

ご利用について
医療専門家向けの本PDQがん情報要約では、小児急性骨髄性白血病とその他の骨髄性悪性疾患の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児急性骨髄性白血病(AML)に関する一般情報
-
小児および青年のがん患者の生存において、劇的な改善が達成されている。[ 1 ]1975年から2010年の間に、小児がんの死亡率は50%を超える低下を示した。急性骨髄白血病(AML)では、同じ期間に5年生存率が15歳未満の小児で20%未満から68%に、15~19歳の青年で20%未満から57%に増加した。[ 1 ]
小児における骨髄性白血病およびその他の骨髄悪性腫瘍の特徴
小児白血病の約20%が骨髄性であり、多種多様な造血器悪性腫瘍を呈する。[ 2 ]大多数の骨髄性白血病が急性であり、残りは慢性骨髄性白血病および若年性骨髄単球性白血病など、慢性および/または亜急性骨髄増殖性疾患が含まれる。骨髄異形成症候群は、小児における発生頻度が成人よりはるかに低く、ファンコニー貧血およびシュバックマン-ダイアモンド症候群のような先天性骨髄不全症候群から進展することがあるクローン性の前白血病状態がほぼ必ずみられる。
骨髄性白血病およびその他の骨髄悪性腫瘍の一般的特徴を以下に記す:
骨髄性悪性疾患に関連する病態
遺伝子異常(がん素因症候群)は、AMLの発症と関連する。一卵性双生児では、AMLの一致率が高い;ただし、これは遺伝リスクと関係しているのではなく、環境を共有することよりも、胎児発育段階で、一方の双生児が他方からの白血病細胞を拒絶できないことに関係していると考えられている。[ 15 ][ 16 ][ 17 ]小児白血病患者の二卵性双生児が約6歳までに白血病を発症するリスクは2~4倍と高いことが推定され、その後のリスクは一般集団と比べてそれほど大きくない。[ 18 ][ 19 ]
AMLの発症は、染色体の不均衡または不安定、DNA修復の欠損、サイトカイン受容体またはシグナル伝達経路活性化の変化、および蛋白合成の変化に起因するさまざまな遺伝性、後天性、および家族性症候群とも関連するとされている。[ 20 ][ 21 ]
AMLに対する非症候性の遺伝的感受性もまた研究されている。例えば、特異的なIKZF1多型のホモ接合性は、乳児AMLのリスク増加と関連するとされている。[ 23 ]
参考文献- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Roberts I, Alford K, Hall G, et al.: GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood 122 (24): 3908-17, 2013.[PUBMED Abstract]
- Zipursky A: Transient leukaemia--a benign form of leukaemia in newborn infants with trisomy 21. Br J Haematol 120 (6): 930-8, 2003.[PUBMED Abstract]
- Gamis AS, Smith FO: Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol 159 (3): 277-87, 2012.[PUBMED Abstract]
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.[PUBMED Abstract]
- Mundschau G, Gurbuxani S, Gamis AS, et al.: Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood 101 (11): 4298-300, 2003.[PUBMED Abstract]
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.[PUBMED Abstract]
- Homans AC, Verissimo AM, Vlacha V: Transient abnormal myelopoiesis of infancy associated with trisomy 21. Am J Pediatr Hematol Oncol 15 (4): 392-9, 1993.[PUBMED Abstract]
- Gamis AS, Alonzo TA, Gerbing RB, et al.: Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood 118 (26): 6752-9; quiz 6996, 2011.[PUBMED Abstract]
- Hasle H, Niemeyer CM: Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154 (2): 185-95, 2011.[PUBMED Abstract]
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.[PUBMED Abstract]
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.[PUBMED Abstract]
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.[PUBMED Abstract]
- Zuelzer WW, Cox DE: Genetic aspects of leukemia. Semin Hematol 6 (3): 228-49, 1969.[PUBMED Abstract]
- Miller RW: Persons with exceptionally high risk of leukemia. Cancer Res 27 (12): 2420-3, 1967.[PUBMED Abstract]
- Inskip PD, Harvey EB, Boice JD, et al.: Incidence of childhood cancer in twins. Cancer Causes Control 2 (5): 315-24, 1991.[PUBMED Abstract]
- Kurita S, Kamei Y, Ota K: Genetic studies on familial leukemia. Cancer 34 (4): 1098-101, 1974.[PUBMED Abstract]
- Greaves M: Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol 7 (3): 233-45, 2003.[PUBMED Abstract]
- Puumala SE, Ross JA, Aplenc R, et al.: Epidemiology of childhood acute myeloid leukemia. Pediatr Blood Cancer 60 (5): 728-33, 2013.[PUBMED Abstract]
- West AH, Godley LA, Churpek JE: Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci 1310: 111-8, 2014.[PUBMED Abstract]
- Tawana K, Wang J, Renneville A, et al.: Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126 (10): 1214-23, 2015.[PUBMED Abstract]
- Ross JA, Linabery AM, Blommer CN, et al.: Genetic variants modify susceptibility to leukemia in infants: a Children's Oncology Group report. Pediatr Blood Cancer 60 (1): 31-4, 2013.[PUBMED Abstract]
- 小児骨髄性悪性疾患の分類
-
小児AMLのFrench-American-British(FAB)分類システム
急性骨髄性白血病(AML)に対する初めての包括的な形態学-組織化学的分類システムが、FAB Cooperative Groupによって開発された。[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ]この分類システムは後述の世界保健機関(WHO)分類システムに置き換えられているが、主として形態学および系列マーカーの免疫組織化学的検出に基づいて、AMLを主要な亜型に分類した。
AMLの主要な亜型には以下のものがある:
このほか、AMLのきわめてまれな亜型には、急性好酸球性白血病および急性好塩基球性白血病がある。
FAB分類は、後述のWHO分類に置き換えられたが、他に特定されないAML(AML、NOS)のWHOサブ分類の基礎になっているため、依然として重要である。
小児AMLに対する世界保健機関(WHO)分類システム
2001年にWHOは、細胞遺伝学的な診断情報を取り入れるとともに、転帰との相関において信頼性を高めた新しい分類システムを提唱した。この分類システムによると、t(8;21)、inv(16)、t(15;17)、またはKMT2A(MLL)転座を認める患者は、小児AML症例のほぼ半数を構成する集団で、頻発性細胞遺伝学的異常を伴うAMLとして分類された。この分類システムでは、AMLの診断に必要な白血病性芽球の骨髄内の割合についても30%から20%に引き下げられた;頻発性細胞遺伝学的異常を認める患者では、AML患者とみなすために最低限必要な芽球数を満たす必要がないように、さらなる明確化が行われた。[ 8 ][ 9 ][ 10 ]
2008年にWHOは、AML分類と関連する細胞遺伝学的異常の数を拡大し、特異的遺伝子変異(CEBPAおよびNPM)を初めてWHO分類システムに含めた。[ 11 ]2016年にWHO分類が改訂され、白血病の診断、予後、および治療にきわめて重要な白血病バイオマーカーに関して蓄積された知識が取り入れられた。[ 12 ]遺伝的分類、エピジェネティック分類、プロテオミック分類、および免疫表現型分類を目指して得られつつある技術によって、AMLの分類は確実に進化を続けており、臨床家および研究者にとって情報的に有益な予後的および生物学的ガイドラインが提供されると予想される。
AMLおよび関連新生物の2016年WHO分類
細胞系列があいまいな急性白血病の2016年WHO分類
AMLと急性リンパ芽球性白血病(ALL)の両方の特徴を有する急性白血病グループ、つまり細胞系列があいまいな急性白血病に関するWHO分類システムを表1に要約する。[ 13 ][ 14 ]混合表現型急性白血病(MPAL)の診断に対して細胞系列を割り当てるための基準が表2で示されている。[ 12 ]
表1.造血器およびリンパ組織腫瘍に関する世界保健機関分類による細胞系列があいまいな急性白血病a 疾患 定義 NOS = 他に特定されない。 aBéné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias!Haematologica 94 (7): 891-3, 2009.[ 13 ]出典:Haematologica/the Hematology Journal website http://www.haematologica.org. 急性未分化型白血病 リンパ系または骨髄系のいずれかに特異的とみなされるマーカーの発現が認められない急性白血病 BCR-ABL1;つまりt(9;22)(q34;q11.2)を有する混合表現型急性白血病 混合表現型急性白血病の診断基準を満たし、(9;22)転座、つまりBCR-ABL1再構成も芽球に認められる急性白血病 t(v;11q23)のKMT2A(MLL)再構成を伴う混合表現型急性白血病 混合表現型急性白血病の診断基準を満たし、KMT2A遺伝子を巻き込んだ転座も芽球に認められる急性白血病 混合表現型急性白血病、B細胞性/骨髄性、NOS B細胞系および骨髄系の両方に割り当てる診断基準を満たし、BCR-ABL1またはKMT2Aを巻き込んだ遺伝子異常が芽球に認められない急性白血病 混合表現型急性白血病、T細胞性/骨髄性、NOS T細胞系および骨髄系の両方に割り当てる診断基準を満たし、BCR-ABL1またはKMT2Aを巻き込んだ遺伝子異常が芽球に認められない急性白血病 混合表現型急性白血病、B細胞性/骨髄性、NOS-まれなタイプ B細胞系およびT細胞系に割り当てる診断基準をいずれも満たす急性白血病 他の細胞系列があいまいな白血病 ナチュラルキラー細胞リンパ芽球性白血病/リンパ腫 表2.骨髄腫瘍と急性白血病の2016年世界保健機関(WHO)分類改訂版による混合表現型急性白血病に対する細胞系列の割り当て基準a 細胞系列 基準 a出典:Arber et al.[ 12 ] b「強い」とは、標本内の正常なBまたはT細胞と比較して同等かまたはより明るいものと定義された。 骨髄性細胞系列 ミエロペルオキシダーゼ(フローサイトメトリー、免疫組織化学、または細胞化学);あるいは単球分化(次のうち少なくとも2つ:非特異的エステラーゼ細胞化学、CD11c、CD14、CD64、リゾチーム) T細胞系列 強いb細胞質CD3(CD3イプシロン鎖に対する抗体を伴う);または細胞表面のCD3 B細胞系列 強いbCD19と次のうち少なくとも1つ以上が強く発現している:CD79a、細胞質CD22、またはCD10;あるいは弱いCD19と次のうち少なくとも2つ以上が強く発現している:CD79a、細胞質CD22、またはCD10 混合表現型の白血病では、以下のようなさまざまな所見がみられることがある:
- 二細胞系列白血病で、通常は1つがリンパ系で1つが骨髄系の2つの異なった細胞集団がみられる。
- 二重表現型白血病で、個々の芽球がリンパ系と骨髄系の両方の特徴を示す。
二重表現型の症例は混合表現型白血病の大多数を占める。[ 15 ] TEL-AML1融合を認めない骨髄系B細胞二重表現型白血病では、前駆B細胞ALL患者と比べて完全寛解(CR)率が低く、イベントフリー生存(EFS)が有意に不良である。[ 15 ]一部の研究によると、二重表現型白血病患者は、骨髄系とは対照的にリンパ系の治療レジメンを用いることで予後がよい可能性が示唆される。[ 16 ][ 17 ][ 18 ][ 19 ];[ 20 ][証拠レベル:3iiiA]国際ベルリン-フランクフルト-ミュンスター(BFM)グループによる大規模なレトロスペクティブ研究で、ALL向けレジメンによる初回治療に伴い、AML向けレジメンまたはALL/AMLの併用レジメンと比較して優れた転帰が得られ、特にCD19陽性例または他のリンパ系抗原発現例で顕著であった。この研究で、初回CR期での造血幹細胞移植(HSCT)は、治療1ヵ月後に骨髄病変が残存する形態学的証拠(芽球が5%以上)がある場合をおそらく除いて、有益でなかった。[ 19 ]
骨髄異形成症候群に関する骨髄および末梢血所見のWHO分類
骨髄異形成症候群(MDS)のFAB分類を小児に完全に適用することはできなかった。[ 21 ][ 22 ]従来のMDS分類システムでは、以下の因子の有無に基づいて、いくつかの異なったカテゴリーに分類している:[ 22 ][ 23 ][ 24 ][ 25 ]
2008年にMDSおよび骨髄増殖性障害(MPD)に対する修正分類法がWHOから公表され、小児MDSおよびMPDに焦点を当てたサブセクションが追加された。[ 26 ]骨髄異形成および骨髄増殖性疾患のWHO分類に対するこの小児アプローチは、2003年に最初に提案された。[ 10 ]2016年WHO分類改訂版では、特異的系列(貧血、血小板減少症、または好中球減少症) への焦点が削除されており、新たに単一系列 vs 複数系列における異形成を有する症例が区別されている。過剰芽球を伴うMDS(MDS-EB)の分類には、以前は過剰芽球を伴う不応性貧血(RAEB)または白血病移行期にあるRAEB(RAEB-T)に分類されていた小児症例が現在は含まれている。[ 27 ]小児不応性血球減少症の分類は、依然として暫定的疾患単位である。2008年のWHO分類スキームに従ったMDSに対する骨髄および末梢血の所見を表3および表4に要約する。[ 12 ][ 26 ]MDS-EBが通常的にAMLに伴う再発性細胞遺伝学的異常と関連している場合は、AMLの診断を行って、患者にはそれに応じた治療を実施すべきである。
MDSと外見が類似した異形成および/または血球減少症の反応性要因との鑑別は困難であると指摘されている。一般に、ある細胞系列で10%を超える異形成の所見は、MDSの診断基準となるが、2016年WHOガイドラインでは、クローン性ではなく反応性病因が10%を超える異形成にみられる可能性があり、特に異形成がわずかな場合および/または単一系列に限定される場合は、除外すべきであると注意している。[ 12 ]
MDSの成人患者において、AMLへの進行リスクおよび転帰を決定するために、International Prognostic Scoring Systemが用いられる。このシステムがMDSまたは若年性骨髄単球性白血病(JMML)の小児に適用されたとき、MDSにおいては5%未満の芽球数および100 x 109/Lを超える血小板数だけがより良好な生存と関連し、JMMLにおいては40 x 109/Lを超える血小板数がより良好な治療成績を予測した。[ 28 ]これらの結果から、小児におけるMDSおよびJMMLは成人型MDSとは明らかに異なる疾患であることが示唆される。
小児MDSはいくつかの一般的なカテゴリーに分類でき、それぞれが以下のように特徴的な臨床的および生物学的特性を有する:[ 27 ]
小児原発性MDSのゲノム特性化によって、選択された遺伝子における変化により定義される特異的サブセットが同定されている(MDSに関する詳しい情報については、本要約の分子的異常のサブセクションを参照のこと)。例えば、GATA2[ 29 ]またはSAMD9/SAMD9L[ 30 ][ 31 ][ 32 ]のいずれかにおける生殖細胞変異は、7番染色体の全部または一部が欠失した小児に特によくみられる。ゲノム特性化によって、小児における原発性MDSは分子レベルで成人のMDSと異なることも示されている。[ 31 ][ 33 ]
表3.骨髄異形成症候群(MDS)の骨髄および末梢血の所見の世界保健機関(WHO)分類a MDSのタイプ 骨髄 末梢血 a出典:Arber et al.[ 12 ] b汎血球減少が認められる症例はMDS-Uに分類されることに注意すること。 c骨髄中の骨髄芽球が5%未満でも、末梢血中の骨髄芽球が2~4%であれば、診断はMDS-EB-1である。 d以下の基準のいずれかに該当すれば、MDS-EB-2と診断すべきである:骨髄中の芽球が10~19%、末梢血中の芽球が5~19%、またはアウエル小体の存在。 eMDSにおける頻発性染色体異常:不均衡型:+8、-7またはdel(7q)、-5またはdel(5q)、del(20q)、-Y、i(17q)またはt(17p)、-13またはdel(13q)、del(11q)、del(12p)またはt(12p)、del(9q)、idic(X)(q13);均衡型:t(11;16)(q23;p13.3)、t(3;21)(q26.2;q22.1)、t(1;3)(p36.3;q21.2)、t(2;11)(p21;q23)、inv(3)(q21q26.2)、t(6;9)(p23;q34)。WHO分類では、原因不明の持続性の血球減少症が認められる状況でのこれらの染色体異常の存在は、形態的特徴が観察されない場合のMDSの推定診断を支持するものとして考えるべきであると指摘されている。 f小児MDS(小児不応性血球減少症[RCC]-暫定的疾患単位)の診断基準には以下を含む:1)骨髄芽球が5%未満、末梢血芽球が2%未満、環状鉄芽球を認めない1~3の細胞系列の持続性血球減少症、および2)1~3の細胞系列における異形成の変化が存在すること。 単一の細胞系列に異形成を伴うMDS 単細胞系列異形成(unilineage dysplasia): 1つの骨髄性細胞系列において10%以上 1~2系列の血球減少症b 芽球が5%未満 芽球が1%未満c 環状鉄芽球が15%未満 環状鉄芽球を伴うMDS(MDS-RS) 赤血球異形成のみ 芽球が5%未満 芽球が認められない 環状鉄芽球が15%以上 多系列細胞異形成(multilineage dysplasia)を伴うMDS 2つ以上の骨髄性細胞系列において10%以上の細胞の異形成 1~3系列の細胞減少症 芽球が5%未満 芽球(認められない、または1%未満)c 環状鉄芽球で±15% アウエル小体が認められない アウエル小体が認められない <1×109/Lの単球 過剰芽球を伴うMDS-1(MDS-EB-1) 単一細胞系列または多系列細胞異形成 血球減少症 芽球が5~9%c 芽球が5%未満c アウエル小体が認められない アウエル小体が認められない <1×109/Lの単球 過剰芽球を伴うMDS-2(MDS-EB-2) 単一細胞系列または多系列細胞異形成 血球減少症 芽球が10~19%d 芽球が5~19%d アウエル小体 ±d アウエル小体 ±d <1×109/Lの単球 孤立性del(5q)を伴うMDS 巨核球が正常~高値(低分葉核:hypolobulated nucleiを伴う) 貧血 芽球が5%未満 芽球(認められない、または1%未満) アウエル小体が認められない 血小板数が正常~高値 孤立性del(5q) MDS-未分類(MDS-U) 1つ以上の骨髄性細胞系列において10%未満の細胞の異形成 血球減少症 MDSの診断に関連した細胞遺伝学的異常e 芽球が1%以下c 芽球が5%未満 暫定的疾患単位:小児不応性血球減少症(RCC)f 詳しい情報については、表4を参照のこと。 表4.小児骨髄異形成症候群(MDS)(暫定的疾患単位:小児不応性血球減少症[RCC])の最小診断基準の定義a 赤血球性細胞系列 骨髄性細胞系列 巨核球性細胞系列 a出典:Baumann et al.[ 34 ] b小児RCCにおける骨髄はしばしば低形成であるため、骨髄トレフィン/生検が必要になることがある。 c特徴としては、異常な核分葉、多核細胞、核架橋(nuclear bridge)の存在が挙げられる。 d偽Pelger-Huet核異常、低または無顆粒の細胞質、巨大な杆状核球の存在。 e巨核球の大きさは不定であり、しばしば丸いまたは分離した核を有する;巨核球が認められないからといって、RCCの診断が除外されるわけではない。 骨髄穿刺b 10%以上の赤血球前駆細胞における異形成および/または巨赤芽球様変化c 10%以上の顆粒球前駆細胞および好中球における異形成 小型巨核球 + 他の異形成の特徴e 芽球が5%未満d 骨髄生検 赤血球前駆細胞の存在 追加の基準なし 小型巨核球 + 他の異形成の特徴e 前赤芽球の増加 免疫組織化学検査でCD61およびCD41が陽性 核分裂像数の増加 末梢血 10%以上の好中球における異形成 芽球が2%未満 参考文献- Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 33 (4): 451-8, 1976.[PUBMED Abstract]
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 103 (4): 620-5, 1985.[PUBMED Abstract]
- Bennett JM, Catovsky D, Daniel MT, et al.: Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-American-British Cooperative Group. Ann Intern Med 103 (3): 460-2, 1985.[PUBMED Abstract]
- Bennett JM, Catovsky D, Daniel MT, et al.: A variant form of hypergranular promyelocytic leukaemia (M3) Br J Haematol 44 (1): 169-70, 1980.[PUBMED Abstract]
- Cheson BD, Bennett JM, Kopecky KJ, et al.: Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 21 (24): 4642-9, 2003.[PUBMED Abstract]
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO) Br J Haematol 78 (3): 325-9, 1991.[PUBMED Abstract]
- Kaleem Z, White G: Diagnostic criteria for minimally differentiated acute myeloid leukemia (AML-M0). Evaluation and a proposal. Am J Clin Pathol 115 (6): 876-84, 2001.[PUBMED Abstract]
- Vardiman JW, Harris NL, Brunning RD: The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100 (7): 2292-302, 2002.[PUBMED Abstract]
- Jaffe ES, Harris NL, Stein H, et al., eds.: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2001. World Health Organization Classification of Tumours, 3.[PUBMED Abstract]
- Hasle H, Niemeyer CM, Chessells JM, et al.: A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 17 (2): 277-82, 2003.[PUBMED Abstract]
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 110-23.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[PUBMED Abstract]
- Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 150-5.[PUBMED Abstract]
- Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.[PUBMED Abstract]
- Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.[PUBMED Abstract]
- Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.[PUBMED Abstract]
- Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.[PUBMED Abstract]
- Hrusak O, de Haas V, Stancikova J, et al.: International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood 132 (3): 264-276, 2018.[PUBMED Abstract]
- Orgel E, Alexander TB, Wood BL, et al.: Mixed-phenotype acute leukemia: A cohort and consensus research strategy from the Children's Oncology Group Acute Leukemia of Ambiguous Lineage Task Force. Cancer 126 (3): 593-601, 2020.[PUBMED Abstract]
- Bennett JM, Catovsky D, Daniel MT, et al.: Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 51 (2): 189-99, 1982.[PUBMED Abstract]
- Mandel K, Dror Y, Poon A, et al.: A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol 24 (7): 596-605, 2002.[PUBMED Abstract]
- Bennett JM: World Health Organization classification of the acute leukemias and myelodysplastic syndrome. Int J Hematol 72 (2): 131-3, 2000.[PUBMED Abstract]
- Head DR: Proposed changes in the definitions of acute myeloid leukemia and myelodysplastic syndrome: are they helpful? Curr Opin Oncol 14 (1): 19-23, 2002.[PUBMED Abstract]
- Nösslinger T, Reisner R, Koller E, et al.: Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98 (10): 2935-41, 2001.[PUBMED Abstract]
- Brunning RD, Porwit A, Orazi A, et al.: Myelodysplastic syndromes/neoplasms overview. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 88-93.[PUBMED Abstract]
- Wlodarski MW, Sahoo SS, Niemeyer CM: Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol Oncol Clin North Am 32 (4): 729-743, 2018.[PUBMED Abstract]
- Hasle H, Baumann I, Bergsträsser E, et al.: The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia 18 (12): 2008-14, 2004.[PUBMED Abstract]
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.[PUBMED Abstract]
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.[PUBMED Abstract]
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.[PUBMED Abstract]
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.[PUBMED Abstract]
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.[PUBMED Abstract]
- Baumann I, Niemeyer CM, Bennett JM, et al.: Childhood myelodysplastic syndrome. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 104-7.[PUBMED Abstract]
