FM, Shiraki M: Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 3 (3): 421-35, 1972.[PUBMED Abstract]
- McGrory JE, Rock MG, Nascimento AG, et al.: Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res (382): 185-90, 2001.[PUBMED Abstract]
- Drilon AD, Popat S, Bhuchar G, et al.: Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 113 (12): 3364-71, 2008.[PUBMED Abstract]
- Stacchiotti S, Pantaleo MA, Astolfi A, et al.: Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer 50 (9): 1657-64, 2014.[PUBMED Abstract]
- Leuschner I, Radig K, Harms D: Desmoplastic small round cell tumor. Semin Diagn Pathol 13 (3): 204-12, 1996.[PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Wollner N, et al.: Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14 (5): 1526-31, 1996.[PUBMED Abstract]
- Saab R, Khoury JD, Krasin M, et al.: Desmoplastic small round cell tumor in childhood: the St. Jude Children's Research Hospital experience. Pediatr Blood Cancer 49 (3): 274-9, 2007.[PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007.[PUBMED Abstract]
- Hayes-Jordan A, LaQuaglia MP, Modak S: Management of desmoplastic small round cell tumor. Semin Pediatr Surg 25 (5): 299-304, 2016.[PUBMED Abstract]
- Arora VC, Price AP, Fleming S, et al.: Characteristic imaging features of desmoplastic small round cell tumour. Pediatr Radiol 43 (1): 93-102, 2013.[PUBMED Abstract]
- Gerald WL, Ladanyi M, de Alava E, et al.: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 16 (9): 3028-36, 1998.[PUBMED Abstract]
- Lal DR, Su WT, Wolden SL, et al.: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40 (1): 251-5, 2005.[PUBMED Abstract]
- Philippe-Chomette P, Kabbara N, Andre N, et al.: Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatr Blood Cancer 58 (6): 891-7, 2012.[PUBMED Abstract]
- Sedig L, Geiger J, Mody R, et al.: Paratesticular desmoplastic small round cell tumors: A case report and review of the literature. Pediatr Blood Cancer 64 (12): , 2017.[PUBMED Abstract]
- Subbiah V, Lamhamedi-Cherradi SE, Cuglievan B, et al.: Multimodality Treatment of Desmoplastic Small Round Cell Tumor: Chemotherapy and Complete Cytoreductive Surgery Improve Patient Survival. Clin Cancer Res 24 (19): 4865-4873, 2018.[PUBMED Abstract]
- Schwarz RE, Gerald WL, Kushner BH, et al.: Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 5 (5): 416-22, 1998 Jul-Aug.[PUBMED Abstract]
- Goodman KA, Wolden SL, La Quaglia MP, et al.: Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys 54 (1): 170-6, 2002.[PUBMED Abstract]
- Osborne EM, Briere TM, Hayes-Jordan A, et al.: Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119 (1): 40-4, 2016.[PUBMED Abstract]
- Atallah V, Honore C, Orbach D, et al.: Role of Adjuvant Radiation Therapy After Surgery for Abdominal Desmoplastic Small Round Cell Tumors. Int J Radiat Oncol Biol Phys 95 (4): 1244-53, 2016.[PUBMED Abstract]
- Hayes-Jordan AA, Coakley BA, Green HL, et al.: Desmoplastic Small Round Cell Tumor Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Results of a Phase 2 Trial. Ann Surg Oncol 25 (4): 872-877, 2018.[PUBMED Abstract]