- 小児AMLの組織化学的、免疫表現型の、および分子的評価
-
組織化学的評価
急性骨髄性白血病(AML)の小児に対する治療は、急性リンパ芽球性白血病(ALL)に対する治療とは大きく異なる。そのため、AMLとALLを鑑別することがきわめて重要である。急性白血病の診断を確定するには、急性白血病患児の骨髄標本に特定の細胞化学染色を行うことが役立つ。最も一般的に用いられている染色法には、ミエロペルオキシダーゼ染色、過ヨウ素酸シッフ染色、ズダンブラックB染色、およびエステラーゼ染色がある。ほとんどの症例において、このような組織化学的染色による染色パターンから、AMLが急性骨髄単球性白血病(AMML)およびALLと鑑別される(表5を参照のこと)。組織化学的染色はフローサイトメトリーを用いた免疫表現型検査にほとんど置き換えられている。
表5.組織化学的染色パターンa M0 AML、APL(M1-M3) AMML(M4) AMoL(M5) AEL(M6) AMKL(M7) ALL AEL = 急性赤白血病;ALL = 急性リンパ芽球性白血病;AML = 急性骨髄性白血病;AMKL = 急性巨核球性白血病;AMML = 急性骨髄単球性白血病;AMoL = 急性単球性白血病;APL = 急性前骨髄球性白血病;PAS = 過ヨウ素酸シッフ。 a AMLの形態学-組織化学的分類システムに関する詳しい情報については、本要約の小児急性骨髄性白血病のフランス-アメリカ-イギリス(FAB)分類のセクションを参照のこと。 b これらの反応はフッ化物で抑制される。 ミエロペルオキシダーゼ - + + - - - - 非特異的エステラーゼ クロロアセテート - + + ± - - - アルファ-ナフトール-アセテート - - + b + b - ± b - ズダンブラックB - + + - - - - PAS - - ± ± + - + 免疫表現型の評価
モノクローナル抗体を用いてAML細胞の細胞表面抗原を明らかにすることは、組織学的診断を補強するのに有用である。最初の診断用精密検査の時点で、AMLをALLおよび細胞系列があいまいな急性白血病と区別するのに有用となる細胞系列に特異的な一連のTリンパ球マーカーおよびBリンパ球マーカーとともに、AML細胞上の抗原を検出する種々の細胞系列に特異的なモノクローナル抗体を用いるべきである。AMLで相対的に細胞系列特異的に発現しているさまざまなクラスター決定因子(CD)蛋白には、CD33、CD13、CD14、CDw41(または血小板抗糖蛋白IIb/IIIa)、CD15、CD11B、CD36、および抗グリコホリンAがある。細胞系列関連のBリンパ球抗原であるCD10、CD19、CD20、CD22、およびCD24は、AML症例の10~20%に認められる場合があるが、通常は単クローン性表面免疫グロブリンおよび細胞質内免疫グロブリンの重鎖はみられない;同様に、CD2、CD3、CD5、およびCD7といった細胞系列関連のTリンパ球抗原は、AML症例の20~40%に認められる。[ 1 ][ 2 ][ 3 ]AML細胞によるリンパ球関連抗原の異常発現は比較的よくみられるが、一般に予後的な意義はない。[ 1 ][ 2 ]
以下のAMLのFrench-American-British(FAB)分類亜型を区別する上でも免疫表現型検査が有用なことがある:
小児における急性白血病症例の5%未満は細胞系列があいまいで、骨髄系およびリンパ系の両細胞系列の特徴を発現している。[ 8 ][ 9 ][ 10 ]これらの症例は、免疫表現型検査および組織化学検査によって主要細胞系例が決定できない点で骨髄系の共発現を伴うALLと区別される。細胞系列があいまいな白血病の定義は、現在では研究者のほとんどがEuropean Group for the Immunological Characterization of Leukemias(EGIL)により確立された基準、またはより厳密な世界保健機関(WHO)基準を採用しているが、研究によってさまざまである。[ 11 ][ 12 ][ 13 ]WHO分類では、骨髄性細胞系列と確定するにはミエロペルオキシダーゼ(MPO)の存在が必要である。EGIL分類ではこれは適用されない。2016年WHO分類改訂版によると、一部の症例で他の点では古典的B細胞ALL免疫表現型を示す白血病は低強度のMPOも発現し、他の骨髄性の特徴がみられないことがあることも示しており、この知見の臨床的意義は不明であるため、これらの症例を混合表現型急性白血病(MPAL)に指定する前に注意すべきである。[ 14 ]
分子的評価
急性骨髄性白血病の分子的特徴
小児および成人AMLの包括的分子プロファイリングにより、AMLは年齢層間で共通性および明確な差の両方を示す疾患であることが明らかにされている。[ 15 ][ 16 ]
白血病芽球細胞の遺伝子解析(従来の細胞遺伝学的方法および分子遺伝学的方法の両方を使用)は、染色体異常および分子的異常がいずれも重要な診断的および予後的マーカーであることから、AMLの小児に対して実施される。[ 17 ][ 18 ][ 19 ][ 20 ][ 21 ][ 22 ][ 23 ]クローン性の染色体異常は、約75%のAML患児の芽球に認められており、予後と治療の両方で重要な亜型を明らかにする上で有用である。
分子的異常の発見はまた、リスク層別化および治療割り当てに役立つ可能性がある。例えば、NPMおよびCEBPAの変異は、良好な転帰に関連する一方で、FLT3の特定の変異では高い再燃リスクが予想されており、後者の変異の同定により標的療法が可能になる場合がある。[ 24 ][ 25 ][ 26 ][ 27 ]
骨髄腫瘍と急性白血病の2016年世界保健機関(WHO)分類改訂版によると、小児AMLにおける頻発性染色体転座は一意的である可能性、または保有率が成人AMLと異なる可能性が強調されている。[ 14 ]従来の染色体分析で発見される小児AMLの染色体転座および潜在性のもの(蛍光in situハイブリダイゼーションまたは分子学的技術によってのみ同定される)は、成人より高い発生率で生じる。これらの頻発性転座を表6に要約する。[ 14 ]表6では、最下部の3行にAML小児で観察される頻発性転座で相対的に多くみられるその他の転座も示している。[ 21 ][ 22 ][ 28 ]
表6.小児急性骨髄性白血病(AML)で多くみられる染色体転座 遺伝子融合産物 染色体転座 小児AMLにおける保有率(%) a潜在性染色体転座。 KMT2A(MLL)転座 11q23.3 25.0 NUP98-NSD1a t(5;11)(q35.3;p15.5) 7.0 CBFA2T3-GLIS2a inv(16)(p13.3;q24.3) 3.0 NUP98-KDM5A4a t(11;12)(p15.5;p13.5) 3.0 DEK-NUP214 t(6;9)(p23;q34.1) 1.7 RBM15(OTT)-MKL1(MAL) t(1;22)(p13.3;q13.1) 0.8 MNX1-ETV6 t(7;12)(q36.3;p13.2) 0.8 KAT6A-CREBBP t(8;16)(p11.2;p13.3) 0.5 RUNX1-RUNX1T1 t(8;21)(q22;q22) 13–14 CBFB-MYH11 inv(16)(p13.1;q22)またはt(16;16)(p13.1;q22) 4–9 PML-RARA t(15;17)(q24;q21) 6–11 小児AML症例におけるゲノムの全体像は、診断から再燃までに変化し、診断時に検出可能な変異が再燃時に減少して、逆に再燃時に新たな変異が現れることがある。診断時および再燃時に塩基配列データが得られた20症例の研究における重要な知見は、診断時の多様体アレル頻度が再燃時の変異の持続と強く相関することであった。[ 29 ]多様体アレル頻度が0.4を超える診断時変異の約90%は再燃まで持続するのに対して、多様体アレル頻度が0.2未満ではわずか28%である(P < 0.001)。この観察結果は、FLT3-ITD変異が存在すると、FLT3-ITDアレル比が高い場合にのみ不良な予後が予想されたことを示す過去の結果と一致している。
特定の再発性の細胞遺伝学的および分子的異常について、以下に簡潔に示す。これらの異常は、臨床用途で患者が予後良好か予後不良かを特定できる異常別に記載しており、それ以外の異常はその後に記載している。骨髄腫瘍と急性白血病の2016年WHO分類改訂版による命名法が重要な疾患単位に組み込まれている。
良好な予後に関係する分子的異常
良好な予後に関係する分子的異常には以下のものがある:
予後不良に関連する分子的異常
予後不良に関連している分子的異常には以下のものがある:
小児AMLに観察される他の分子的異常
小児AMLに認められる他の分子的異常には以下のものがある:
参考文献- Kuerbitz SJ, Civin CI, Krischer JP, et al.: Expression of myeloid-associated and lymphoid-associated cell-surface antigens in acute myeloid leukemia of childhood: a Pediatric Oncology Group study. J Clin Oncol 10 (9): 1419-29, 1992.[PUBMED Abstract]
- Smith FO, Lampkin BC, Versteeg C, et al.: Expression of lymphoid-associated cell surface antigens by childhood acute myeloid leukemia cells lacks prognostic significance. Blood 79 (9): 2415-22, 1992.[PUBMED Abstract]
- Dinndorf PA, Andrews RG, Benjamin D, et al.: Expression of normal myeloid-associated antigens by acute leukemia cells. Blood 67 (4): 1048-53, 1986.[PUBMED Abstract]
- Zhou Y, Jorgensen JL, Wang SA, et al.: Usefulness of CD11a and CD18 in flow cytometric immunophenotypic analysis for diagnosis of acute promyelocytic leukemia. Am J Clin Pathol 138 (5): 744-50, 2012.[PUBMED Abstract]
- Paietta E, Goloubeva O, Neuberg D, et al.: A surrogate marker profile for PML/RAR alpha expressing acute promyelocytic leukemia and the association of immunophenotypic markers with morphologic and molecular subtypes. Cytometry B Clin Cytom 59B (1): 1-9, 2004.[PUBMED Abstract]
- Lin P, Hao S, Medeiros LJ, et al.: Expression of CD2 in acute promyelocytic leukemia correlates with short form of PML-RARalpha transcripts and poorer prognosis. Am J Clin Pathol 121 (3): 402-7, 2004.[PUBMED Abstract]
- Creutzig U, Ritter J, Schellong G: Identification of two risk groups in childhood acute myelogenous leukemia after therapy intensification in study AML-BFM-83 as compared with study AML-BFM-78. AML-BFM Study Group. Blood 75 (10): 1932-40, 1990.[PUBMED Abstract]
- Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.[PUBMED Abstract]
- Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.[PUBMED Abstract]
- Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.[PUBMED Abstract]
- Bene MC, Castoldi G, Knapp W, et al.: Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 9 (10): 1783-6, 1995.[PUBMED Abstract]
- Vardiman JW, Thiele J, Arber DA, et al.: The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114 (5): 937-51, 2009.[PUBMED Abstract]
- Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 150-5.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.[PUBMED Abstract]
- Bolouri H, Farrar JE, Triche T, et al.: The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24 (1): 103-112, 2018.[PUBMED Abstract]
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.[PUBMED Abstract]
- Grimwade D, Walker H, Oliver F, et al.: The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood 92 (7): 2322-33, 1998.[PUBMED Abstract]
- Gilliland DG: Targeted therapies in myeloid leukemias. Ann Hematol 83 (Suppl 1): S75-6, 2004.[PUBMED Abstract]
- Avivi I, Rowe JM: Prognostic factors in acute myeloid leukemia. Curr Opin Hematol 12 (1): 62-7, 2005.[PUBMED Abstract]
- Harrison CJ, Hills RK, Moorman AV, et al.: Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol 28 (16): 2674-81, 2010.[PUBMED Abstract]
- von Neuhoff C, Reinhardt D, Sander A, et al.: Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol 28 (16): 2682-9, 2010.[PUBMED Abstract]
- Grimwade D, Hills RK, Moorman AV, et al.: Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116 (3): 354-65, 2010.[PUBMED Abstract]
- Brown P, McIntyre E, Rau R, et al.: The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 110 (3): 979-85, 2007.[PUBMED Abstract]
- Hollink IH, Zwaan CM, Zimmermann M, et al.: Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 23 (2): 262-70, 2009.[PUBMED Abstract]
- Ho PA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 113 (26): 6558-66, 2009.[PUBMED Abstract]
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.[PUBMED Abstract]
- Struski S, Lagarde S, Bories P, et al.: NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia 31 (3): 565-572, 2017.[PUBMED Abstract]
- Farrar JE, Schuback HL, Ries RE, et al.: Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res 76 (8): 2197-205, 2016.[PUBMED Abstract]
- Rubnitz JE, Raimondi SC, Halbert AR, et al.: Characteristics and outcome of t(8;21)-positive childhood acute myeloid leukemia: a single institution's experience. Leukemia 16 (10): 2072-7, 2002.[PUBMED Abstract]
- Tallman MS, Hakimian D, Shaw JM, et al.: Granulocytic sarcoma is associated with the 8;21 translocation in acute myeloid leukemia. J Clin Oncol 11 (4): 690-7, 1993.[PUBMED Abstract]
- Mrózek K, Heerema NA, Bloomfield CD: Cytogenetics in acute leukemia. Blood Rev 18 (2): 115-36, 2004.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.[PUBMED Abstract]
- Raimondi SC, Chang MN, Ravindranath Y, et al.: Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study-POG 8821. Blood 94 (11): 3707-16, 1999.[PUBMED Abstract]
- Lie SO, Abrahamsson J, Clausen N, et al.: Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down's syndrome: results of NOPHO-AML trials. Br J Haematol 122 (2): 217-25, 2003.[PUBMED Abstract]
- Klein K, Kaspers G, Harrison CJ, et al.: Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Münster Study Group. J Clin Oncol 33 (36): 4247-58, 2015.[PUBMED Abstract]
- Larson RA, Williams SF, Le Beau MM, et al.: Acute myelomonocytic leukemia with abnormal eosinophils and inv(16) or t(16;16) has a favorable prognosis. Blood 68 (6): 1242-9, 1986.[PUBMED Abstract]
- Duployez N, Marceau-Renaut A, Boissel N, et al.: Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127 (20): 2451-9, 2016.[PUBMED Abstract]
- Faber ZJ, Chen X, Gedman AL, et al.: The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 48 (12): 1551-1556, 2016.[PUBMED Abstract]
- Noort S, Zimmermann M, Reinhardt D, et al.: Prognostic impact of t(16;21)(p11;q22) and t(16;21)(q24;q22) in pediatric AML: a retrospective study by the I-BFM Study Group. Blood 132 (15): 1584-1592, 2018.[PUBMED Abstract]
- Jahn N, Agrawal M, Bullinger L, et al.: Incidence and prognostic impact of ASXL2 mutations in adult acute myeloid leukemia patients with t(8;21)(q22;q22): a study of the German-Austrian AML Study Group. Leukemia 31 (4): 1012-1015, 2017.[PUBMED Abstract]
- Yamato G, Shiba N, Yoshida K, et al.: ASXL2 mutations are frequently found in pediatric AML patients with t(8;21)/ RUNX1-RUNX1T1 and associated with a better prognosis. Genes Chromosomes Cancer 56 (5): 382-393, 2017.[PUBMED Abstract]
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed March 16, 2020.[PUBMED Abstract]
- Mistry AR, Pedersen EW, Solomon E, et al.: The molecular pathogenesis of acute promyelocytic leukaemia: implications for the clinical management of the disease. Blood Rev 17 (2): 71-97, 2003.[PUBMED Abstract]
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.[PUBMED Abstract]
- Grimwade D, Lo Coco F: Acute promyelocytic leukemia: a model for the role of molecular diagnosis and residual disease monitoring in directing treatment approach in acute myeloid leukemia. Leukemia 16 (10): 1959-73, 2002.[PUBMED Abstract]
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.[PUBMED Abstract]
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.[PUBMED Abstract]
- Grimwade D, Biondi A, Mozziconacci MJ, et al.: Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood 96 (4): 1297-308, 2000.[PUBMED Abstract]
- Falini B, Martelli MP, Bolli N, et al.: Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood 108 (6): 1999-2005, 2006.[PUBMED Abstract]
- Falini B, Mecucci C, Tiacci E, et al.: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352 (3): 254-66, 2005.[PUBMED Abstract]
- Döhner K, Schlenk RF, Habdank M, et al.: Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106 (12): 3740-6, 2005.[PUBMED Abstract]
- Verhaak RG, Goudswaard CS, van Putten W, et al.: Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106 (12): 3747-54, 2005.[PUBMED Abstract]
- Schnittger S, Schoch C, Kern W, et al.: Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106 (12): 3733-9, 2005.[PUBMED Abstract]
- Schlenk RF, Döhner K, Krauter J, et al.: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358 (18): 1909-18, 2008.[PUBMED Abstract]
- Gale RE, Green C, Allen C, et al.: The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111 (5): 2776-84, 2008.[PUBMED Abstract]
- Cazzaniga G, Dell'Oro MG, Mecucci C, et al.: Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 106 (4): 1419-22, 2005.[PUBMED Abstract]
- Balgobind BV, Hollink IH, Arentsen-Peters ST, et al.: Integrative analysis of type-I and type-II aberrations underscores the genetic heterogeneity of pediatric acute myeloid leukemia. Haematologica 96 (10): 1478-87, 2011.[PUBMED Abstract]
- Staffas A, Kanduri M, Hovland R, et al.: Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood 118 (22): 5905-13, 2011.[PUBMED Abstract]
- Tawana K, Wang J, Renneville A, et al.: Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 126 (10): 1214-23, 2015.[PUBMED Abstract]
- Marcucci G, Maharry K, Radmacher MD, et al.: Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol 26 (31): 5078-87, 2008.[PUBMED Abstract]
- Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al.: Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113 (13): 3088-91, 2009.[PUBMED Abstract]
- Dufour A, Schneider F, Metzeler KH, et al.: Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 28 (4): 570-7, 2010.[PUBMED Abstract]
- Taskesen E, Bullinger L, Corbacioglu A, et al.: Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 117 (8): 2469-75, 2011.[PUBMED Abstract]
- Fasan A, Haferlach C, Alpermann T, et al.: The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28 (4): 794-803, 2014.[PUBMED Abstract]
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 96 (3): 384-92, 2011.[PUBMED Abstract]
- Groet J, McElwaine S, Spinelli M, et al.: Acquired mutations in GATA1 in neonates with Down's syndrome with transient myeloid disorder. Lancet 361 (9369): 1617-20, 2003.[PUBMED Abstract]
- Hitzler JK, Cheung J, Li Y, et al.: GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101 (11): 4301-4, 2003.[PUBMED Abstract]
- Rainis L, Bercovich D, Strehl S, et al.: Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102 (3): 981-6, 2003.[PUBMED Abstract]
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.[PUBMED Abstract]
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.[PUBMED Abstract]
- Gurbuxani S, Vyas P, Crispino JD: Recent insights into the mechanisms of myeloid leukemogenesis in Down syndrome. Blood 103 (2): 399-406, 2004.[PUBMED Abstract]