- Scalabre A, Philippe-Chomette P, Passot G, et al.: Cytoreductive surgery and hyperthermic intraperitoneal perfusion with chemotherapy in children with peritoneal tumor spread: A French nationwide study over 14 years. Pediatr Blood Cancer 65 (4): , 2018.[PUBMED Abstract]
- Honoré C, Atallah V, Mir O, et al.: Abdominal desmoplastic small round cell tumor without extraperitoneal metastases: Is there a benefit for HIPEC after macroscopically complete cytoreductive surgery? PLoS One 12 (2): e0171639, 2017.[PUBMED Abstract]
- Cook RJ, Wang Z, Arora M, et al.: Clinical outcomes of patients with desmoplastic small round cell tumor of the peritoneum undergoing autologous HCT: a CIBMTR retrospective analysis. Bone Marrow Transplant 47 (11): 1455-8, 2012.[PUBMED Abstract]
- Tarek N, Hayes-Jordan A, Salvador L, et al.: Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer 65 (1): , 2018.[PUBMED Abstract]
- Kodet R, Newton WA, Sachs N, et al.: Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol 22 (7): 674-84, 1991.[PUBMED Abstract]
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999.[PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.[PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.[PUBMED Abstract]
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al.: Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 54 (1): 35-40, 2010.[PUBMED Abstract]
- Puri DR, Meyers PA, Kraus DH, et al.: Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 50 (1): 167-9, 2008.[PUBMED Abstract]
- Madigan CE, Armenian SH, Malogolowkin MH, et al.: Extracranial malignant rhabdoid tumors in childhood: the Childrens Hospital Los Angeles experience. Cancer 110 (9): 2061-6, 2007.[PUBMED Abstract]
- Bourdeaut F, Fréneaux P, Thuille B, et al.: Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51 (3): 363-8, 2008.[PUBMED Abstract]
- Wetmore C, Boyett J, Li S, et al.: Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol 17 (6): 882-8, 2015.[PUBMED Abstract]
- Martignoni G, Pea M, Reghellin D, et al.: Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med 134 (1): 33-40, 2010.[PUBMED Abstract]
- Bissler JJ, McCormack FX, Young LR, et al.: Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358 (2): 140-51, 2008.[PUBMED Abstract]
- Davies DM, Johnson SR, Tattersfield AE, et al.: Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358 (2): 200-3, 2008.[PUBMED Abstract]
- Agaram NP, Sung YS, Zhang L, et al.: Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39 (6): 813-25, 2015.[PUBMED Abstract]
- Folpe A, Inwards C, eds.: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology. Philadelphia, Pa: WB Saunders Co, 2010.[PUBMED Abstract]
- Armah HB, Parwani AV: Perivascular epithelioid cell tumor. Arch Pathol Lab Med 133 (4): 648-54, 2009.[PUBMED Abstract]
- Alaggio R, Cecchetto G, Martignoni G, et al.: Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47 (6): e31-40, 2012.[PUBMED Abstract]
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al.: Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835-40, 2010.[PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children's Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed February 06, 2020.[PUBMED Abstract]