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.[PUBMED Abstract]
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Outcome of pediatric patients with acute myeloid leukemia (AML) and -5/5q- abnormalities from five pediatric AML treatment protocols: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2073-8, 2013.[PUBMED Abstract]
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.[PUBMED Abstract]
- Wells RJ, Arthur DC, Srivastava A, et al.: Prognostic variables in newly diagnosed children and adolescents with acute myeloid leukemia: Children's Cancer Group Study 213. Leukemia 16 (4): 601-7, 2002.[PUBMED Abstract]
- Hasle H, Alonzo TA, Auvrignon A, et al.: Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood 109 (11): 4641-7, 2007.[PUBMED Abstract]
- Rasche M, von Neuhoff C, Dworzak M, et al.: Genotype-outcome correlations in pediatric AML: the impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia 31 (12): 2807-2814, 2017.[PUBMED Abstract]
- Swansbury GJ, Lawler SD, Alimena G, et al.: Long-term survival in acute myelogenous leukemia: a second follow-up of the Fourth International Workshop on Chromosomes in Leukemia. Cancer Genet Cytogenet 73 (1): 1-7, 1994.[PUBMED Abstract]
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.[PUBMED Abstract]
- Gröschel S, Sanders MA, Hoogenboezem R, et al.: A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157 (2): 369-81, 2014.[PUBMED Abstract]
- Yamazaki H, Suzuki M, Otsuki A, et al.: A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25 (4): 415-27, 2014.[PUBMED Abstract]
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.[PUBMED Abstract]
- Balgobind BV, Lugthart S, Hollink IH, et al.: EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 24 (5): 942-9, 2010.[PUBMED Abstract]
- Schnittger S, Schoch C, Dugas M, et al.: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100 (1): 59-66, 2002.[PUBMED Abstract]
- Thiede C, Steudel C, Mohr B, et al.: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99 (12): 4326-35, 2002.[PUBMED Abstract]
- Whitman SP, Archer KJ, Feng L, et al.: Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res 61 (19): 7233-9, 2001.[PUBMED Abstract]
- Iwai T, Yokota S, Nakao M, et al.: Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children's Cancer and Leukemia Study Group, Japan. Leukemia 13 (1): 38-43, 1999.[PUBMED Abstract]
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.[PUBMED Abstract]
- Meshinchi S, Stirewalt DL, Alonzo TA, et al.: Activating mutations of RTK/ras signal transduction pathway in pediatric acute myeloid leukemia. Blood 102 (4): 1474-9, 2003.[PUBMED Abstract]
- Zwaan CM, Meshinchi S, Radich JP, et al.: FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood 102 (7): 2387-94, 2003.[PUBMED Abstract]
- Chang P, Kang M, Xiao A, et al.: FLT3 mutation incidence and timing of origin in a population case series of pediatric leukemia. BMC Cancer 10: 513, 2010.[PUBMED Abstract]
- Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, et al.: NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 118 (13): 3645-56, 2011.[PUBMED Abstract]
- Ostronoff F, Othus M, Gerbing RB, et al.: NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood 124 (15): 2400-7, 2014.[PUBMED Abstract]
- Shih LY, Kuo MC, Liang DC, et al.: Internal tandem duplication and Asp835 mutations of the FMS-like tyrosine kinase 3 (FLT3) gene in acute promyelocytic leukemia. Cancer 98 (6): 1206-16, 2003.[PUBMED Abstract]
- Noguera NI, Breccia M, Divona M, et al.: Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 16 (11): 2185-9, 2002.[PUBMED Abstract]
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.[PUBMED Abstract]
- Abu-Duhier FM, Goodeve AC, Wilson GA, et al.: Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol 113 (4): 983-8, 2001.[PUBMED Abstract]
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.[PUBMED Abstract]
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.[PUBMED Abstract]
- Sung L, Aplenc R, Alonzo TA, et al.: Predictors and short-term outcomes of hyperleukocytosis in children with acute myeloid leukemia: a report from the Children's Oncology Group. Haematologica 97 (11): 1770-3, 2012.[PUBMED Abstract]
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.[PUBMED Abstract]
- Schnittger S, Bacher U, Haferlach C, et al.: Clinical impact of FLT3 mutation load in acute promyelocytic leukemia with t(15;17)/PML-RARA. Haematologica 96 (12): 1799-807, 2011.[PUBMED Abstract]
- Breccia M, Loglisci G, Loglisci MG, et al.: FLT3-ITD confers poor prognosis in patients with acute promyelocytic leukemia treated with AIDA protocols: long-term follow-up analysis. Haematologica 98 (12): e161-3, 2013.[PUBMED Abstract]
- Poiré X, Moser BK, Gallagher RE, et al.: Arsenic trioxide in front-line therapy of acute promyelocytic leukemia (C9710): prognostic significance of FLT3 mutations and complex karyotype. Leuk Lymphoma 55 (7): 1523-32, 2014.[PUBMED Abstract]
- Pui CH, Relling MV, Rivera GK, et al.: Epipodophyllotoxin-related acute myeloid leukemia: a study of 35 cases. Leukemia 9 (12): 1990-6, 1995.[PUBMED Abstract]
- Inaba H, Zhou Y, Abla O, et al.: Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood 126 (13): 1575-84, 2015.[PUBMED Abstract]
- Balgobind BV, Raimondi SC, Harbott J, et al.: Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 114 (12): 2489-96, 2009.[PUBMED Abstract]
- Swansbury GJ, Slater R, Bain BJ, et al.: Hematological malignancies with t(9;11)(p21-22;q23)--a laboratory and clinical study of 125 cases. European 11q23 Workshop participants. Leukemia 12 (5): 792-800, 1998.[PUBMED Abstract]
- Rubnitz JE, Raimondi SC, Tong X, et al.: Favorable impact of the t(9;11) in childhood acute myeloid leukemia. J Clin Oncol 20 (9): 2302-9, 2002.[PUBMED Abstract]
- Mrózek K, Heinonen K, Lawrence D, et al.: Adult patients with de novo acute myeloid leukemia and t(9; 11)(p22; q23) have a superior outcome to patients with other translocations involving band 11q23: a Cancer and Leukemia Group B study. Blood 90 (11): 4532-8, 1997.[PUBMED Abstract]
- Martinez-Climent JA, Espinosa R, Thirman MJ, et al.: Abnormalities of chromosome band 11q23 and the MLL gene in pediatric myelomonocytic and monoblastic leukemias. Identification of the t(9;11) as an indicator of long survival. J Pediatr Hematol Oncol 17 (4): 277-83, 1995.[PUBMED Abstract]
- Casillas JN, Woods WG, Hunger SP, et al.: Prognostic implications of t(10;11) translocations in childhood acute myelogenous leukemia: a report from the Children's Cancer Group. J Pediatr Hematol Oncol 25 (8): 594-600, 2003.[PUBMED Abstract]
- Morerio C, Rosanda C, Rapella A, et al.: Is t(10;11)(p11.2;q23) involving MLL and ABI-1 genes associated with congenital acute monocytic leukemia? Cancer Genet Cytogenet 139 (1): 57-9, 2002.[PUBMED Abstract]
- Taki T, Shibuya N, Taniwaki M, et al.: ABI-1, a human homolog to mouse Abl-interactor 1, fuses the MLL gene in acute myeloid leukemia with t(10;11)(p11.2;q23). Blood 92 (4): 1125-30, 1998.[PUBMED Abstract]
- Coenen EA, Raimondi SC, Harbott J, et al.: Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood 117 (26): 7102-11, 2011.[PUBMED Abstract]
- Ageberg M, Drott K, Olofsson T, et al.: Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer 47 (4): 276-87, 2008.[PUBMED Abstract]
- Shiba N, Ichikawa H, Taki T, et al.: NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 52 (7): 683-93, 2013.[PUBMED Abstract]
- Slovak ML, Gundacker H, Bloomfield CD, et al.: A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective, multicenter initiative for rare 'poor prognosis' myeloid malignancies. Leukemia 20 (7): 1295-7, 2006.[PUBMED Abstract]
- Alsabeh R, Brynes RK, Slovak ML, et al.: Acute myeloid leukemia with t(6;9) (p23;q34): association with myelodysplasia, basophilia, and initial CD34 negative immunophenotype. Am J Clin Pathol 107 (4): 430-7, 1997.[PUBMED Abstract]
- Sandahl JD, Coenen EA, Forestier E, et al.: t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica 99 (5): 865-72, 2014.[PUBMED Abstract]
- Tarlock K, Alonzo TA, Moraleda PP, et al.: Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: a report from the Children's Oncology Group. Br J Haematol 166 (2): 254-9, 2014.[PUBMED Abstract]
- Gruber TA, Larson Gedman A, Zhang J, et al.: An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22 (5): 683-97, 2012.[PUBMED Abstract]
- Thiollier C, Lopez CK, Gerby B, et al.: Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med 209 (11): 2017-31, 2012.[PUBMED Abstract]
- de Rooij JD, Hollink IH, Arentsen-Peters ST, et al.: NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia 27 (12): 2280-8, 2013.[PUBMED Abstract]
- Masetti R, Pigazzi M, Togni M, et al.: CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 121 (17): 3469-72, 2013.[PUBMED Abstract]
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.[PUBMED Abstract]
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.[PUBMED Abstract]
- Hara Y, Shiba N, Ohki K, et al.: Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer 56 (5): 394-404, 2017.[PUBMED Abstract]
- Carroll A, Civin C, Schneider N, et al.: The t(1;22) (p13;q13) is nonrandom and restricted to infants with acute megakaryoblastic leukemia: a Pediatric Oncology Group Study. Blood 78 (3): 748-52, 1991.[PUBMED Abstract]
- Lion T, Haas OA: Acute megakaryocytic leukemia with the t(1;22)(p13;q13). Leuk Lymphoma 11 (1-2): 15-20, 1993.[PUBMED Abstract]
- Duchayne E, Fenneteau O, Pages MP, et al.: Acute megakaryoblastic leukaemia: a national clinical and biological study of 53 adult and childhood cases by the Groupe Français d'Hématologie Cellulaire (GFHC). Leuk Lymphoma 44 (1): 49-58, 2003.[PUBMED Abstract]
- Ma Z, Morris SW, Valentine V, et al.: Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet 28 (3): 220-1, 2001.[PUBMED Abstract]
- Mercher T, Coniat MB, Monni R, et al.: Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc Natl Acad Sci U S A 98 (10): 5776-9, 2001.[PUBMED Abstract]
- Bernstein J, Dastugue N, Haas OA, et al.: Nineteen cases of the t(1;22)(p13;q13) acute megakaryblastic leukaemia of infants/children and a review of 39 cases: report from a t(1;22) study group. Leukemia 14 (1): 216-8, 2000.[PUBMED Abstract]
- Coenen EA, Zwaan CM, Reinhardt D, et al.: Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood 122 (15): 2704-13, 2013.[PUBMED Abstract]
- Wong KF, Yuen HL, Siu LL, et al.: t(8;16)(p11;p13) predisposes to a transient but potentially recurring neonatal leukemia. Hum Pathol 39 (11): 1702-7, 2008.[PUBMED Abstract]
- Wu X, Sulavik D, Roulston D, et al.: Spontaneous remission of congenital acute myeloid leukemia with t(8;16)(p11;13). Pediatr Blood Cancer 56 (2): 331-2, 2011.[PUBMED Abstract]
- Terui K, Sato T, Sasaki S, et al.: Two novel variants of MOZ-CBP fusion transcripts in spontaneously remitted infant leukemia with t(1;16;8)(p13;p13;p11), a new variant of t(8;16)(p11;p13). Haematologica 93 (10): 1591-3, 2008.[PUBMED Abstract]
- Sainati L, Bolcato S, Cocito MG, et al.: Transient acute monoblastic leukemia with reciprocal (8;16)(p11;p13) translocation. Pediatr Hematol Oncol 13 (2): 151-7, 1996 Mar-Apr.[PUBMED Abstract]
- Weintraub M, Kaplinsky C, Amariglio N, et al.: Spontaneous regression of congenital leukaemia with an 8;16 translocation. Br J Haematol 111 (2): 641-3, 2000.[PUBMED Abstract]
- Classen CF, Behnisch W, Reinhardt D, et al.: Spontaneous complete and sustained remission of a rearrangement CBP (16p13)-positive disseminated congenital myelosarcoma. Ann Hematol 84 (4): 274-5, 2005.[PUBMED Abstract]
- Beverloo HB, Panagopoulos I, Isaksson M, et al.: Fusion of the homeobox gene HLXB9 and the ETV6 gene in infant acute myeloid leukemias with the t(7;12)(q36;p13). Cancer Res 61 (14): 5374-7, 2001.[PUBMED Abstract]
- Slater RM, von Drunen E, Kroes WG, et al.: t(7;12)(q36;p13) and t(7;12)(q32;p13)--translocations involving ETV6 in children 18 months of age or younger with myeloid disorders. Leukemia 15 (6): 915-20, 2001.[PUBMED Abstract]
- von Bergh AR, van Drunen E, van Wering ER, et al.: High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer 45 (8): 731-9, 2006.[PUBMED Abstract]
- Tosi S, Harbott J, Teigler-Schlegel A, et al.: t(7;12)(q36;p13), a new recurrent translocation involving ETV6 in infant leukemia. Genes Chromosomes Cancer 29 (4): 325-32, 2000.[PUBMED Abstract]
- Park J, Kim M, Lim J, et al.: Three-way complex translocations in infant acute myeloid leukemia with t(7;12)(q36;p13): the incidence and correlation of a HLXB9 overexpression. Cancer Genet Cytogenet 191 (2): 102-5, 2009.[PUBMED Abstract]
- Takeda A, Yaseen NR: Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol 27: 3-10, 2014.[PUBMED Abstract]
- Brown J, Jawad M, Twigg SR, et al.: A cryptic t(5;11)(q35;p15.5) in 2 children with acute myeloid leukemia with apparently normal karyotypes, identified by a multiplex fluorescence in situ hybridization telomere assay. Blood 99 (7): 2526-31, 2002.[PUBMED Abstract]
- Panarello C, Rosanda C, Morerio C: Cryptic translocation t(5;11)(q35;p15.5) with involvement of the NSD1 and NUP98 genes without 5q deletion in childhood acute myeloid leukemia. Genes Chromosomes Cancer 35 (3): 277-81, 2002.[PUBMED Abstract]
- Cerveira N, Correia C, Dória S, et al.: Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17 (11): 2244-7, 2003.[PUBMED Abstract]
- Jaju RJ, Fidler C, Haas OA, et al.: A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 98 (4): 1264-7, 2001.[PUBMED Abstract]
- McNeer NA, Philip J, Geiger H, et al.: Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33 (8): 1934-1943, 2019.[PUBMED Abstract]
- Yamato G, Shiba N, Yoshida K, et al.: RUNX1 mutations in pediatric acute myeloid leukemia are associated with distinct genetic features and an inferior prognosis. Blood 131 (20): 2266-2270, 2018.[PUBMED Abstract]
- Radich JP, Kopecky KJ, Willman CL, et al.: N-ras mutations in adult de novo acute myelogenous leukemia: prevalence and clinical significance. Blood 76 (4): 801-7, 1990.[PUBMED Abstract]
- Farr C, Gill R, Katz F, et al.: Analysis of ras gene mutations in childhood myeloid leukaemia. Br J Haematol 77 (3): 323-7, 1991.[PUBMED Abstract]