- Laetsch TW, Roy A, Xu L, et al.: Undifferentiated Sarcomas in Children Harbor Clinically Relevant Oncogenic Fusions and Gene Copy-Number Alterations: A Report from the Children's Oncology Group. Clin Cancer Res 24 (16): 3888-3897, 2018.[PUBMED Abstract]
- Randall RL, Albritton KH, Ferney BJ, et al.: Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am J Orthop 33 (12): 602-8, 2004.[PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun.[PUBMED Abstract]
- Le Guellec S, Chibon F, Ouali M, et al.: Are peripheral purely undifferentiated pleomorphic sarcomas with MDM2 amplification dedifferentiated liposarcomas? Am J Surg Pathol 38 (3): 293-304, 2014.[PUBMED Abstract]
- Bjerkehagen B, Smeland S, Walberg L, et al.: Radiation-induced sarcoma: 25-year experience from the Norwegian Radium Hospital. Acta Oncol 47 (8): 1475-82, 2008.[PUBMED Abstract]
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.[PUBMED Abstract]
- Coffin CM, Dehner LP: Vascular tumors in children and adolescents: a clinicopathologic study of 228 tumors in 222 patients. Pathol Annu 28 Pt 1: 97-120, 1993.[PUBMED Abstract]
- Mehrabi A, Kashfi A, Fonouni H, et al.: Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer 107 (9): 2108-21, 2006.[PUBMED Abstract]
- Haro A, Saitoh G, Tamiya S, et al.: Four-year natural clinical course of pulmonary epithelioid hemangioendothelioma without therapy. Thorac Cancer 6 (4): 544-7, 2015.[PUBMED Abstract]
- Sardaro A, Bardoscia L, Petruzzelli MF, et al.: Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev 8 (2): 259, 2014.[PUBMED Abstract]
- Dong K, Wang XX, Feng JL, et al.: Pathological characteristics of liver biopsies in eight patients with hepatic epithelioid hemangioendothelioma. Int J Clin Exp Pathol 8 (9): 11015-23, 2015.[PUBMED Abstract]
- Adams DM, Hammill A: Other vascular tumors. Semin Pediatr Surg 23 (4): 173-7, 2014.[PUBMED Abstract]
- Xiao Y, Wang C, Song Y, et al.: Primary epithelioid hemangioendothelioma of the kidney: the first case report in a child and literature review. Urology 82 (4): 925-7, 2013.[PUBMED Abstract]
- Reich S, Ringe H, Uhlenberg B, et al.: Epithelioid hemangioendothelioma of the lung presenting with pneumonia and heart rhythm disturbances in a teenage girl. J Pediatr Hematol Oncol 32 (4): 274-6, 2010.[PUBMED Abstract]
- Cournoyer E, Al-Ibraheemi A, Engel E, et al.: Clinical characterization and long-term outcomes in pediatric epithelioid hemangioendothelioma. Pediatr Blood Cancer 67 (2): e28045, 2020.[PUBMED Abstract]
- Daller JA, Bueno J, Gutierrez J, et al.: Hepatic hemangioendothelioma: clinical experience and management strategy. J Pediatr Surg 34 (1): 98-105; discussion 105-6, 1999.[PUBMED Abstract]
- Ackermann O, Fabre M, Franchi S, et al.: Widening spectrum of liver angiosarcoma in children. J Pediatr Gastroenterol Nutr 53 (6): 615-9, 2011.[PUBMED Abstract]
- Stacchiotti S, Provenzano S, Dagrada G, et al.: Sirolimus in Advanced Epithelioid Hemangioendothelioma: A Retrospective Case-Series Analysis from the Italian Rare Cancer Network Database. Ann Surg Oncol 23 (9): 2735-44, 2016.[PUBMED Abstract]
- Semenisty V, Naroditsky I, Keidar Z, et al.: Pazopanib for metastatic pulmonary epithelioid hemangioendothelioma-a suitable treatment option: case report and review of anti-angiogenic treatment options. BMC Cancer 15: 402, 2015.[PUBMED Abstract]
- Raheja A, Suri A, Singh S, et al.: Multimodality management of a giant skull base hemangioendothelioma of the sphenopetroclival region. J Clin Neurosci 22 (9): 1495-8, 2015.[PUBMED Abstract]
- Ahmad N, Adams DM, Wang J, et al.: Hepatic epithelioid hemangioendothelioma in a patient with hemochromatosis. J Natl Compr Canc Netw 12 (9): 1203-7, 2014.[PUBMED Abstract]
- Otte JB, Zimmerman A: The role of liver transplantation for pediatric epithelioid hemangioendothelioma. Pediatr Transplant 14 (3): 295-7, 2010.[PUBMED Abstract]
- Cioffi A, Reichert S, Antonescu CR, et al.: Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am 27 (5): 975-88, 2013.[PUBMED Abstract]
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014.[PUBMED Abstract]
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971.[PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol 39 (2): 109-14, 2002.[PUBMED Abstract]
- Deyrup AT, Miettinen M, North PE, et al.: Pediatric cutaneous angiosarcomas: a clinicopathologic study of 10 cases. Am J Surg Pathol 35 (1): 70-5, 2011.[PUBMED Abstract]
- Elliott P, Kleinschmidt I: Angiosarcoma of the liver in Great Britain in proximity to vinyl chloride sites. Occup Environ Med 54 (1): 14-8, 1997.[PUBMED Abstract]
- Lezama-del Valle P, Gerald WL, Tsai J, et al.: Malignant vascular tumors in young patients. Cancer 83 (8): 1634-9, 1998.[PUBMED Abstract]
- Fata F, O'Reilly E, Ilson D, et al.: Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer 86 (10): 2034-7, 1999.[PUBMED Abstract]
- Lahat G, Dhuka AR, Hallevi H, et al.: Angiosarcoma: clinical and molecular insights. Ann Surg 251 (6): 1098-106, 2010.[PUBMED Abstract]
- Orlando G, Adam R, Mirza D, et al.: Hepatic hemangiosarcoma: an absolute contraindication to liver transplantation--the European Liver Transplant Registry experience. Transplantation 95 (6): 872-7, 2013.[PUBMED Abstract]
- Sanada T, Nakayama H, Irisawa R, et al.: Clinical outcome and dose volume evaluation in patients who undergo brachytherapy for angiosarcoma of the scalp and face. Mol Clin Oncol 6 (3): 334-340, 2017.[PUBMED Abstract]
- Dickson MA, D'Adamo DR, Keohan ML, et al.: Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma 2015: 532478, 2015.[PUBMED Abstract]
- Scott MT, Portnow LH, Morris CG, et al.: Radiation therapy for angiosarcoma: the 35-year University of Florida experience. Am J Clin Oncol 36 (2): 174-80, 2013.[PUBMED Abstract]
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001.[PUBMED Abstract]
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001.[PUBMED Abstract]
- Ravi V, Patel S: Vascular sarcomas. Curr Oncol Rep 15 (4): 347-55, 2013.[PUBMED Abstract]
- Grassia KL, Peterman CM, Iacobas I, et al.: Clinical case series of pediatric hepatic angiosarcoma. Pediatr Blood Cancer 64 (11): , 2017.[PUBMED Abstract]
- 転移性小児軟部肉腫の治療
-
転移性の小児軟部肉腫に対する標準治療法の選択肢には以下のものがある:
- 化学療法、放射線療法、肺転移巣の外科的切除を用いる集学的治療。
治療法の選択肢については、本要約の個々の腫瘍型のセクションを参照のこと。
転移性軟部肉腫の小児患者の予後は不良であり[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ][ 6 ]、このような小児には化学療法、放射線療法および肺転移巣の外科的切除を組み合わせた治療が行われるべきである。プロスペクティブ・ランダム化試験では、ビンクリスチン、ダクチノマイシン、ドキソルビシン、およびシクロホスファミドによる化学療法にダカルバジンを併用または併用しないで、切除不能または転移性の病変を認める患者の3分の1において腫瘍反応が得られた。しかしながら、推定4年生存率は不良で、生存者は3分の1未満であった。[ 6 ][ 7 ][ 8 ]