- Berman JN, Gerbing RB, Alonzo TA, et al.: Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children's Oncology Group. Leukemia 25 (6): 1039-42, 2011.[PUBMED Abstract]
- Kühn MW, Radtke I, Bullinger L, et al.: High-resolution genomic profiling of adult and pediatric core-binding factor acute myeloid leukemia reveals new recurrent genomic alterations. Blood 119 (10): e67-75, 2012.[PUBMED Abstract]
- Schnittger S, Kohl TM, Haferlach T, et al.: KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 107 (5): 1791-9, 2006.[PUBMED Abstract]
- Tokumasu M, Murata C, Shimada A, et al.: Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial. Leukemia 29 (12): 2438-41, 2015.[PUBMED Abstract]
- Cairoli R, Beghini A, Grillo G, et al.: Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 107 (9): 3463-8, 2006.[PUBMED Abstract]
- Paschka P, Marcucci G, Ruppert AS, et al.: Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol 24 (24): 3904-11, 2006.[PUBMED Abstract]
- Shimada A, Taki T, Tabuchi K, et al.: KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 107 (5): 1806-9, 2006.[PUBMED Abstract]
- Shih LY, Liang DC, Huang CF, et al.: Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 22 (2): 303-7, 2008.[PUBMED Abstract]
- Goemans BF, Zwaan CM, Miller M, et al.: Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 19 (9): 1536-42, 2005.[PUBMED Abstract]
- Boissel N, Leroy H, Brethon B, et al.: Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 20 (6): 965-70, 2006.[PUBMED Abstract]
- Pollard JA, Alonzo TA, Gerbing RB, et al.: Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 115 (12): 2372-9, 2010.[PUBMED Abstract]
- Paschka P, Marcucci G, Ruppert AS, et al.: Wilms' tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 26 (28): 4595-602, 2008.[PUBMED Abstract]
- Virappane P, Gale R, Hills R, et al.: Mutation of the Wilms' tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 26 (33): 5429-35, 2008.[PUBMED Abstract]
- Gaidzik VI, Schlenk RF, Moschny S, et al.: Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 113 (19): 4505-11, 2009.[PUBMED Abstract]
- Renneville A, Boissel N, Zurawski V, et al.: Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 115 (16): 3719-27, 2009.[PUBMED Abstract]
- Ho PA, Zeng R, Alonzo TA, et al.: Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children's Oncology Group. Blood 116 (5): 702-10, 2010.[PUBMED Abstract]
- Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, et al.: Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 113 (23): 5951-60, 2009.[PUBMED Abstract]
- Ley TJ, Ding L, Walter MJ, et al.: DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363 (25): 2424-33, 2010.[PUBMED Abstract]
- Yan XJ, Xu J, Gu ZH, et al.: Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 43 (4): 309-15, 2011.[PUBMED Abstract]
- Thol F, Damm F, Lüdeking A, et al.: Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 29 (21): 2889-96, 2011.[PUBMED Abstract]
- Ho PA, Kutny MA, Alonzo TA, et al.: Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (2): 204-9, 2011.[PUBMED Abstract]
- Green CL, Evans CM, Hills RK, et al.: The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 116 (15): 2779-82, 2010.[PUBMED Abstract]
- Paschka P, Schlenk RF, Gaidzik VI, et al.: IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28 (22): 3636-43, 2010.[PUBMED Abstract]
- Abbas S, Lugthart S, Kavelaars FG, et al.: Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116 (12): 2122-6, 2010.[PUBMED Abstract]
- Marcucci G, Maharry K, Wu YZ, et al.: IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28 (14): 2348-55, 2010.[PUBMED Abstract]
- Wagner K, Damm F, Göhring G, et al.: Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 28 (14): 2356-64, 2010.[PUBMED Abstract]
- Figueroa ME, Abdel-Wahab O, Lu C, et al.: Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18 (6): 553-67, 2010.[PUBMED Abstract]
- Ward PS, Patel J, Wise DR, et al.: The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17 (3): 225-34, 2010.[PUBMED Abstract]
- Dang L, White DW, Gross S, et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274): 739-44, 2009.[PUBMED Abstract]
- Damm F, Thol F, Hollink I, et al.: Prevalence and prognostic value of IDH1 and IDH2 mutations in childhood AML: a study of the AML-BFM and DCOG study groups. Leukemia 25 (11): 1704-10, 2011.[PUBMED Abstract]
- Oki K, Takita J, Hiwatari M, et al.: IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia 25 (2): 382-4, 2011.[PUBMED Abstract]
- Pigazzi M, Ferrari G, Masetti R, et al.: Low prevalence of IDH1 gene mutation in childhood AML in Italy. Leukemia 25 (1): 173-4, 2011.[PUBMED Abstract]
- Ho PA, Alonzo TA, Kopecky KJ, et al.: Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 24 (5): 909-13, 2010.[PUBMED Abstract]
- Andersson AK, Miller DW, Lynch JA, et al.: IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25 (10): 1570-7, 2011.[PUBMED Abstract]
- Maxson JE, Ries RE, Wang YC, et al.: CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 127 (24): 3094-8, 2016.[PUBMED Abstract]
- Germeshausen M, Kratz CP, Ballmaier M, et al.: RAS and CSF3R mutations in severe congenital neutropenia. Blood 114 (16): 3504-5, 2009.[PUBMED Abstract]
- Skokowa J, Steinemann D, Katsman-Kuipers JE, et al.: Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood 123 (14): 2229-37, 2014.[PUBMED Abstract]
- 小児AMLに対する治療法選択肢の概要
-
白血病は診断時に造血系に拡がっていると考えられ、孤立性緑色腫(顆粒球肉腫または骨髄性肉腫とも呼ばれる)を呈する急性骨髄性白血病(AML)患児の場合も例外ではない。このような患児は、全身化学療法を受けなければ、必ず数ヵ月後あるいは数年後にAMLへと移行する。AMLは髄膜、脳実質、精巣、卵巣、または皮膚(皮膚白血病)といった非造血(髄外性)組織に浸潤することがある。髄外白血病は、年長のAML患児より乳児によくみられる。[ 1 ]
骨髄中の芽球が20%以上の場合に、小児AMLと診断される。芽球は、フランス-アメリカ-イギリス(FAB)のいずれかのAML亜型の形態学的、および組織化学的特徴を認める。緑色腫の生検によっても診断が可能である。治療目的のために、典型的にAMLに伴うクローン性細胞遺伝学的異常として、t(8;21)(RUNX1-RUNX1T1)、inv(16)(CBFB-MYH11)、t(9;11)(MLLT3-KMT2A)、またはt(15;17)(PML-RARA)などを認める患者、および骨髄芽球が20%を下回る患者は、骨髄異形成症候ではなく、むしろAMLであるとみなされる。[ 2 ]
米国では、伝統的に完全寛解(CR)が以下のような形態学的基準を用いて定義されている:
AMLでは、強力な化学療法により引き起こされる持続的な骨髄抑制のために、形態学的検査を用いた寛解の代替定義が用いられ、血小板回復が不完全なCRおよび骨髄回復(典型的に好中球絶対数)が不完全なCRなどがある。不完全な血小板回復を使用することで、臨床的に意味のある反応が得られるが、CR中の患者は、血小板回復が不完全な患者より生存期間が長い可能性がはるかに高いことが明らかにされたことから、伝統的なCRの定義が依然としてゴールドスタンダードである。[ 4 ]
AMLで寛解を得る際に、M3亜型(急性前骨髄球性白血病[APL])を除いて、通常は骨髄低形成(形態学的検査を用いる)を達成することが第一ステップである;APLでは、寛解達成前に骨髄低形成の段階が必要ないことが多い。さらに、いずれのAML亜型でも、早期の骨髄再生は白血病残存と区別することが困難な場合があるが、フローサイトメトリーによる免疫表現型検査および細胞遺伝学的/分子的検査の適用により、この問題は少なくなっている。血球数と臨床状態の相関性の検討は、AMLで早期の骨髄所見の結果に対して最終的な判断を下す際に不可欠である。[ 5 ]得られた所見について疑わしい点があれば、1~2週間後に再び骨髄穿刺を行うべきである。[ 1 ]
形態学的検査に加えて、反応評価にはより正確な検査法(例、マルチパラメータフローサイトメトリーまたは定量的逆転写ポリメラーゼ連鎖反応[RT-PCR])が使用され、形態学的検査より予後的意義が大きいことが示されている。(これらの検査法に関する詳しい情報については、本要約の小児AMLの予後因子のセクションを参照のこと。)
治療アプローチ
治療アプローチの中心は、全身投与する多剤併用化学療法である。[ 6 ]正常組織を温存しながら抗白血病治療を改善するために、リスクグループの層別化および生物学的標的療法を含むアプローチが検証されている。[ 7 ]AMLの至適治療を行うには、骨髄および全身の病状をコントロールすることが必要とされる。ほとんどの小児AML治療プロトコルで、CNSの治療に通常は髄腔内への薬剤投与が用いられるが、生存期間延長に直接寄与するかはまだ示されていない。CNSへの放射線照射は、予防としても、脳脊髄液白血病を呈するものの髄腔内および全身性化学療法により消失する患者に対しても必要ない。
通常は、以下の2つの相に治療が分けられる:
寛解後療法がさまざまなコース数の強化化学療法および/または同種造血幹細胞移植(HSCT)により行われる場合がある。例えば、小児腫瘍学グループ(COG)および英国医学研究審議会(MRC)で進行中の試験では、2コースの寛解導入化学療法の後に強化化学療法を2~3コース追加する同様な化学療法レジメンが用いられている。[ 8 ][ 9 ]
2件のランダム化臨床試験で、現代の強化化学療法後に維持療法を行った場合に有益性が示されなかったため、ほとんどの小児AMLプロトコルに維持療法は含まれていない。[ 10 ][ 11 ]APLでは、このような一般化に対して例外とされたが、これは、維持療法でオールトランス(all-trans)レチノイン酸(ATRA)を化学療法と併用した場合に、イベントフリー生存(EFS)および全生存(OS)の改善が示されたためである。[ 12 ]成人APL患者を対象として三酸化ヒ素による治療を組み入れた研究を含むいくつかの研究で、維持療法の有益性は示されていない。[ 13 ][ 14 ]
AML小児では、急性および長期合併症の両方への注意が不可欠である。現代のAMLの治療アプローチは通常、関連合併症を伴う重度かつ長期化する骨髄抑制と関係している。AMLの小児では、適切な支持療法設備(例、特殊な血液製剤、小児集中治療室、精神面および発育面での支援提供)を有するがんセンターまたは病院において小児がん専門医の指示の下で医療を行うべきである。支持療法の改善により、毒性による死亡のうち、初回治療失敗が占める割合は以前より低くなっている。[ 8 ]つい最近のCOG試験では、抵抗性疾患のために寛解導入失敗の発生率が11~13%で、2つの導入コースで毒性による死亡に起因する失敗はわずか2~3%であったことが報告された。[ 15 ][ 16 ]
AMLに対する治療を受けた小児は、長く生存し、治療から数ヵ月または数年経過後まで持続またはその後に発現することがあるがん治療の副作用について綿密なモニタリングが必要である。アントラサイクリン系薬剤の累積用量が高いと、心機能の長期モニタリングが必要である。HSCTに伴う全身放射線照射などの一部の治療法では、成長不全、性腺および甲状腺機能障害、白内障形成、二次悪性腫瘍のリスクが高まるため、その使用が減少している。[ 17 ](詳しい情報については、本要約の生存者と有害な晩期続発症のセクションまたは小児がん治療の晩期合併症(晩期障害)に関するPDQ要約を参照のこと。)
小児AMLの予後因子
小児AMLにおける予後因子は、以下のような分類が可能である:
宿主の危険因子
白血病の危険因子
治療効果の危険因子
リスク分類システム
治療割り付けに対するリスク分類システムがAML小児を対象とした臨床試験を実施しているいくつかの共同研究グループで使用されている。COGでは、危険因子に基づく治療選択の層別化が非APLで非ダウン症候群の患者に対する比較的最近のアプローチである。分類は、ほとんどがEFSおよびOSに関するMRC AML 10試験[ 57 ]の観察から直接もたらされ、さらに小児患者が再寛解導入を受け、二回目の完全寛解を得る能力およびその後の初回再燃からのOSに基づいて適用される。[ 77 ]
以下のCOG試験では、治療選択の層別化にリスク分類システムを用いている:
- COG AAML0531(NCT00372593)は、リスク群により治療を層別化した最初のCOG試験で、診断用細胞遺伝学的検査および寛解導入1後の反応に基づいて3つのリスク群に患者を層別化した。[ 16 ]
- その後のCOG試験であるCOG-AAML1031(NCT01371981)では、マルチパラメータフローサイトメトリーによるMRDの使用を追加することで、中リスク分類の患者がより特異的かつ予後的に定義できることが明らかになったことに基づき、これらのリスク群が2つに減らされた。[ 78 ]
- COG-AAML1031で、研究の層別化は、さらに細胞遺伝学、分子マーカー、および寛解導入1後の骨髄回復時のMRDに基づいたもので、以下のように低リスク群または高リスク群に患者が分類された:
- 低リスク群は、患者の約73%を占め、OS率が約75%と予測され、以下によって定義される:
- 高リスク群は、患者の残り27%を占め、OS率が35%未満と予想され、以下によって定義される:
危険因子が互いに矛盾する場合、証拠に基づいて作成された下の表が用いられる(表7を参照)。
表7.AAML1031におけるリスク割り当てa,b リスク割り当て: 低リスク 高リスク 低リスク群1 低リスク群2 高リスク群1 高リスク群2 高リスク群3 aこれらの群は、危険因子の組み合わせに基づいており、個々の患者で明らかにできる。 b太字は、リスク群の割り当てで最も重要な危険因子を示す。 cNPM1、CEBPA、t(8;21)、inv(16)。 d7モノソミー、5モノソミー、del(5q)。 FLT3-ITDのアレル比 低/陰性 低/陰性 高 低/陰性 低/陰性 リスク良好の分子マーカーc 有 無 任意 無 無 リスク不良の細胞遺伝学的マーカーd 任意 無 任意 有 無 微小残存病変 任意 陰性 任意 任意 陽性
高リスク群の患者では、第一寛解期に最も適切な利用可能ドナーによる移植が指針となる。低リスク群の患者では、再燃した場合に移植を受けるよう指導する。このアプローチの妥当性評価についての解析が待たれる。[ 62 ][ 79 ]
層別化に用いられる危険因子は、小児および成人を対象とした共同臨床試験グループにより異なり、所定の危険因子の予後への影響は、その意義において、使用される治療の基本骨格に応じて異なることがある。他の小児共同研究グループは、これらと同じ因子の一部または全部を用いており、一般に多数の試験で再現可能となっている危険因子が選択され、ときにリスク群層別化アプローチで以前に用いたことのある他の危険因子を含めている。
参考文献- Ebb DH, Weinstein HJ: Diagnosis and treatment of childhood acute myelogenous leukemia. Pediatr Clin North Am 44 (4): 847-62, 1997.[PUBMED Abstract]
- Chan GC, Wang WC, Raimondi SC, et al.: Myelodysplastic syndrome in children: differentiation from acute myeloid leukemia with a low blast count. Leukemia 11 (2): 206-11, 1997.[PUBMED Abstract]
- Cheson BD, Bennett JM, Kopecky KJ, et al.: Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 21 (24): 4642-9, 2003.[PUBMED Abstract]
- Walter RB, Kantarjian HM, Huang X, et al.: Effect of complete remission and responses less than complete remission on survival in acute myeloid leukemia: a combined Eastern Cooperative Oncology Group, Southwest Oncology Group, and M. D. Anderson Cancer Center Study. J Clin Oncol 28 (10): 1766-71, 2010.[PUBMED Abstract]
- Konopleva M, Cheng SC, Cortes JE, et al.: Independent prognostic significance of day 21 cytogenetic findings in newly-diagnosed acute myeloid leukemia or refractory anemia with excess blasts. Haematologica 88 (7): 733-6, 2003.[PUBMED Abstract]
- Loeb DM, Arceci RJ: What is the optimal therapy for childhood AML? Oncology (Huntingt) 16 (8): 1057-66; discussion 1066, 1068-70, 2002.[PUBMED Abstract]
- Arceci RJ: Progress and controversies in the treatment of pediatric acute myelogenous leukemia. Curr Opin Hematol 9 (4): 353-60, 2002.[PUBMED Abstract]
- Hann IM, Webb DK, Gibson BE, et al.: MRC trials in childhood acute myeloid leukaemia. Ann Hematol 83 (Suppl 1): S108-12, 2004.[PUBMED Abstract]
- Gibson BE, Webb DK, Howman AJ, et al.: Results of a randomized trial in children with Acute Myeloid Leukaemia: medical research council AML12 trial. Br J Haematol 155 (3): 366-76, 2011.[PUBMED Abstract]
- Wells RJ, Woods WG, Buckley JD, et al.: Treatment of newly diagnosed children and adolescents with acute myeloid leukemia: a Childrens Cancer Group study. J Clin Oncol 12 (11): 2367-77, 1994.[PUBMED Abstract]
- Perel Y, Auvrignon A, Leblanc T, et al.: Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of a prospective randomized trial, LAME 89/91. Leucámie Aiqüe Myéloïde Enfant. J Clin Oncol 20 (12): 2774-82, 2002.[PUBMED Abstract]
- Fenaux P, Chastang C, Chevret S, et al.: A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood 94 (4): 1192-200, 1999.[PUBMED Abstract]
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.[PUBMED Abstract]
- Avvisati G, Lo-Coco F, Paoloni FP, et al.: AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood 117 (18): 4716-25, 2011.[PUBMED Abstract]
- Cooper TM, Franklin J, Gerbing RB, et al.: AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's Oncology Group. Cancer 118 (3): 761-9, 2012.[PUBMED Abstract]
- Gamis AS, Alonzo TA, Meshinchi S, et al.: Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 32 (27): 3021-32, 2014.[PUBMED Abstract]
- Leung W, Hudson MM, Strickland DK, et al.: Late effects of treatment in survivors of childhood acute myeloid leukemia. J Clin Oncol 18 (18): 3273-9, 2000.[PUBMED Abstract]
- Webb DK, Harrison G, Stevens RF, et al.: Relationships between age at diagnosis, clinical features, and outcome of therapy in children treated in the Medical Research Council AML 10 and 12 trials for acute myeloid leukemia. Blood 98 (6): 1714-20, 2001.[PUBMED Abstract]
- Razzouk BI, Estey E, Pounds S, et al.: Impact of age on outcome of pediatric acute myeloid leukemia: a report from 2 institutions. Cancer 106 (11): 2495-502, 2006.[PUBMED Abstract]
- Lange BJ, Smith FO, Feusner J, et al.: Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood 111 (3): 1044-53, 2008.[PUBMED Abstract]
- Creutzig U, Büchner T, Sauerland MC, et al.: Significance of age in acute myeloid leukemia patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112 (3): 562-71, 2008.[PUBMED Abstract]
- Woods WG, Franklin AR, Alonzo TA, et al.: Outcome of adolescents and young adults with acute myeloid leukemia treated on COG trials compared to CALGB and SWOG trials. Cancer 119 (23): 4170-9, 2013.[PUBMED Abstract]
- Canner J, Alonzo TA, Franklin J, et al.: Differences in outcomes of newly diagnosed acute myeloid leukemia for adolescent/young adult and younger patients: a report from the Children's Oncology Group. Cancer 119 (23): 4162-9, 2013.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Bourquin JP, et al.: Favorable outcome in infants with AML after intensive first- and second-line treatment: an AML-BFM study group report. Leukemia 26 (4): 654-61, 2012.[PUBMED Abstract]
- Kawasaki H, Isoyama K, Eguchi M, et al.: Superior outcome of infant acute myeloid leukemia with intensive chemotherapy: results of the Japan Infant Leukemia Study Group. Blood 98 (13): 3589-94, 2001.[PUBMED Abstract]
- Masetti R, Rondelli R, Fagioli F, et al.: Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 99 (8): e127-9, 2014.[PUBMED Abstract]