肺転移
一般的に、孤立性の肺転移巣が認められる小児には、肉眼的病変をすべて切除することを目的に外科的手技を検討すべきである。[ 9 ]多発性または反復性の肺転移を認める患者に対しては、合併症が認容可能と考えられる場合は、外科手術を追加施行することがある。1件のレトロスペクティブ・レビューにおいて、滑膜肉腫および複数の肺転移が認められたが、すべての転移性肺病変を完全切除できた患者は、完全切除を達成できなかった患者よりも生存が優れていた。[ 9 ][証拠レベル:3iiiA]正式の肺区域切除術、肺葉切除術、および縦隔リンパ節郭清術は不要である。[ 10 ]
代替アプローチとして集中的な放射線療法(分割定位放射線治療)があり、成人患者に使用され、病変の制御に成功している。成人を対象とした諸研究では、肺転移巣切除目的の開胸が実施された後の推定5年生存率は10~58%とされている。[ 11 ]
参考文献- Demetri GD, Elias AD: Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas. Implications for decision making in the clinic. Hematol Oncol Clin North Am 9 (4): 765-85, 1995.[PUBMED Abstract]
- Elias A, Ryan L, Sulkes A, et al.: Response to mesna, doxorubicin, ifosfamide, and dacarbazine in 108 patients with metastatic or unresectable sarcoma and no prior chemotherapy. J Clin Oncol 7 (9): 1208-16, 1989.[PUBMED Abstract]
- Edmonson JH, Ryan LM, Blum RH, et al.: Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 11 (7): 1269-75, 1993.[PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec.[PUBMED Abstract]
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995.[PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999.[PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999.[PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998.[PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013.[PUBMED Abstract]
- Putnam JB, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995.[PUBMED Abstract]
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012.[PUBMED Abstract]
- 進行性/再発性小児軟部肉腫の治療
-
乳児型線維肉腫の乳児は除外される可能性があるが、腫瘍が進行性または再発性の患者における予後は不良である。小児軟部肉腫の局所制御を強化することが最終的な生存率改善につながることを実証したプロスペクティブ試験は存在しない。このため、治療は再発部位、腫瘍の生物学的特徴(例、悪性度、浸潤度、大きさ)、以前の治療法、および個々の患者の考慮事項に応じて個別に行っていくべきである。再発腫瘍が認められる患者はすべて、臨床試験を検討すべきである。
進行性または再発性疾患に対する治療法の選択肢には以下のものがある:
- 手術。
- 化学療法。
- チロシンキナーゼ阻害薬。
- 免疫チェックポイント阻害薬。
- 放射線療法。
再発した小児非横紋筋肉腫性軟部肉腫の標準治療は外科的切除である。それまでに放射線療法を受けたことのない患者であれば、再発腫瘍の局所切除実施後に術後放射線療法を検討すべきである。成人では術後密封小線源治療による患肢温存療法が評価済みであるが、小児では大々的な研究は実施されていない。四肢に肉腫が認められ以前に放射線療法を受けたことのある小児では、切断が唯一の治療法の選択肢となる場合もある。
滑膜肉腫の再燃を来した小児の治療成績に関して2件の研究の結果が発表された。1件の研究ではほとんどの患児に遠隔部位の再燃があり(44例中29例)[ 14 ]、もう一方の研究では、ほとんどの患児に局所再燃が認められた(37例中27例)。[ 15 ]遠隔再発は予後不良変数であった一方で、四肢への再燃のように、再燃時に腫瘍が切除可能であることが両試験において良好な治療成績に関連していた。
臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、現在実施されている全米および/または施設の臨床試験の例である:
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018.[PUBMED Abstract]
- Belal A, Salah E, Hajjar W, et al.: Pulmonary metastatectomy for soft tissue sarcomas: is it valuable? J Cardiovasc Surg (Torino) 42 (6): 835-40, 2001.[PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Prognostic factors for disease-specific survival after first relapse of soft-tissue sarcoma: analysis of 402 patients with disease relapse after initial conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys 57 (3): 739-47, 2003.[PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013.[PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007.[PUBMED Abstract]