- Guest EM, Aplenc R, Sung L, et al.: Gemtuzumab ozogamicin in infants with AML: results from the Children's Oncology Group trials AAML03P1 and AAML0531. Blood 130 (7): 943-945, 2017.[PUBMED Abstract]
- Aplenc R, Alonzo TA, Gerbing RB, et al.: Ethnicity and survival in childhood acute myeloid leukemia: a report from the Children's Oncology Group. Blood 108 (1): 74-80, 2006.[PUBMED Abstract]
- Rubnitz JE, Lensing S, Razzouk BI, et al.: Effect of race on outcome of white and black children with acute myeloid leukemia: the St. Jude experience. Pediatr Blood Cancer 48 (1): 10-5, 2007.[PUBMED Abstract]
- Lange BJ, Kobrinsky N, Barnard DR, et al.: Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2): 608-15, 1998.[PUBMED Abstract]
- Sorrell AD, Alonzo TA, Hilden JM, et al.: Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children's Oncology Group trial A2971: a report from the Children's Oncology Group. Cancer 118 (19): 4806-14, 2012.[PUBMED Abstract]
- Taub JW, Berman JN, Hitzler JK, et al.: Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children's Oncology Group AAML0431 trial. Blood 129 (25): 3304-3313, 2017.[PUBMED Abstract]
- Creutzig U, Reinhardt D, Diekamp S, et al.: AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia 19 (8): 1355-60, 2005.[PUBMED Abstract]
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.[PUBMED Abstract]
- Gamis AS, Woods WG, Alonzo TA, et al.: Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol 21 (18): 3415-22, 2003.[PUBMED Abstract]
- Lange BJ, Gerbing RB, Feusner J, et al.: Mortality in overweight and underweight children with acute myeloid leukemia. JAMA 293 (2): 203-11, 2005.[PUBMED Abstract]
- Inaba H, Surprise HC, Pounds S, et al.: Effect of body mass index on the outcome of children with acute myeloid leukemia. Cancer 118 (23): 5989-96, 2012.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Ritter J, et al.: Definition of a standard-risk group in children with AML. Br J Haematol 104 (3): 630-9, 1999.[PUBMED Abstract]
- Chang M, Raimondi SC, Ravindranath Y, et al.: Prognostic factors in children and adolescents with acute myeloid leukemia (excluding children with Down syndrome and acute promyelocytic leukemia): univariate and recursive partitioning analysis of patients treated on Pediatric Oncology Group (POG) Study 8821. Leukemia 14 (7): 1201-7, 2000.[PUBMED Abstract]
- Pession A, Masetti R, Rizzari C, et al.: Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122 (2): 170-8, 2013.[PUBMED Abstract]
- Sung L, Aplenc R, Alonzo TA, et al.: Predictors and short-term outcomes of hyperleukocytosis in children with acute myeloid leukemia: a report from the Children's Oncology Group. Haematologica 97 (11): 1770-3, 2012.[PUBMED Abstract]
- Testi AM, Biondi A, Lo Coco F, et al.: GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 106 (2): 447-53, 2005.[PUBMED Abstract]
- de Botton S, Coiteux V, Chevret S, et al.: Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol 22 (8): 1404-12, 2004.[PUBMED Abstract]
- Ortega JJ, Madero L, Martín G, et al.: Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol 23 (30): 7632-40, 2005.[PUBMED Abstract]
- Kutny MA, Alonzo TA, Gerbing RB, et al.: Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients With Acute Promyelocytic Leukemia: Report From the Children's Oncology Group Phase III Historically Controlled Trial AAML0631. J Clin Oncol 35 (26): 3021-3029, 2017.[PUBMED Abstract]
- Athale UH, Razzouk BI, Raimondi SC, et al.: Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution's experience. Blood 97 (12): 3727-32, 2001.[PUBMED Abstract]
- Reinhardt D, Diekamp S, Langebrake C, et al.: Acute megakaryoblastic leukemia in children and adolescents, excluding Down's syndrome: improved outcome with intensified induction treatment. Leukemia 19 (8): 1495-6, 2005.[PUBMED Abstract]
- Schweitzer J, Zimmermann M, Rasche M, et al.: Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol 94 (8): 1327-36, 2015.[PUBMED Abstract]
- de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al.: Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood 127 (26): 3424-30, 2016.[PUBMED Abstract]
- de Rooij JD, Branstetter C, Ma J, et al.: Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet 49 (3): 451-456, 2017.[PUBMED Abstract]
- Barbaric D, Alonzo TA, Gerbing RB, et al.: Minimally differentiated acute myeloid leukemia (FAB AML-M0) is associated with an adverse outcome in children: a report from the Children's Oncology Group, studies CCG-2891 and CCG-2961. Blood 109 (6): 2314-21, 2007.[PUBMED Abstract]
- Abbott BL, Rubnitz JE, Tong X, et al.: Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: a single institution's experience. Leukemia 17 (11): 2090-6, 2003.[PUBMED Abstract]
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Central nervous system disease in pediatric acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 64 (12): , 2017.[PUBMED Abstract]
- Johnston DL, Alonzo TA, Gerbing RB, et al.: The presence of central nervous system disease at diagnosis in pediatric acute myeloid leukemia does not affect survival: a Children's Oncology Group study. Pediatr Blood Cancer 55 (3): 414-20, 2010.[PUBMED Abstract]
- Lugthart S, Gröschel S, Beverloo HB, et al.: Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol 28 (24): 3890-8, 2010.[PUBMED Abstract]
- Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al.: Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 120 (16): 3187-205, 2012.[PUBMED Abstract]
- Wheatley K, Burnett AK, Goldstone AH, et al.: A simple, robust, validated and highly predictive index for the determination of risk-directed therapy in acute myeloid leukaemia derived from the MRC AML 10 trial. United Kingdom Medical Research Council's Adult and Childhood Leukaemia Working Parties. Br J Haematol 107 (1): 69-79, 1999.[PUBMED Abstract]
- Marcucci G, Mrózek K, Ruppert AS, et al.: Abnormal cytogenetics at date of morphologic complete remission predicts short overall and disease-free survival, and higher relapse rate in adult acute myeloid leukemia: results from Cancer and Leukemia Group B study 8461. J Clin Oncol 22 (12): 2410-8, 2004.[PUBMED Abstract]
- Sievers EL, Lange BJ, Alonzo TA, et al.: Immunophenotypic evidence of leukemia after induction therapy predicts relapse: results from a prospective Children's Cancer Group study of 252 patients with acute myeloid leukemia. Blood 101 (9): 3398-406, 2003.[PUBMED Abstract]
- Weisser M, Kern W, Rauhut S, et al.: Prognostic impact of RT-PCR-based quantification of WT1 gene expression during MRD monitoring of acute myeloid leukemia. Leukemia 19 (8): 1416-23, 2005.[PUBMED Abstract]
- van der Velden VH, van der Sluijs-Geling A, Gibson BE, et al.: Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia 24 (9): 1599-606, 2010.[PUBMED Abstract]
- Loken MR, Alonzo TA, Pardo L, et al.: Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children's Oncology Group. Blood 120 (8): 1581-8, 2012.[PUBMED Abstract]
- Buldini B, Rizzati F, Masetti R, et al.: Prognostic significance of flow-cytometry evaluation of minimal residual disease in children with acute myeloid leukaemia treated according to the AIEOP-AML 2002/01 study protocol. Br J Haematol 177 (1): 116-126, 2017.[PUBMED Abstract]
- Rubnitz JE, Inaba H, Dahl G, et al.: Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 11 (6): 543-52, 2010.[PUBMED Abstract]
- Tierens A, Bjørklund E, Siitonen S, et al.: Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia; results of the NOPHO-AML 2004 study. Br J Haematol 174 (4): 600-9, 2016.[PUBMED Abstract]
- Buonamici S, Ottaviani E, Testoni N, et al.: Real-time quantitation of minimal residual disease in inv(16)-positive acute myeloid leukemia may indicate risk for clinical relapse and may identify patients in a curable state. Blood 99 (2): 443-9, 2002.[PUBMED Abstract]
- Viehmann S, Teigler-Schlegel A, Bruch J, et al.: Monitoring of minimal residual disease (MRD) by real-time quantitative reverse transcription PCR (RQ-RT-PCR) in childhood acute myeloid leukemia with AML1/ETO rearrangement. Leukemia 17 (6): 1130-6, 2003.[PUBMED Abstract]
- Weisser M, Haferlach C, Hiddemann W, et al.: The quality of molecular response to chemotherapy is predictive for the outcome of AML1-ETO-positive AML and is independent of pretreatment risk factors. Leukemia 21 (6): 1177-82, 2007.[PUBMED Abstract]
- Zhang L, Cao Z, Ruan M, et al.: Monitoring the AML1/ETO fusion transcript to predict outcome in childhood acute myeloid leukemia. Pediatr Blood Cancer 61 (10): 1761-6, 2014.[PUBMED Abstract]
- Krönke J, Schlenk RF, Jensen KO, et al.: Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol 29 (19): 2709-16, 2011.[PUBMED Abstract]
- Corbacioglu A, Scholl C, Schlenk RF, et al.: Prognostic impact of minimal residual disease in CBFB-MYH11-positive acute myeloid leukemia. J Clin Oncol 28 (23): 3724-9, 2010.[PUBMED Abstract]
- Cloos J, Goemans BF, Hess CJ, et al.: Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia 20 (7): 1217-20, 2006.[PUBMED Abstract]
- Mandelli F, Diverio D, Avvisati G, et al.: Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche Maligne dell'Adulto and Associazione Italiana di Ematologia ed Oncologia Pediatrica Cooperative Groups. Blood 90 (3): 1014-21, 1997.[PUBMED Abstract]
- Burnett AK, Grimwade D, Solomon E, et al.: Presenting white blood cell count and kinetics of molecular remission predict prognosis in acute promyelocytic leukemia treated with all-trans retinoic acid: result of the Randomized MRC Trial. Blood 93 (12): 4131-43, 1999.[PUBMED Abstract]
- Diverio D, Rossi V, Avvisati G, et al.: Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter "AIDA" trial. GIMEMA-AIEOP Multicenter "AIDA" Trial. Blood 92 (3): 784-9, 1998.[PUBMED Abstract]
- Martinelli G, Ottaviani E, Testoni N, et al.: Disappearance of PML/RAR alpha acute promyelocytic leukemia-associated transcript during consolidation chemotherapy. Haematologica 83 (11): 985-8, 1998.[PUBMED Abstract]
- Webb DK, Wheatley K, Harrison G, et al.: Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia 13 (1): 25-31, 1999.[PUBMED Abstract]
- Tarlock K, Meshinchi S: Pediatric acute myeloid leukemia: biology and therapeutic implications of genomic variants. Pediatr Clin North Am 62 (1): 75-93, 2015.[PUBMED Abstract]
- Pui CH, Carroll WL, Meshinchi S, et al.: Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 29 (5): 551-65, 2011.[PUBMED Abstract]
- 小児AMLの治療
-
小児および青年の急性骨髄性白血病(AML)に対する治療の一般的な原則を以下に述べ、続いてダウン症候群および急性前骨髄球性白血病(APL)の小児の治療についてさらに具体的に検討する。
過去30年間、AML小児の全生存(OS)率は改善しており、現在の5年生存率は55~65%である。[ 1 ][ 2 ][ 3 ][ 4 ][ 5 ]全寛解導入率は約85~90%で、診断時からのイベントフリー生存(EFS)率は45~55%の範囲である。[ 2 ][ 3 ][ 4 ][ 5 ]しかしながら、AMLの異なる生物学的亜型の転帰は広範囲にわたる(詳しい情報については、本要約の分子的評価およびリスク分類システムのセクションを参照のこと);小児の白血病の特異的な生物学的因子を考慮に入れた上で、個々の患者について予測される転帰はAML小児の一般集団に対する全体の転帰と比較して、はるかに良好である場合もあれば、はるかに不良である場合もある。
寛解導入療法
小児AMLに対する最新の治療プロトコルによって、85~90%の完全寛解(CR)率が得られる。[ 6 ][ 7 ][ 8 ]約2~3%の患者が寛解導入期に死亡し、治療関連合併症によるものが最も多い。[ 6 ][ 7 ][ 8 ][ 9 ]CRを得るには、通常、現在使用されている併用化学療法レジメンによる高度の骨髄低形成を惹起する必要がある(ただし、M3のAPL亜型は除く)。寛解導入化学療法により重度の骨髄抑制が引き起こされるため、寛解導入期の感染症または出血による合併症および死亡が大きな問題となる場合がある。
AML小児に対する寛解導入期の治療法選択肢には以下を含めてもよい:
化学療法
AML患児の寛解導入に用いられる最も有効で不可欠な薬剤には、シタラビンとアントラサイクリン系薬剤の2つがある。一般に用いられる小児の寛解導入療法のレジメンでは、シタラビンおよびアントラサイクリンをエトポシドおよび/またはthioguanineのような別の薬物と併用する。[ 3 ][ 10 ][ 11 ]
証拠(寛解導入化学療法レジメン):
- 英国医学研究審議会(MRC)AML10試験では、シタラビン、ダウノルビシン、およびエトポシド(ADE)に対して、シタラビン、ダウノルビシン、およびthioguanine(DAT)による寛解導入療法が比較された。[ 12 ]
- MRC AML15試験で、ダウノルビシンおよびシタラビン(DA)による寛解導入では、ADEによる寛解導入と比較して同程度の生存率が得られることが実証された。[ 13 ]
AML患児に対する寛解導入レジメンで最もよく用いられているアントラサイクリン系薬剤はダウノルビシンであるが[ 3 ][ 10 ][ 11 ]、イダルビシンおよびアントラセンジオンのミトキサントロンも用いられている。[ 6 ][ 14 ][ 15 ]AML患児に対する寛解導入療法の一部として、他のアントラサイクリン系またはアントラセンジオン系の薬剤がダウノルビシンより優れているかどうかを判定するために、ランダム化試験が行われている。等毒性用量で投与した場合、他のアントラサイクリン系薬剤またはミトキサントロンがダウノルビシンよりも優れた結果をもたらすという説得力のあるデータがないため、米国では依然としてダウノルビシンがAML患児に対する寛解導入療法で最もよく用いられているアントラサイクリン系薬剤である。
証拠(アントラサイクリン系薬剤):
- ドイツのBerlin-Frankfurt-Münster(BFM)グループのAML-BFM 93研究では、シタラビン + エトポシド + ダウノルビシンまたはイダルビシン(ADEまたはAIE)が評価された。[ 11 ][ 14 ]
- MRC-LEUK-AML12(NCT00002658)臨床試験では、AMLの小児および成人を対象にシタラビン、ミトキサントロン、およびエトポシド(MAE)による寛解導入療法が検討され、ダウノルビシンを用いた同様なレジメン(ADE)と比較された。[ 6 ][ 16 ]
- AML-BFM 2004(NCT00111345)臨床試験では、寛解導入療法としてリポソーマルダウノルビシン(L-DNR)とイダルビシンが同等量より高い用量(1日当たり80mg/m2 vs 12mg/m2を3日間投与)で比較された。[ 17 ]
証拠(アントラサイクリン系薬物を低下させた寛解導入レジメン):
- アントラサイクリン系薬物とシタラビンの併用は成人と小児に対する標準の初回寛解導入療法の基礎であるが、必要な場合にはアントラサイクリン系薬物の使用を低下させるために代替の薬物が使用できるという証拠がある。St. Jude Children's Research Hospital(SJCRH)のAML08(NCT00703820)プロトコルでは、患者が寛解導入Iにクロファラビン/シタラビン(CA)または高用量シタラビンとダウノルビシンおよびエトポシドの併用(HD-ADE)のいずれかを受ける群にランダムに割り付けられた;すべての患者が続いて、寛解導入IIにアントラサイクリン系薬物を含む標準用量のADEレジメンを受けた。[ 18 ]
寛解導入療法の強度は、全体的な治療成績に影響を及ぼす。CCG -2891研究では、投与時期が標準的な寛解導入療法(4日間投与し、2週間以上休薬するコース)よりも投与時期が集中的な寛解導入療法(4日間投与し、6日間のみ休薬するコース)の方が良好なEFSが得られたことを明らかにした。[ 19 ]MRCは、シタラビンの投与期間を10日間に延長することにより、寛解導入療法を強化している。[ 10 ]
成人で、この他の寛解導入の強化方法として、高用量シタラビンの使用がある。非高齢者を対象とした研究では、高用量シタラビン(2~3g/m2/回)による強化寛解導入療法を行った方が、標準用量のシタラビンを投与するよりも有利なことが示唆されるが[ 20 ][ 21 ]、小児では、シタラビン1g/m2を1日2回、7日間にわたってダウノルビシンおよびthioguanineと併用投与した場合、標準用量に比して高用量シタラビンを用いる有益性は認められなかった。[ 22 ]別の小児研究でも、高用量シタラビンを導入療法期間に使用した場合の標準用量シタラビンを超える有益性は明らかにならなかった。[ 23 ]
ゲムツズマブオゾガマイシン
寛解導入レジメンをさらに強化しても、毒性が増加し、EFSまたはOSの改善がほとんど得られないため、ゲムツズマブオゾガマイシンの使用など、代替アプローチが検討されている。
証拠(ゲムツズマブオゾガマイシン):
- 小児腫瘍学グループ(COG)は、抗CD33結合抗体のゲムツズマブオゾガマイシンの寛解導入療法への組み込みについて検討した一連の試験-AAML03P1(NCT00070174)のパイロット研究およびAAML0531(NCT00372593)のランダム化試験-を完了した。[ 8 ][ 9 ]
- 比較的高齢の成人を対象にしたALFA-0701(NCT00927498)試験のレトロスペクティブ解析において、CD33の発現量が多いとゲムツズマブオゾガマイシンを用いた治療でより大きな有益性が得られた。[ 26 ]
- AML細胞におけるCD33受容体は構造的なばらつき(多型性)を示し、その結果51%の患者が一塩基多型(SNP)、rs12459419(CCを指定)を発現しており、これらの患者ではゲムツズマブオゾガマイシンを使用した場合に、使用しなかった患者と比較して再燃が有意に減少した(26% vs 49%;P < 0.001)。このSNPの変化により、ゲムツズマブオゾガマイシンが結合するCD33 IgV領域が存在しないCD33アイソフォームが生じており、診断的免疫表現型検査に利用される。[ 27 ]
- AMLの成人に対するゲムツズマブオゾガマイシンについて評価した5件のランダム化臨床試験のメタアナリシスでは、以下が観察された:[ 28 ]
支持療法
現代の強化療法を受けるAMLの小児では、重症細菌感染症の推定発生率が50~60%で、侵襲性真菌感染症の推定発生率が7.0~12.5%である。[ 30 ][ 31 ][ 32 ]AMLの小児において感染による合併症および死亡を低減することに関して、いくつかのアプローチが検討されている。
造血成長因子
AMLの成人を対象とした多施設プラセボ対照研究では、長期間の骨髄抑制に伴う毒性を低下させようと、AML寛解導入療法中の顆粒球マクロファージコロニー刺激因子(GM-CSF)または顆粒球コロニー刺激因子(G-CSF)といった造血成長因子が評価されている。[ 7 ][ 33 ]これらの研究では、G-CSFまたはGM-CSFを用いることにより、一般に好中球減少症の持続期間が数日間短縮されることが示されているが[ 33 ]、治療関連死亡またはOSに対する有意な効果は示されていない。[ 33 ](詳しい情報については、成人急性骨髄性白血病の治療のPDQ要約におけるAMLに対する治療法選択肢の概要のセクションを参照のこと。)
AML小児に対して、造血成長因子のルーチンでの予防的使用は推奨されない。
証拠(造血成長因子):
抗生物質の予防的投与
AML向けの治療を受ける小児では、抗生物質の予防的投与の使用がいくつかの研究で支持されている。1件のプロスペクティブ・ランダム化試験を含む複数の研究で、抗生物質の予防的投与を実施する有益性が示唆されている。
証拠(抗菌薬の予防的投与):
- SJCRHのAML患者を対象としたレトロスペクティブ研究では、経口シプロフロキサシンまたはセファロスポリンと併用した静注(IV)セフェピムまたはバンコマイシンの使用により、細菌感染および敗血症の発生率が経口抗生物質のみの予防的投与を受けた患者または全く受けなかった患者と比較して有意に低下したことが報告された。[ 36 ]
- このSJCRHの結果は、その後の研究で確認された。[ 37 ]
- COG AAML0531(NCT00372593)試験からのレトロスペクティブ報告では、無菌部位細菌感染に加え、特にグラム陽性菌無菌部位感染における有意な低下は、いずれも抗菌薬の予防的投与の使用と関連していることが実証された。[ 38 ]この研究では、G-CSFの予防的投与によって、細菌感染およびクロストリジウム・ディフィシレ(Clostridium difficile)感染が減少したことも報告された。[ 38 ]
- 化学療法とともに抗菌薬または抗真菌薬の予防的投与を受けた急性リンパ芽球性白血病(ALL)またはAMLの小児を対象に、血流感染症または侵襲性真菌感染症の割合を比較した研究では、予防的投与を受けなかった歴史的対照群と比較して、両変数に有意な減少が観察された。[ 39 ]
- 強化化学療法を受ける小児を対象にしたCOGのプロスペクティブACCL0934試験では、患者が異なる2つのグループ-急性白血病患者(AMLまたは再燃したALL患者で構成された)および幹細胞移植を受ける患者に登録された。急性白血病患者は、1~2サイクルの化学療法の好中球減少期間中にレボフロキサシン(n = 96)を受ける群または抗生物質予防投与なしの群(n = 99)にランダムに割り付けられた。[ 40 ]
抗真菌薬の予防的投与
抗真菌薬の予防的投与の役割は、ランダム化プロスペクティブ研究を用いて、AMLの小児を対象に検討されたことがない。
証拠(抗真菌薬の予防的投与):
- 2件のメタアナリシスの報告によると、治療誘発性好中球減少期間中または骨髄移植中のAMLの小児患者における抗真菌薬の予防的投与により、侵襲性真菌感染症の頻度に加え、ある症例では非再燃死亡率が確かに低下することが示唆されている。[ 42 ][ 43 ]
- COG AAML0531(NCT00372593)臨床試験で治療を受けたAML患者1,024人を解析した別の研究では、真菌感染症または非再燃死亡率に対する抗真菌薬の予防的投与の有益性がみられないことが報告された。[ 38 ]
- ただし、AML成人を対象としたいくつかのランダム化試験では、抗真菌薬の予防的投与により侵襲性真菌感染症が減少する点で、有意な有益性が報告されている。このような研究では、有害な副作用と均等した費用となっている;侵襲性真菌感染症を低下させる有効性がこれらの他の因子と均等している場合、ポサコナゾール、ボリコナゾール、カスポファンギン、およびミカファンギンは、妥当な選択と考えられる。[ 39 ][ 44 ][ 45 ][ 46 ][ 47 ][ 48 ]
心臓モニタリング
菌血症または敗血症とアントラサイクリン系薬剤の使用は、左室機能低下として明らかにされる心毒性発生の有意な危険因子として同定されている。[ 49 ][ 50 ]治療中に連続的検査を実施する心機能のモニタリングは、心毒性を発見し、適応であれば治療を調整するための有効な方法である。アントラサイクリン系薬剤のボーラス投与と併用するデクスラゾキサンの使用は、治療中の心機能不全のリスクを低下させる有効な方法となりうる。[ 51 ]
証拠(心臓モニタリング):
入院
治療関連死亡率を低下させるために、顆粒球(好中球または食細胞の絶対数)が十分に回復するまで入院が行われている。COG-2961(NCT00002798)試験では、治療関連死亡率の有意な低下(本試験で他の支持療法の変更とともに入院を必須とする前が19%であったのに対して、その後は12%)が最初に認められた;本試験でOSも改善した(P < 0.001)。[ 3 ]施設でのルーチン医療の調査を用いた入院の効果に関する別の解析によると、入院を必須とした施設では、方針を設定しなかった施設と比較して患者の治療関連死亡率が有意ではないものの低下した(調整後HR、0.60[0.26-1.36、P = 0.22])ことが明らかになった。[ 38 ]この研究で有意な有益性はみられなかったが、著者らは、その方法論(調査方法)、症例の検証不能、および治療関連死亡率の差を判定する検出力の限界を含め、いくつかの限界を認めた。血球数回復までの入院の長期化を避けるために、一部の施設では、外来での静注抗生物質の予防的投与を効果的に使用している。[ 37 ]
寛解導入失敗(不応性AML)
寛解導入失敗(寛解導入の全コース終了時点で形態学的に骨髄芽球が5%以上存在)は、AML小児の10~15%にみられる。寛解導入に失敗した患者のその後の転帰は早期(寛解後12ヵ月未満)に再燃したAML患者の転帰に類似している。[ 52 ][ 53 ]
顆粒球肉腫/緑色腫
顆粒球肉腫(緑色腫)は、白血病細胞の髄外集合巣を示す。まれにではあるが、これらの集合巣が白血病の唯一の証拠として発現することがある。旧Children's Cancer Groupにより実施された3件のAML研究を対象としたレビューでは、孤立性の顆粒球肉腫を認めた患者は1%未満で、診断時の骨髄病変とともに顆粒球肉腫を認めた患者は11%であった。[ 54 ]この発生率は、NOPHO-AML 2004(NCT00476541)試験でもみられた。[ 55 ]
重要な点として、孤立性腫瘍を呈し、骨髄病変の証拠が認められない患者は、全身性疾患を有するものとして治療を受けなければならない。孤立性顆粒球肉腫を認める患者が現在のAML療法による治療を受けた場合は、予後良好である。[ 54 ]
新たにAMLと診断された小児1,459人を対象とした研究によると、眼窩顆粒球肉腫の患者および中枢神経系(CNS)顆粒球肉腫の患者は、これらの部位以外に顆粒球肉腫があり骨髄病変を伴う患者および髄外病変がないAML患者よりも生存が良好であった。[ 55 ][ 56 ]眼窩に顆粒球肉腫を認める患者のほとんどがt(8;21)の異常を有し、予後良好との関係が認められている。化学療法に対して完全奏効を示す顆粒球肉腫患者では、放射線療法を使用しても生存は改善されないが、顆粒球肉腫の部位が化学療法に対して完全奏効を示さない場合、または局所再発する病変に対しては放射線療法が必要な場合がある。[ 54 ]
AMLに対する中枢神経系(CNS)予防法
AMLによるCNS浸潤およびその予後への影響については、本要約の小児AMLの予後因子のセクションで既に考察している。診断時に認められるCNS白血病の治療およびその後のCNS白血病発症の予防に放射線療法または髄腔内化学療法のいずれかによる治療が使用されている。放射線療法の使用は、実証された有益性の欠如および長期の続発症のために、予防手段として基本的に廃止されている。[ 57 ]COGは、CNS予防および治療にいずれもシタラビン単剤を使用している。他のグループは、その他の髄腔内投与薬剤を用いてCNS再燃の予防を試みている。
証拠(CNS予防):
AMLに対する寛解後療法
AML小児の治療における主な課題は、化学療法または造血幹細胞移植(HSCT)を追加することによって、初回の寛解持続期間を延長することである。
寛解後のAML小児の治療法選択肢には以下を含めてもよい:
化学療法
寛解後化学療法には、寛解導入時に用いた薬物の一部を含め、同時に交叉耐性のない薬物に加え、高用量シタラビンも多く再寛解導入に使用される。成人AMLを対象とした研究では、特にinv(16)およびt(8;21)を伴うAML亜型の患者において、標準用量よりも高用量シタラビンを用いる地固め療法レジメンの方が、治療成績を改善することが明らかにされている。[ 60 ][ 61 ](詳しい情報については、成人急性骨髄性白血病の治療のPDQ要約における寛解期の成人AMLのセクションを参照のこと。)小児では寛解後療法における高用量シタラビンの効果を評価するランダム化研究は実施されていないが、歴史的対照を用いた研究によると、高用量シタラビンを用いたレジメンによる地固め療法では、それほど強力ではない地固め療法と比べて転帰が改善することが示唆される。[ 11 ][ 62 ][ 63 ]
寛解後の至適治療コース数は、依然として明らかになっていないが、寛解導入コースを含めて、少なくとも3コースの強化療法が必要と考えられている。[ 3 ]
証拠(寛解後化学療法のコース数):
- 英国医学研究審議会(MRC)の研究では、4または5コースの強化療法に成人および小児患者をランダムに割り付けた。5コース群では、無再燃生存およびOSにおける優位性が認められなかった。[ 6 ][ 16 ][証拠レベル:1iiA]
- このMRCのデータに基づいて、COG AAML1031(NCT01371981)試験では、HSCTを併用せずに治療され第一CR期にある高リスク以外の患者(全患者の73%)が5サイクル(寛解導入2サイクルおよび地固め3サイクル)ではなく4サイクルの化学療法(寛解導入2サイクルおよび地固め2サイクル)を受けた;移植を受けない患者は以前のCOG AAML0531(NCT00372593)およびAAML03P1(NCT00070174)試験では5サイクルの化学療法を受けていた。[
64
]
強化コースの数および使用される特異的薬物に関する追加の研究では、この問題がより適切に扱われるであろうが、これらのデータから、4コースの化学療法は上述の予後良好な集団にのみ実施すべきであり、他のすべての移植を受けない患者は5コースの化学療法を受けるべきであると示唆されている。
HSCT
1970年代後半以降から、第一寛解期におけるHSCTの実施についての評価が行われており、自家および同種HSCTの適応に関して、証拠に基づく評価結果が発表されている。[ 65 ]AML患児を対象とした移植のプロスペクティブ試験によると、HLA適合ドナーが得られ、第一寛解中に同種HSCTを受けた小児全体の60~70%で長期寛解が得られることが示唆されるが[ 10 ][ 66 ]、同種HSCT後の転帰はリスク分類状態に応じて異なるとのただし書きが付されている。[ 67 ]
成人および小児を対象に同種HSCTを化学療法および/または自家HSCTと比較したプロスペクティブ試験によると、家族に6/6座または5/6座のHLA適合ドナーが得られるかどうかで同種骨髄移植群に割り付けられた患者で優れたDFSが認められている。[ 10 ][ 66 ][ 68 ][ 69 ][ 70 ][ 71 ][ 72 ]しかしながら、同種HSCTでは、化学療法を超える優位性が常に観察されているわけではない。[ 73 ]AML患児を対象とした数件の大規模な共同研究グループ臨床試験では、自家HSCTに強化化学療法を上回る有益性は認められなかった。[ 10 ][ 66 ][ 68 ][ 70 ]
現在の同種HSCTの適用には、移植を第一寛解期に追及すべきかどうかの判断にリスク分類を組み入れることが含まれる。予後良好の特徴(低リスクの細胞遺伝学的または分子的変異)が認められる患者が現代的な化学療法レジメンによる治療を受けた場合に転帰が改善すること、さらにこの患者集団におけるHSCTの優位性が実証されていないことから、この患者群には典型的に、最初の再燃後および第二CR達成後にのみ適合家族ドナー(MFD)によるHSCTが実施される。[ 65 ][ 67 ][ 74 ][ 75 ]
中リスクの特徴(低リスクまたは高リスクの細胞遺伝学的または分子的変異がいずれもない)がみられる患者に対する第一寛解期での同種HSCTの役割に関しては、相反するエビデンスがある。
証拠(中リスクのAML患者に対する第一寛解期での同種HSCT):
- POG-8821、CCG-2891、COG-2961(NCT00002798)、およびMRC AML10の研究結果を併合した研究では、中リスクのAML患者において同種HSCTのDFSおよびOSの優位性が確認されたが、良好リスク(inv(16)およびt(8;21))または不良リスク(del(5q)、5または7モノソミー、またはPOG/CCG研究で初回寛解導入後の芽球比率が15%を超える)のAMLでは認められなかった;MRC研究では、高リスク分類に3q異常および複雑な細胞遺伝学的異常が含められた。[ 67 ]この研究の弱点としては、大きな割合の患者がリスク群に割り当てられていないことに加え、最近の臨床試験で観察された結果と比較すると、化学療法に割り付けられた中リスクのAML患者におけるEFSおよびOS率が相対的に低いことが挙げられる。[ 6 ][ 17 ]
- 日本小児AML共同研究グループによるAML99臨床試験では、MFDによるHSCTに割り付けられた中リスク患者においてDFSに有意差が観察されたが、OSでは有意差が認められなかった。[ 76 ]
- このAML-BFM 99臨床試験では、中リスク患者でMFDによるHSCTに割り付けられた患者と化学療法に割り付けられた患者において、DFSまたはOSのいずれにも有意差はないことが明らかになった。[ 73 ]
最近の臨床試験における中リスクAML患者における転帰の改善に加え、同種移植に伴う急性および慢性毒性の負担を考慮して、小児AML治療グループ(COGなど)の多くは、第一寛解期の中リスク患者に化学療法を採用し、以降に考えられる再燃で使用するために同種HSCTを残している。[ 6 ][ 76 ][ 77 ]
病態が高リスクの患者に対する第一寛解期の同種HSCTの役割に関しては、相反するデータが得られており、さまざまな研究グループで使用された高リスクの定義が異なっているために複雑化している。
証拠(高リスクのAML患者に対する第一寛解期での同種HSCT):
- COGおよびCenter for International Blood and Marrow Transplant Research(CIBMTR)からのレトロスペクティブ解析では、7モノソミー/del(7q)、5モノソミー/del(5q)、3q異常、t(6;9)、または複雑な核型として定義される高リスクの細胞遺伝学的異常がみられるAML患者に対する化学療法単独と適合血縁者ドナーおよび適合非血縁者ドナーによるHSCTが比較された。[ 78 ]
- Nordic Society for Pediatric Hematology and Oncology研究では、寛解導入療法に反応がみられなかったAML患者に対して時間集中的な再寛解導入療法を行った後に利用可能な最善のドナーによる移植を実施し、追跡期間中央値2.6年で70%の生存率が得られたことが報告された。[ 79 ][証拠レベル:2A]
- 単一施設のレトロスペクティブ研究で、高リスクAML(FLT3-ITD、11q23のKMT2A[MLL]再構成、5番または7番染色体異常の存在、寛解導入失敗、持続性病変)の連続登録患者36人(0~30歳)は、同種移植前に形態学的に第一寛解であった。[ 80 ]
- AML-BFM 98臨床試験からのサブグループ解析によると、同種HSCTに割り付けられた患者で11q23異常がある場合は生存率が改善するが、11q23異常がない場合は改善がみられないことが明らかになった。[ 73 ]
- FLT3-ITD(高アレル比)の小児で、MFDによるHSCTを受けた患者(n = 6)は、標準化学療法を受けた患者(n = 28)よりOS率が高かった;しかしながら、この研究対象の症例数で結論を下すには力不足である。[ 81 ]
- その後のAML若年成人を対象とした3件の連続した試験からのレトロスペクティブ報告によると、FLT3-ITDの高アレル比の患者は、同種HSCTから利益が得られる(P = 0.03)が、低アレル比の患者は、利益が得られない(P = 0.64)ことが明らかになった。[ 82 ]
- COGの第III相試験のサブセット解析では、新たにAMLと診断された小児における寛解導入療法で、ゲムツズマブオゾガマイシンが評価された。[ 24 ]
すべてではないが、多くの小児臨床試験グループが高リスク患者に対して第一寛解期での同種HSCTを用いている。[ 75 ]例えば、COGのフロントラインAML臨床試験(COG-AAML1031)では、細胞遺伝学的および分子的特徴で予後不良および導入療法終了時のMRD高値を基に治療失敗リスクが高いと予測される患者のみを対象に、第一寛解期で同種HSCTを用いている。一方で、AML-BFM試験では、同種HSCTを第二CR期の患者および不応性AMLに限定している。これは、AML-BFM 98研究からの結果に基づいており、この研究では、第一CR期に同種HSCTを受けた高リスク患者でDFSまたはOSに改善がみられなかった上に、第二CR期に達した相当な割合の患者に対するHSCTを用いた治療が成功したことが明らかになった。[ 73 ][ 83 ]さらに、AML-BFM 98研究において第一寛解期に同種HSCTを受けた小児では、晩期続発症(例えば、心筋症、骨格異常、および肝機能障害または肝硬変)が増加した。[ 73 ]
腫瘍の分子的特徴(例えば、FLT3遺伝子内縦列重複、WT1変異、およびNPM1変異)が転帰および治療への反応(例えば、導入療法後のMRD評価)にも関係していることが続けて認められ、高、中、低リスクAMLの定義が進化していることから、現在および将来の臨床試験では、同種HSCTによる治療を受けた患者亜集団の詳細な解析が引き続き必要となる。
ハプロタイプ一致ドナーを含む代替のドナーソースが研究中ではあるが、第一CR期に移植を選択する場合の移植前処置レジメンの至適タイプおよびドナー細胞のソースは未だ確定していない。[ 72 ][ 84 ][ 85 ]ブスルファンをベースとした骨髄破壊的レジメンよりも全身放射線照射(TBI)が優れていることを示唆するデータはない。[ 73 ][ 74 ]また、treosulfanをベースとしたレジメンで治療された患者で著明な治療成績が示されている;しかしながら、treosulfanとブスルファンまたはTBIを比較した試験は不足している。[ 86 ]
証拠(骨髄破壊的レジメン):
- 第一CR期のAMLに対する移植前処置レジメンとして、ブスルファン + フルダラビンとブスルファン + シクロホスファミドとを比較したランダム化試験により、前者のレジメンの方が毒性作用が少なく、DFSおよびOSが同程度であったことが実証された。[ 87 ]
- また、AML、骨髄異形成症候群(MDS)、および慢性骨髄性白血病(CML)の小児および成人を対象としたCIBMTRによる大規模プロスペクティブ・コホート研究では、早期疾患(慢性期のCML、第一CR期のAML、およびMDS-不応性貧血)の患者がブスルファンをベースとしたレジメンによる治療を受けた場合にTBIよりも優れた生存を示した。[ 88 ]
APL亜型以外で、強力な寛解後療法後の維持療法による寛解持続期間の有意な延長を示すデータはない。現代の強化地固め療法を用いた2件のランダム化研究で維持化学療法の有益性は示されず[ 62 ][ 89 ]、インターロイキン-2による維持療法でも効果がないことが明らかにされた。[ 3 ]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
再発または不応性の小児AMLおよびその他の骨髄性悪性疾患
COGの基準に従ったAMLの再発または再燃の診断は、基本的にAMLの診断を下すための基準と同じである。通常これは、世界保健機関(WHO)分類基準に従ったAMLの診断で、以前に治療後に寛解中であった骨髄芽球が5%を超える患者として定義される。[ 90 ][ 91 ]
初回の寛解導入療法で用いたものと同様な薬剤により治療を行った場合、2分の1を超えるAML患児が第二寛解に誘導されるにもかかわらず、AMLの再発または進行が認められる患児の予後は一般に不良である。[ 52 ][ 92 ]
再発小児AML
再燃の約50~60%は診断後1年以内に認められ、ほとんどの再燃が診断から4年までに生じる。[ 92 ]大半の再燃は骨髄にみられ、CNS再燃はきわめてまれである。[ 92 ]
予後および予後因子
第二寛解の達成能に影響を及ぼす因子には以下のものがある:
その他の予後因子が以下の研究で特定された:
再発AMLの治療
再発AML患児に対する治療法選択肢には以下のものがある:
化学療法
再発AML患児における寛解導入に使用されて効果が認められているレジメンには、以下の薬剤と併用投与される高用量シタラビンが含まれていることが多い:
クロファラビンをベースに構築されたレジメンも使用されている[ 106 ][ 107 ][ 108 ][証拠レベル:2Div];2-クロロアデノシンのレジメンも同様である。[ 109 ]COG AAML0523(NCT00372619)試験では、再燃AML患者を対象にクロファラビン + 高用量シタラビンの併用が評価された;奏効率は48%で、奏効者23人中21人がHSCTを受けて、OS率は46%であった。HSCT前のMRDは、生存の強力な予測因子であった。[ 110 ][証拠レベル:2Di]
新たにAMLと診断された小児を対象とした英国のMRC AML10研究で用いられた標準用量シタラビンのレジメン(シタラビン + ダウノルビシン + エトポシドまたはthioguanine)では、再燃の設定で使用した場合、高用量シタラビンのレジメンで得られたものと同程度の寛解率が得られた。[ 94 ]COG第II相研究では、イダルビシン + 低用量シタラビンへボルテゾミブを追加することで、全CR率が57%となり、エトポシド + 高用量シタラビンへボルテゾミブを追加することで、全CR率が48%となった。[ 111 ]
HSCT
第二完全寛解達成後の追加治療法の選択は、以前の治療および個々の患者に対する考慮事項に依存する。地固め化学療法とその後のHSCTは、従来から推奨されているものの、第二完全寛解が得られた後に治療コースを追加して効果が得られるかに関して、対照を設けたプロスペクティブなデータはない。[ 92 ]
証拠(第二完全寛解後のHSCT):
- BFMグループにより、AMLの小児の治療成績が35年にわたって調査され、全体的な転帰における最大の改善は再燃後の生存における改善であったことが明らかにされた。再燃後または不応性疾患後のこのEFSの改善は、救助療法の一部として幹細胞移植を受けた患者においてのみ示された。[ 112 ]
- 非血縁ドナーによるHSCTでは、第二完全寛解期、再燃確定時、および初回寛解導入失敗時に移植を受けたAML患者の5年無白血病生存率が、それぞれ45%、20%、12%であったことが報告されている。[ 113 ][証拠レベル:3iiA]
- 骨髄性疾患の小児および成人を対象とした大規模なプロスペクティブCIBMTRコホート研究など、多くの研究で、ブスルファンをベースにしたレジメンによりTBIと比較して同程度か、それ以上の生存が示されている。[ 88 ][ 114 ][ 115 ]
- 適合同胞ドナー移植では、一般に最善の転帰が得られているが、1抗原不一致の血縁者または適合非血縁者ドナーでも、GVHDおよび非再燃死亡の発生率増加と引き換えに、きわめて類似した生存が得られる。[ 116 ]臍帯血による転帰は、他の非血縁者ドナーによる転帰と同程度であるが、最低限7/8アレル(HLA A、B、C、DRB1)で一致した患者では、非再燃死亡が少なくなる。[ 117 ]ハプロタイプを一致させるアプローチを使用する頻度が増加しており、小児における他の幹細胞ソースと同程度の転帰が示されている。[ 118 ]小児においてハプロタイプ一致と他の非血縁者ドナーのソースの直接比較は実施されていないが、成人における研究では、同程度の転帰が示されている。[ 119 ]
- 強度縮小アプローチは、小児で使用されて成功しているが、主に骨髄破壊的アプローチが受けられない小児で使用される。[ 120 ]成人を対象としたランダム化試験では、強度縮小レジメンと比較して骨髄破壊的アプローチで優れた転帰が示された。[ 121 ]
CNS再燃
孤立性CNS再燃は、小児AML患者の3~6%にみられる。[ 58 ][ 125 ][ 126 ]孤立性CNS再燃のリスク増加に関連する因子には以下のものがある:[ 125 ]
CNS再燃のリスクは、初回AML診断時のCNS白血病浸潤増加とともに上昇する(孤立性CNS再燃の発生率がCNS1:0.6%、CNS2:2.6%、CNS3:5.8%、P < 0.001;CNS3に対する多変量HR:7.82、P = 0.0003)。[ 58 ]孤立性CNS再燃の転帰は、全身性再燃として治療を実施した場合、骨髄再燃と同程度である。ある研究で、孤立性CNS再燃が認められる小児コホートの8年OS率は26%±16%であった。[ 125 ]CNS再燃は、骨髄再燃の設定でもみられることがあり、その可能性は、診断時のCNS浸潤とともに高まる(同時性CNS再燃の発生率がCNS1:2.7%、CNS2:8.5%、CNS3:9.2%、P < 0.001)。[ 58 ]
不応性小児AML(寛解導入失敗)
不応性AML患児に対する治療法選択肢には以下を含めてもよい:
- 化学療法。
- ゲムツズマブオゾガマイシン。
AMLが再燃した患者と同様に、寛解導入に失敗した患者は、寛解を達成した時点でHSCTに向かうのが通例であり、その理由は、化学療法単独による治療を受けた患者よりもEFSが良好なこと(31.2% vs 5%、P < 0.0001)が諸研究により示唆されるためである。これらの患者にとって形態学的CRの達成は、HSCT後のDFS(46% vs 0%;P = 0.02)の有意な予後因子であり、治療失敗は主に再燃が原因である(再燃リスク、53.9% vs 88.9%;P = 0.02)。[ 127 ]
証拠(ゲムツズマブオゾガマイシンによる不応性AMLの治療):
- SJCRH試験のAML02(NCT00136084)では、初回寛解導入サイクル後に典型的にMRDが少ない(0.1~5.6%)ものの検出可能な場合にゲムツズマブオゾガマイシン単独(n = 17)、または残存するMRDが多い(1~97%)患者に化学療法との併用(n = 29)で投与された。[ 128 ]
- 以前の寛解再導入の試みが失敗した再発/不応性AMLの小児を対象としたゲムツズマブオゾガマイシン単独の第II相試験では、患者30人中11人がCRまたは部分的CRに達し、奏効者と非奏効者で3年OSは27% vs 0%(P = 0.001)であった。[ 129 ]
臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、全米および/または各施設で現在実施されている臨床試験の例である。
- NCT03071276(不応性または再燃AML、ALL、またはMDSの若年患者の治療におけるselinexor、リン酸フルダラビン、およびシタラビン):選択的核エクスポート阻害剤selinexorの追加、つまり一般的なAMLの寛解再導入の基本骨格へ追加した場合に研究のエンドポイントである完全奏効が改善されるかどうかを検討するSJCRHがスポンサーとなった単群、オープンラベル、第II相試験。
- NCT02538965(再燃または不応性AMLの小児被験者におけるレナリドミド研究):この業界共同/COG研究のAAML1522は、初回転帰として最大4サイクル以内に完全奏効が認められた再燃または不応性AMLの小児に対する単剤としてのレナリドミドの活性、安全性、および薬物動態について評価する単群、オープンラベル、第II相試験である。
- NCT02642965(再燃または不応性AMLの若年患者の治療におけるリポソーマルシタラビン-ダウノルビシンのCPX-351、リン酸フルダラビン、シタラビン、およびフィルグラスチム):この初回AML再燃の小児に対する第I/II相COG試験のAAML1421では、シタラビンとダウノルビシンの2つの薬物の新規リポソーム製剤をサイクル1で5:1の固定モル濃度で使用し、これらの従来からAMLに使用されている2つの薬物を製剤化するこの方法では、毒性および全奏効率の主要転帰により判定して、毒性が低下し、より有効であるかどうかを検討する。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Ries LAG, Melbert D, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2005. Bethesda, Md: National Cancer Institute, 2007. Also available online. Last accessed March 25, 2020.[PUBMED Abstract]
- Gibson BE, Wheatley K, Hann IM, et al.: Treatment strategy and long-term results in paediatric patients treated in consecutive UK AML trials. Leukemia 19 (12): 2130-8, 2005.[PUBMED Abstract]
- Lange BJ, Smith FO, Feusner J, et al.: Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood 111 (3): 1044-53, 2008.[PUBMED Abstract]
- Creutzig U, Büchner T, Sauerland MC, et al.: Significance of age in acute myeloid leukemia patients younger than 30 years: a common analysis of the pediatric trials AML-BFM 93/98 and the adult trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 112 (3): 562-71, 2008.[PUBMED Abstract]
- Kaspers GJ, Creutzig U: Pediatric acute myeloid leukemia: international progress and future directions. Leukemia 19 (12): 2025-9, 2005.[PUBMED Abstract]
- Gibson BE, Webb DK, Howman AJ, et al.: Results of a randomized trial in children with Acute Myeloid Leukaemia: medical research council AML12 trial. Br J Haematol 155 (3): 366-76, 2011.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Lehrnbecher T, et al.: Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AML-BFM 98. J Clin Oncol 24 (27): 4499-506, 2006.[PUBMED Abstract]
- Cooper TM, Franklin J, Gerbing RB, et al.: AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's Oncology Group. Cancer 118 (3): 761-9, 2012.[PUBMED Abstract]
- Gamis AS, Alonzo TA, Meshinchi S, et al.: Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol 32 (27): 3021-32, 2014.[PUBMED Abstract]