- Le Cesne A, Cresta S, Maki RG, et al.: A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur J Cancer 48 (16): 3036-44, 2012.[PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Maki RG, et al.: Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: multicenter phase II and pharmacokinetic study. J Clin Oncol 23 (24): 5484-92, 2005.[PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Manola J, et al.: Phase II and pharmacokinetic study of ecteinascidin 743 in patients with progressive sarcomas of soft tissues refractory to chemotherapy. J Clin Oncol 22 (8): 1480-90, 2004.[PUBMED Abstract]
- Glade Bender JL, Lee A, Reid JM, et al.: Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children's oncology group phase I consortium report. J Clin Oncol 31 (24): 3034-43, 2013.[PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012.[PUBMED Abstract]
- Casanova M, Basso E, Magni C, et al.: Response to pazopanib in two pediatric patients with pretreated relapsing synovial sarcoma. Tumori 103 (1): e1-e3, 2017.[PUBMED Abstract]
- Tawbi HA, Burgess M, Bolejack V, et al.: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18 (11): 1493-1501, 2017.[PUBMED Abstract]
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012.[PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012.[PUBMED Abstract]
- Soole F, Maupain C, Defachelles AS, et al.: Synovial sarcoma relapses in children and adolescents: prognostic factors, treatment, and outcome. Pediatr Blood Cancer 61 (8): 1387-93, 2014.[PUBMED Abstract]
- 本要約の変更点(06/09/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
小児軟部肉腫に関する一般情報
小児腫瘍学グループ(COG)のARST0332試験でのリスクグループおよび治療割り付けについて図4が追加された(引用、参考文献75としてSpunt et al.)。
本文に、COG ARST0332試験について3つのリスクグループの記述が追加された。
本文に、COG ARST0332試験について治療グループの記述が追加された。
本文に以下の記述が追加された;COG ARST0332試験で用いられた化学療法には、6サイクルのイホスファミド、3週間ごとの1~3日目に静脈内投与および5サイクルのドキソルビシン、3週間ごとの1~2日目に静脈内投与が含まれ、順序は手術または放射線療法のタイミングに応じて調整された。
COG ARST0332試験の生存の結果をまとめた表3が追加された。
小児軟部肉腫に対する治療法選択肢の概要
本文に、リスクグループ、治療グループ、および転帰の結果の記述を含めてCOG ARST0332試験に関する記述が追加された(引用、参考文献1としてSpunt et al.)。
本文に以下の記述が追加された;放射線技術は正常組織の温存に影響を与える可能性がある;三次元原体照射療法と比較して、強度変調放射線療法では、皮膚および骨端(四肢肉腫に照射する場合)への放射線量を低下できる可能性がある(引用、参考文献24としてRao et al.)。
本文で以下の記述が改訂された;これはランダム化研究ではなかったが、2.6年後の結果から、高リスクで、肉眼的切除を受けた非転移性腫瘍で、放射線療法およびドキソルビシンとイホスファミドによる術後化学療法を受けた患者は5年イベントフリー生存(EFS)率が67.2%および全生存(OS)率が78%であったことが示された。また本文で以下の記述が改訂された;術前化学療法および放射線療法による治療を受けた転移病変患者では、推定5年EFS率は21.2%、OS率は35.5%であった。
新規診断小児軟部肉腫の治療
本文に、21歳未満の炎症性筋線維芽細胞性腫瘍患者37人を確認したドイツの研究のレビュー結果に関する記述が追加された(引用、参考文献89としてKube et al.および証拠レベル:3iiA)。
本文に、悪性末梢神経鞘腫瘍患者784人を確認したオランダのがん登録データのレトロスペクティブ解析の結果に関する記述が追加された(引用、参考文献154としてMartin et al.および証拠レベル:3iA)。
本文に、悪性末梢神経鞘腫瘍を有する21歳以下の患者を対象にしたEuropean Pediatric Soft Tissue Sarcoma Group(EpSSG)研究の結果に関する記述が追加された(引用、参考文献157としてvan Noesel et al.)。
本文に、類上皮肉腫を有する30歳未満の患者が登録されたCOGおよびEpSSGのプロスペクティブ臨床試験がレビューされた1件のレトロスペクティブ解析の結果に関する記述が追加された(引用、参考文献221としてSpunt et al.および証拠レベル:2A)。
本文に以下の記述が追加された;1件の試験において、進行した肉腫を有する患者が、アキシチニブおよびペムブロリズマブの併用で治療された。胞巣状軟部肉腫を有する12人の患者について、3ヵ月無増悪生存率は73%であった。評価可能な病変を有した患者11人中6人で、アキシチニブに対して部分奏効が得られた。
線維形成性小円形細胞腫瘍のセクションの治療のサブセクションは広範囲にわたって改訂された。
脈管腫瘍のサブセクションは包括的に見直された。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児軟部肉腫の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Childhood Soft Tissue Sarcoma Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/child-soft-tissue-treatment-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389361]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。