- Stevens RF, Hann IM, Wheatley K, et al.: Marked improvements in outcome with chemotherapy alone in paediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council's 10th AML trial. MRC Childhood Leukaemia Working Party. Br J Haematol 101 (1): 130-40, 1998.[PUBMED Abstract]
- Creutzig U, Ritter J, Zimmermann M, et al.: Improved treatment results in high-risk pediatric acute myeloid leukemia patients after intensification with high-dose cytarabine and mitoxantrone: results of Study Acute Myeloid Leukemia-Berlin-Frankfurt-Münster 93. J Clin Oncol 19 (10): 2705-13, 2001.[PUBMED Abstract]
- Hann IM, Stevens RF, Goldstone AH, et al.: Randomized comparison of DAT versus ADE as induction chemotherapy in children and younger adults with acute myeloid leukemia. Results of the Medical Research Council's 10th AML trial (MRC AML10). Adult and Childhood Leukaemia Working Parties of the Medical Research Council. Blood 89 (7): 2311-8, 1997.[PUBMED Abstract]
- Burnett AK, Russell NH, Hills RK, et al.: Optimization of chemotherapy for younger patients with acute myeloid leukemia: results of the medical research council AML15 trial. J Clin Oncol 31 (27): 3360-8, 2013.[PUBMED Abstract]
- Creutzig U, Ritter J, Zimmermann M, et al.: Idarubicin improves blast cell clearance during induction therapy in children with AML: results of study AML-BFM 93. AML-BFM Study Group. Leukemia 15 (3): 348-54, 2001.[PUBMED Abstract]
- Pession A, Masetti R, Rizzari C, et al.: Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 122 (2): 170-8, 2013.[PUBMED Abstract]
- Burnett AK, Hills RK, Milligan DW, et al.: Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol 28 (4): 586-95, 2010.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Bourquin JP, et al.: Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from Study AML-BFM 2004. Blood 122 (1): 37-43, 2013.[PUBMED Abstract]
- Rubnitz JE, Lacayo NJ, Inaba H, et al.: Clofarabine Can Replace Anthracyclines and Etoposide in Remission Induction Therapy for Childhood Acute Myeloid Leukemia: The AML08 Multicenter, Randomized Phase III Trial. J Clin Oncol 37 (23): 2072-2081, 2019.[PUBMED Abstract]
- Woods WG, Kobrinsky N, Buckley JD, et al.: Timed-sequential induction therapy improves postremission outcome in acute myeloid leukemia: a report from the Children's Cancer Group. Blood 87 (12): 4979-89, 1996.[PUBMED Abstract]
- Weick JK, Kopecky KJ, Appelbaum FR, et al.: A randomized investigation of high-dose versus standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute myeloid leukemia: a Southwest Oncology Group study. Blood 88 (8): 2841-51, 1996.[PUBMED Abstract]
- Bishop JF, Matthews JP, Young GA, et al.: A randomized study of high-dose cytarabine in induction in acute myeloid leukemia. Blood 87 (5): 1710-7, 1996.[PUBMED Abstract]
- Becton D, Dahl GV, Ravindranath Y, et al.: Randomized use of cyclosporin A (CsA) to modulate P-glycoprotein in children with AML in remission: Pediatric Oncology Group Study 9421. Blood 107 (4): 1315-24, 2006.[PUBMED Abstract]
- Rubnitz JE, Inaba H, Dahl G, et al.: Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol 11 (6): 543-52, 2010.[PUBMED Abstract]
- Tarlock K, Alonzo TA, Gerbing RB, et al.: Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (8): 1951-7, 2016.[PUBMED Abstract]
- Pollard JA, Loken M, Gerbing RB, et al.: CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 34 (7): 747-55, 2016.[PUBMED Abstract]
- Olombel G, Guerin E, Guy J, et al.: The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood 127 (17): 2157-60, 2016.[PUBMED Abstract]
- Lamba JK, Chauhan L, Shin M, et al.: CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children's Oncology Group Trial AAML0531. J Clin Oncol 35 (23): 2674-2682, 2017.[PUBMED Abstract]
- Hills RK, Castaigne S, Appelbaum FR, et al.: Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 15 (9): 986-96, 2014.[PUBMED Abstract]
- Castaigne S, Pautas C, Terré C, et al.: Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 379 (9825): 1508-16, 2012.[PUBMED Abstract]
- Sung L, Gamis A, Alonzo TA, et al.: Infections and association with different intensity of chemotherapy in children with acute myeloid leukemia. Cancer 115 (5): 1100-8, 2009.[PUBMED Abstract]
- Kaya Z, Gursel T, Kocak U, et al.: Invasive fungal infections in pediatric leukemia patients receiving fluconazole prophylaxis. Pediatr Blood Cancer 52 (4): 470-5, 2009.[PUBMED Abstract]
- Kobayashi R, Kaneda M, Sato T, et al.: The clinical feature of invasive fungal infection in pediatric patients with hematologic and malignant diseases: a 10-year analysis at a single institution at Japan. J Pediatr Hematol Oncol 30 (12): 886-90, 2008.[PUBMED Abstract]
- Ozer H, Armitage JO, Bennett CL, et al.: 2000 update of recommendations for the use of hematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. American Society of Clinical Oncology Growth Factors Expert Panel. J Clin Oncol 18 (20): 3558-85, 2000.[PUBMED Abstract]
- Lehrnbecher T, Zimmermann M, Reinhardt D, et al.: Prophylactic human granulocyte colony-stimulating factor after induction therapy in pediatric acute myeloid leukemia. Blood 109 (3): 936-43, 2007.[PUBMED Abstract]
- Ehlers S, Herbst C, Zimmermann M, et al.: Granulocyte colony-stimulating factor (G-CSF) treatment of childhood acute myeloid leukemias that overexpress the differentiation-defective G-CSF receptor isoform IV is associated with a higher incidence of relapse. J Clin Oncol 28 (15): 2591-7, 2010.[PUBMED Abstract]
- Kurt B, Flynn P, Shenep JL, et al.: Prophylactic antibiotics reduce morbidity due to septicemia during intensive treatment for pediatric acute myeloid leukemia. Cancer 113 (2): 376-82, 2008.[PUBMED Abstract]
- Inaba H, Gaur AH, Cao X, et al.: Feasibility, efficacy, and adverse effects of outpatient antibacterial prophylaxis in children with acute myeloid leukemia. Cancer 120 (13): 1985-92, 2014.[PUBMED Abstract]
- Sung L, Aplenc R, Alonzo TA, et al.: Effectiveness of supportive care measures to reduce infections in pediatric AML: a report from the Children's Oncology Group. Blood 121 (18): 3573-7, 2013.[PUBMED Abstract]
- Yeh TC, Liu HC, Hou JY, et al.: Severe infections in children with acute leukemia undergoing intensive chemotherapy can successfully be prevented by ciprofloxacin, voriconazole, or micafungin prophylaxis. Cancer 120 (8): 1255-62, 2014.[PUBMED Abstract]
- Alexander S, Fisher BT, Gaur AH, et al.: Effect of Levofloxacin Prophylaxis on Bacteremia in Children With Acute Leukemia or Undergoing Hematopoietic Stem Cell Transplantation: A Randomized Clinical Trial. JAMA 320 (10): 995-1004, 2018.[PUBMED Abstract]
- Taplitz RA, Kennedy EB, Bow EJ, et al.: Antimicrobial Prophylaxis for Adult Patients With Cancer-Related Immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. J Clin Oncol 36 (30): 3043-3054, 2018.[PUBMED Abstract]
- Ethier MC, Science M, Beyene J, et al.: Mould-active compared with fluconazole prophylaxis to prevent invasive fungal diseases in cancer patients receiving chemotherapy or haematopoietic stem-cell transplantation: a systematic review and meta-analysis of randomised controlled trials. Br J Cancer 106 (10): 1626-37, 2012.[PUBMED Abstract]
- Robenshtok E, Gafter-Gvili A, Goldberg E, et al.: Antifungal prophylaxis in cancer patients after chemotherapy or hematopoietic stem-cell transplantation: systematic review and meta-analysis. J Clin Oncol 25 (34): 5471-89, 2007.[PUBMED Abstract]
- Mandhaniya S, Swaroop C, Thulkar S, et al.: Oral voriconazole versus intravenous low dose amphotericin B for primary antifungal prophylaxis in pediatric acute leukemia induction: a prospective, randomized, clinical study. J Pediatr Hematol Oncol 33 (8): e333-41, 2011.[PUBMED Abstract]
- Mattiuzzi GN, Kantarjian H, Faderl S, et al.: Amphotericin B lipid complex as prophylaxis of invasive fungal infections in patients with acute myelogenous leukemia and myelodysplastic syndrome undergoing induction chemotherapy. Cancer 100 (3): 581-9, 2004.[PUBMED Abstract]
- Mattiuzzi GN, Kantarjian H, O'Brien S, et al.: Intravenous itraconazole for prophylaxis of systemic fungal infections in patients with acute myelogenous leukemia and high-risk myelodysplastic syndrome undergoing induction chemotherapy. Cancer 100 (3): 568-73, 2004.[PUBMED Abstract]
- Tacke D, Buchheidt D, Karthaus M, et al.: Primary prophylaxis of invasive fungal infections in patients with haematologic malignancies. 2014 update of the recommendations of the Infectious Diseases Working Party of the German Society for Haematology and Oncology. Ann Hematol 93 (9): 1449-56, 2014.[PUBMED Abstract]
- Grau S, de la Cámara R, Sabater FJ, et al.: Cost-effectiveness of posaconazole versus fluconazole or itraconazole in the prevention of invasive fungal infections among high-risk neutropenic patients in Spain. BMC Infect Dis 12: 83, 2012.[PUBMED Abstract]
- Getz KD, Sung L, Ky B, et al.: Occurrence of Treatment-Related Cardiotoxicity and Its Impact on Outcomes Among Children Treated in the AAML0531 Clinical Trial: A Report From the Children's Oncology Group. J Clin Oncol 37 (1): 12-21, 2019.[PUBMED Abstract]
- Feijen EAM, Leisenring WM, Stratton KL, et al.: Derivation of Anthracycline and Anthraquinone Equivalence Ratios to Doxorubicin for Late-Onset Cardiotoxicity. JAMA Oncol 5 (6): 864-871, 2019.[PUBMED Abstract]
- Getz KD, Sung L, Leger K, et al.: Effect of dexrazoxane on left ventricular function and treatment outcomes in patients with acute myeloid leukemia: a Children's Oncology Group report. [Abstract] J Clin Oncol 36 (Suppl 18): A-10501, 2018. Also available online. Last accessed March 25, 2020.[PUBMED Abstract]
- Wells RJ, Adams MT, Alonzo TA, et al.: Mitoxantrone and cytarabine induction, high-dose cytarabine, and etoposide intensification for pediatric patients with relapsed or refractory acute myeloid leukemia: Children's Cancer Group Study 2951. J Clin Oncol 21 (15): 2940-7, 2003.[PUBMED Abstract]
- Aplenc R, Alonzo TA, Gerbing RB, et al.: Safety and efficacy of gemtuzumab ozogamicin in combination with chemotherapy for pediatric acute myeloid leukemia: a report from the Children's Oncology Group. J Clin Oncol 26 (14): 2390-3295, 2008.[PUBMED Abstract]
- Dusenbery KE, Howells WB, Arthur DC, et al.: Extramedullary leukemia in children with newly diagnosed acute myeloid leukemia: a report from the Children's Cancer Group. J Pediatr Hematol Oncol 25 (10): 760-8, 2003.[PUBMED Abstract]
- Støve HK, Sandahl JD, Abrahamsson J, et al.: Extramedullary leukemia in children with acute myeloid leukemia: A population-based cohort study from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Pediatr Blood Cancer 64 (12): , 2017.[PUBMED Abstract]
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Superior outcome of pediatric acute myeloid leukemia patients with orbital and CNS myeloid sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 58 (4): 519-24, 2012.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Bourquin JP, et al.: CNS irradiation in pediatric acute myleoid leukemia: equal results by 12 or 18 Gy in studies AML-BFM98 and 2004. Pediatr Blood Cancer 57 (6): 986-92, 2011.[PUBMED Abstract]
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Central nervous system disease in pediatric acute myeloid leukemia: A report from the Children's Oncology Group. Pediatr Blood Cancer 64 (12): , 2017.[PUBMED Abstract]
- Pui CH, Howard SC: Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol 9 (3): 257-68, 2008.[PUBMED Abstract]
- Mayer RJ, Davis RB, Schiffer CA, et al.: Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med 331 (14): 896-903, 1994.[PUBMED Abstract]
- Cassileth PA, Lynch E, Hines JD, et al.: Varying intensity of postremission therapy in acute myeloid leukemia. Blood 79 (8): 1924-30, 1992.[PUBMED Abstract]
- Wells RJ, Woods WG, Buckley JD, et al.: Treatment of newly diagnosed children and adolescents with acute myeloid leukemia: a Childrens Cancer Group study. J Clin Oncol 12 (11): 2367-77, 1994.[PUBMED Abstract]
- Wells RJ, Woods WG, Lampkin BC, et al.: Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Childrens Cancer Group. J Clin Oncol 11 (3): 538-45, 1993.[PUBMED Abstract]
- Getz KD, Alonzo TA, Sung L, et al.: Four versus five chemotherapy courses in patients with low risk acute myeloid leukemia: a Children's Oncology Group report. [Abstract] J Clin Oncol 35 (Suppl 15): A-10515, 2017. Also available online. Last accessed March 25, 2020.[PUBMED Abstract]
- Oliansky DM, Rizzo JD, Aplan PD, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute myeloid leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 13 (1): 1-25, 2007.[PUBMED Abstract]
- Woods WG, Neudorf S, Gold S, et al.: A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission. Blood 97 (1): 56-62, 2001.[PUBMED Abstract]
- Horan JT, Alonzo TA, Lyman GH, et al.: Impact of disease risk on efficacy of matched related bone marrow transplantation for pediatric acute myeloid leukemia: the Children's Oncology Group. J Clin Oncol 26 (35): 5797-801, 2008.[PUBMED Abstract]
- Ravindranath Y, Yeager AM, Chang MN, et al.: Autologous bone marrow transplantation versus intensive consolidation chemotherapy for acute myeloid leukemia in childhood. Pediatric Oncology Group. N Engl J Med 334 (22): 1428-34, 1996.[PUBMED Abstract]
- Feig SA, Lampkin B, Nesbit ME, et al.: Outcome of BMT during first complete remission of AML: a comparison of two sequential studies by the Children's Cancer Group. Bone Marrow Transplant 12 (1): 65-71, 1993.[PUBMED Abstract]
- Amadori S, Testi AM, Aricò M, et al.: Prospective comparative study of bone marrow transplantation and postremission chemotherapy for childhood acute myelogenous leukemia. The Associazione Italiana Ematologia ed Oncologia Pediatrica Cooperative Group. J Clin Oncol 11 (6): 1046-54, 1993.[PUBMED Abstract]
- Bleakley M, Lau L, Shaw PJ, et al.: Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant 29 (10): 843-52, 2002.[PUBMED Abstract]
- Koreth J, Schlenk R, Kopecky KJ, et al.: Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA 301 (22): 2349-61, 2009.[PUBMED Abstract]
- Klusmann JH, Reinhardt D, Zimmermann M, et al.: The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica 97 (1): 21-9, 2012.[PUBMED Abstract]
- Creutzig U, Reinhardt D: Current controversies: which patients with acute myeloid leukaemia should receive a bone marrow transplantation?--a European view. Br J Haematol 118 (2): 365-77, 2002.[PUBMED Abstract]
- Niewerth D, Creutzig U, Bierings MB, et al.: A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia. Blood 116 (13): 2205-14, 2010.[PUBMED Abstract]
- Tsukimoto I, Tawa A, Horibe K, et al.: Risk-stratified therapy and the intensive use of cytarabine improves the outcome in childhood acute myeloid leukemia: the AML99 trial from the Japanese Childhood AML Cooperative Study Group. J Clin Oncol 27 (24): 4007-13, 2009.[PUBMED Abstract]
- Abrahamsson J, Forestier E, Heldrup J, et al.: Response-guided induction therapy in pediatric acute myeloid leukemia with excellent remission rate. J Clin Oncol 29 (3): 310-5, 2011.[PUBMED Abstract]
- Kelly MJ, Horan JT, Alonzo TA, et al.: Comparable survival for pediatric acute myeloid leukemia with poor-risk cytogenetics following chemotherapy, matched related donor, or unrelated donor transplantation. Pediatr Blood Cancer 61 (2): 269-75, 2014.[PUBMED Abstract]
- Wareham NE, Heilmann C, Abrahamsson J, et al.: Outcome of poor response paediatric AML using early SCT. Eur J Haematol 90 (3): 187-94, 2013.[PUBMED Abstract]
- Burke MJ, Wagner JE, Cao Q, et al.: Allogeneic hematopoietic cell transplantation in first remission abrogates poor outcomes associated with high-risk pediatric acute myeloid leukemia. Biol Blood Marrow Transplant 19 (7): 1021-5, 2013.[PUBMED Abstract]
- Meshinchi S, Alonzo TA, Stirewalt DL, et al.: Clinical implications of FLT3 mutations in pediatric AML. Blood 108 (12): 3654-61, 2006.[PUBMED Abstract]
- Schlenk RF, Kayser S, Bullinger L, et al.: Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood 124 (23): 3441-9, 2014.[PUBMED Abstract]
- Beier R, Albert MH, Bader P, et al.: Allo-SCT using BU, CY and melphalan for children with AML in second CR. Bone Marrow Transplant 48 (5): 651-6, 2013.[PUBMED Abstract]
- Liu DH, Xu LP, Liu KY, et al.: Long-term outcomes of unmanipulated haploidentical HSCT for paediatric patients with acute leukaemia. Bone Marrow Transplant 48 (12): 1519-24, 2013.[PUBMED Abstract]
- Locatelli F, Masetti R, Rondelli R, et al.: Outcome of children with high-risk acute myeloid leukemia given autologous or allogeneic hematopoietic cell transplantation in the aieop AML-2002/01 study. Bone Marrow Transplant 50 (2): 181-8, 2015.[PUBMED Abstract]
- Nemecek ER, Hilger RA, Adams A, et al.: Treosulfan, Fludarabine, and Low-Dose Total Body Irradiation for Children and Young Adults with Acute Myeloid Leukemia or Myelodysplastic Syndrome Undergoing Allogeneic Hematopoietic Cell Transplantation: Prospective Phase II Trial of the Pediatric Blood and Marrow Transplant Consortium. Biol Blood Marrow Transplant 24 (8): 1651-1656, 2018.[PUBMED Abstract]
- Liu H, Zhai X, Song Z, et al.: Busulfan plus fludarabine as a myeloablative conditioning regimen compared with busulfan plus cyclophosphamide for acute myeloid leukemia in first complete remission undergoing allogeneic hematopoietic stem cell transplantation: a prospective and multicenter study. J Hematol Oncol 6: 15, 2013.[PUBMED Abstract]
- Bredeson C, LeRademacher J, Kato K, et al.: Prospective cohort study comparing intravenous busulfan to total body irradiation in hematopoietic cell transplantation. Blood 122 (24): 3871-8, 2013.[PUBMED Abstract]
- Perel Y, Auvrignon A, Leblanc T, et al.: Treatment of childhood acute myeloblastic leukemia: dose intensification improves outcome and maintenance therapy is of no benefit--multicenter studies of the French LAME (Leucémie Aiguë Myéloblastique Enfant) Cooperative Group. Leukemia 19 (12): 2082-9, 2005.[PUBMED Abstract]
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 110-23.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Webb DK: Management of relapsed acute myeloid leukaemia. Br J Haematol 106 (4): 851-9, 1999.[PUBMED Abstract]
- Stahnke K, Boos J, Bender-Götze C, et al.: Duration of first remission predicts remission rates and long-term survival in children with relapsed acute myelogenous leukemia. Leukemia 12 (10): 1534-8, 1998.[PUBMED Abstract]
- Webb DK, Wheatley K, Harrison G, et al.: Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia 13 (1): 25-31, 1999.[PUBMED Abstract]
- Nakayama H, Tabuchi K, Tawa A, et al.: Outcome of children with relapsed acute myeloid leukemia following initial therapy under the AML99 protocol. Int J Hematol 100 (2): 171-9, 2014.[PUBMED Abstract]
- Gorman MF, Ji L, Ko RH, et al.: Outcome for children treated for relapsed or refractory acute myelogenous leukemia (rAML): a Therapeutic Advances in Childhood Leukemia (TACL) Consortium study. Pediatr Blood Cancer 55 (3): 421-9, 2010.[PUBMED Abstract]
- Bachas C, Schuurhuis GJ, Reinhardt D, et al.: Clinical relevance of molecular aberrations in paediatric acute myeloid leukaemia at first relapse. Br J Haematol 166 (6): 902-10, 2014.[PUBMED Abstract]
- Sander A, Zimmermann M, Dworzak M, et al.: Consequent and intensified relapse therapy improved survival in pediatric AML: results of relapse treatment in 379 patients of three consecutive AML-BFM trials. Leukemia 24 (8): 1422-8, 2010.[PUBMED Abstract]
- Creutzig U, Zimmermann M, Dworzak MN, et al.: The prognostic significance of early treatment response in pediatric relapsed acute myeloid leukemia: results of the international study Relapsed AML 2001/01. Haematologica 99 (9): 1472-8, 2014.[PUBMED Abstract]
- Karlsson L, Forestier E, Hasle H, et al.: Outcome after intensive reinduction therapy and allogeneic stem cell transplant in paediatric relapsed acute myeloid leukaemia. Br J Haematol 178 (4): 592-602, 2017.[PUBMED Abstract]
- Dinndorf PA, Avramis VI, Wiersma S, et al.: Phase I/II study of idarubicin given with continuous infusion fludarabine followed by continuous infusion cytarabine in children with acute leukemia: a report from the Children's Cancer Group. J Clin Oncol 15 (8): 2780-5, 1997.[PUBMED Abstract]
- Fleischhack G, Hasan C, Graf N, et al.: IDA-FLAG (idarubicin, fludarabine, cytarabine, G-CSF), an effective remission-induction therapy for poor-prognosis AML of childhood prior to allogeneic or autologous bone marrow transplantation: experiences of a phase II trial. Br J Haematol 102 (3): 647-55, 1998.[PUBMED Abstract]
- Tavil B, Aytac S, Balci YI, et al.: Fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) for the treatment of children with poor-prognosis acute leukemia: the Hacettepe experience. Pediatr Hematol Oncol 27 (7): 517-28, 2010.[PUBMED Abstract]
- Capizzi RL, Davis R, Powell B, et al.: Synergy between high-dose cytarabine and asparaginase in the treatment of adults with refractory and relapsed acute myelogenous leukemia--a Cancer and Leukemia Group B Study. J Clin Oncol 6 (3): 499-508, 1988.[PUBMED Abstract]
- Kaspers GJ, Zimmermann M, Reinhardt D, et al.: Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J Clin Oncol 31 (5): 599-607, 2013.[PUBMED Abstract]
- Hijiya N, Gaynon P, Barry E, et al.: A multi-center phase I study of clofarabine, etoposide and cyclophosphamide in combination in pediatric patients with refractory or relapsed acute leukemia. Leukemia 23 (12): 2259-64, 2009.[PUBMED Abstract]
- Jeha S, Razzouk B, Rytting M, et al.: Phase II study of clofarabine in pediatric patients with refractory or relapsed acute myeloid leukemia. J Clin Oncol 27 (26): 4392-7, 2009.[PUBMED Abstract]
- Shukla N, Kobos R, Renaud T, et al.: Phase II trial of clofarabine with topotecan, vinorelbine, and thiotepa in pediatric patients with relapsed or refractory acute leukemia. Pediatr Blood Cancer 61 (3): 431-5, 2014.[PUBMED Abstract]
- Chaleff S, Hurwitz CA, Chang M, et al.: Phase II study of 2-chlorodeoxyadenosine plus idarubicin for children with acute myeloid leukaemia in first relapse: a paediatric oncology group study. Br J Haematol 156 (5): 649-55, 2012.[PUBMED Abstract]
- Cooper TM, Alonzo TA, Gerbing RB, et al.: AAML0523: a report from the Children's Oncology Group on the efficacy of clofarabine in combination with cytarabine in pediatric patients with recurrent acute myeloid leukemia. Cancer 120 (16): 2482-9, 2014.[PUBMED Abstract]
- Horton TM, Perentesis JP, Gamis AS, et al.: A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 61 (10): 1754-60, 2014.[PUBMED Abstract]
- Rasche M, Zimmermann M, Borschel L, et al.: Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 32 (10): 2167-2177, 2018.[PUBMED Abstract]
- Bunin NJ, Davies SM, Aplenc R, et al.: Unrelated donor bone marrow transplantation for children with acute myeloid leukemia beyond first remission or refractory to chemotherapy. J Clin Oncol 26 (26): 4326-32, 2008.[PUBMED Abstract]
- Woodard P, Carpenter PA, Davies SM, et al.: Unrelated donor bone marrow transplantation for myelodysplastic syndrome in children. Biol Blood Marrow Transplant 17 (5): 723-8, 2011.[PUBMED Abstract]
- Uberti JP, Agovi MA, Tarima S, et al.: Comparative analysis of BU and CY versus CY and TBI in full intensity unrelated marrow donor transplantation for AML, CML and myelodysplasia. Bone Marrow Transplant 46 (1): 34-43, 2011.[PUBMED Abstract]
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010.[PUBMED Abstract]
- Eapen M, Klein JP, Ruggeri A, et al.: Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood 123 (1): 133-40, 2014.[PUBMED Abstract]
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.[PUBMED Abstract]
- Rashidi A, DiPersio JF, Westervelt P, et al.: Comparison of Outcomes after Peripheral Blood Haploidentical versus Matched Unrelated Donor Allogeneic Hematopoietic Cell Transplantation in Patients with Acute Myeloid Leukemia: A Retrospective Single-Center Review. Biol Blood Marrow Transplant 22 (9): 1696-1701, 2016.[PUBMED Abstract]
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.[PUBMED Abstract]
- Scott BL, Pasquini MC, Logan BR, et al.: Myeloablative Versus Reduced-Intensity Hematopoietic Cell Transplantation for Acute Myeloid Leukemia and Myelodysplastic Syndromes. J Clin Oncol 35 (11): 1154-1161, 2017.[PUBMED Abstract]
- Meshinchi S, Leisenring WM, Carpenter PA, et al.: Survival after second hematopoietic stem cell transplantation for recurrent pediatric acute myeloid leukemia. Biol Blood Marrow Transplant 9 (11): 706-13, 2003.[PUBMED Abstract]
- Nishikawa T, Inagaki J, Nagatoshi Y, et al.: The second therapeutic trial for children with hematological malignancies who relapsed after their first allogeneic SCT: long-term outcomes. Pediatr Transplant 16 (7): 722-8, 2012.[PUBMED Abstract]
- Yaniv I, Krauss AC, Beohou E, et al.: Second Hematopoietic Stem Cell Transplantation for Post-Transplantation Relapsed Acute Leukemia in Children: A Retrospective EBMT-PDWP Study. Biol Blood Marrow Transplant 24 (8): 1629-1642, 2018.[PUBMED Abstract]
- Johnston DL, Alonzo TA, Gerbing RB, et al.: Risk factors and therapy for isolated central nervous system relapse of pediatric acute myeloid leukemia. J Clin Oncol 23 (36): 9172-8, 2005.[PUBMED Abstract]
- Abbott BL, Rubnitz JE, Tong X, et al.: Clinical significance of central nervous system involvement at diagnosis of pediatric acute myeloid leukemia: a single institution's experience. Leukemia 17 (11): 2090-6, 2003.[PUBMED Abstract]
- Quarello P, Fagioli F, Basso G, et al.: Outcome of children with acute myeloid leukaemia (AML) experiencing primary induction failure in the AIEOP AML 2002/01 clinical trial. Br J Haematol 171 (4): 566-73, 2015.[PUBMED Abstract]
- O'Hear C, Inaba H, Pounds S, et al.: Gemtuzumab ozogamicin can reduce minimal residual disease in patients with childhood acute myeloid leukemia. Cancer 119 (22): 4036-43, 2013.[PUBMED Abstract]
- Zwaan CM, Reinhardt D, Zimmerman M, et al.: Salvage treatment for children with refractory first or second relapse of acute myeloid leukaemia with gemtuzumab ozogamicin: results of a phase II study. Br J Haematol 148 (5): 768-76, 2010.[PUBMED Abstract]
- 急性前骨髄球性白血病(APL)
-
急性前骨髄球性白血病(APL)は、異なる急性骨髄性白血病(AML)の亜型であり、その理由として以下のいくつかの要因がある:
これらのAPLに特有な特徴により、治療開始から最初の数日間における凝固障害性合併症を避けるために適切な支持療法手段が開始できるように、診断時に疑念指数を高めることが避けられない。凝固障害性合併症のリスクを最小化する治療の異なる導入レジメンを策定すること、およびAPLに対する過去のアプローチよりも、また他のAML型の患者に対する転帰と比較しても、はるかに改善された長期の無再燃生存および全生存(OS)を提供することも不可欠である。[ 2 ][ 3 ]
分子的異常
APLに特徴的にみられる染色体異常は、t(15;17)である。この転座には、レチノイン酸受容体遺伝子を含む切断点が含まれているため、前骨髄球性白血病(PML)-レチノイン酸受容体α(RARA)の融合蛋白が産生される。[ 1 ]
APLの診断が疑われる患者は、PML-RARA融合の検出(例、蛍光in situハイブリダイゼーション[FISH]、逆転写ポリメラーゼ連鎖反応[RT-PCR]、または従来の細胞遺伝学検査)によって診断を確定することができる。抗PMLモノクローナル抗体を用いる免疫蛍光法では、融合蛋白の存在下で生じるPMLの特徴的な分布パターンを基に、PML-RARA融合蛋白の存在を短時間で判定可能である。[ 4 ][ 5 ][ 6 ]
臨床所見
臨床的なAPLの特徴は、重度の凝固障害が診断時によくみられることである。[ 7 ]これは、典型的に血小板減少症、プロトロンビン時間延長、部分トロンボプラスチン時間延長、Dダイマー上昇、および低フィブリノゲン血症を伴って現れる。[ 8 ]この亜型では寛解導入期(特に細胞毒性薬を単独で使用した場合)の出血合併症による死亡が他のFAB分類または世界保健機関(WHO)分類よりも多くみられる。[ 9 ][ 10 ]ATRAおよび化学療法による治療を受けたAPL小児を対象とした多施設共同研究グループの解析によると、小児683人中25人(3.7%)で寛解導入初期の凝固障害による死亡が発生したことが報告された;23人は出血(19人がCNS、4人が肺)による死亡で、2人がCNS血栓症による死亡であった。[ 11 ]凝固障害の証拠がみられなくなるまで、診断用の腰椎穿刺は実施すべきではない。
形態学的症状および臨床像に基づいてAPLが疑われた場合は、直ちにATRA療法を開始するが[ 2 ][ 12 ]、これは、APL患者でATRAにより出血リスクが改善することが示されているためである。[ 13 ]1件のレトロスペクティブ分析により、ATRA寛解導入療法が遅れたAPL患者において出血による早期死亡の増加が確認された。[ 8 ]さらに、凝固障害の改善で指示される補充輸血のような支持療法手段の開始は、診断および治療におけるこのような最初の数日間できわめて重要である。凝固障害性合併症のリスクが最も高い患者は、白血球(WBC)数が高値、高い肥満指数、低フィブリノゲン血症、APLの分子的多様体、およびFLT3-ITD変異の存在を認める患者である。[ 8 ][ 11 ]
小児のAPLは、一般に成人のAPLと類似しているが、小児では白血球増加症(白血球数が10×109/Lを超える場合として定義)の発生率が高く、微小顆粒の形態学的亜型の発生率が高い。[ 14 ][ 15 ][ 16 ][ 17 ]成人と同様に、診断時のWBC数が10×109/L未満の小児は、WBC数が多い患児より転帰が有意に良好である。[ 15 ][ 16 ][ 18 ]
治療層別化のためのリスク分類
WBC数の予後的意義は、高リスクおよび低リスク患者集団の定義および導入後療法の割り付けに使用され、高リスク患者はWBC数が10×109/L以上により定義されることが最も一般的である。[ 19 ][ 20 ]FLT3変異(遺伝子内の縦列重複またはキナーゼドメインの変異のいずれか)は、APL症例の40~50%に認められ、FLT3変異の存在は、高いWBC数および微小顆粒多様体(M3v)の亜型と相関する。[ 21 ][ 22 ][ 23 ][ 24 ][ 25 ]このFLT3変異は、寛解導入期の死亡リスク増加、および一部の報告で治療失敗リスク増加と関係するとされている。[ 21 ][ 22 ][ 23 ][ 24 ][ 25 ][ 26 ][ 27 ]
COG AAML0631(NCT00866918)試験には化学療法、ATRA、および三酸化ヒ素による治療が含まれ、リスク分類では、再燃リスクではなく、主に早期死亡リスクが規定された(標準リスク、患者66人中0人 vs 高リスク、患者35人中4人)。寛解導入後の再燃リスクは全体で4%であり、標準リスクの小児で1人の再燃および高リスクの小児で2人の再燃がみられた。本試験では、高リスク患者に対してイダルビシンの開始を早め、白血病腫瘍量を急速に減少させるために初回投与を3日目ではなく1日目に行うとともに、地固め化学療法(高用量シタラビンおよびイダルビシン)およびATRAを1サイクル追加した。[ 28 ]
中枢神経系(CNS)およびAPL
診断時点におけるCNS浸潤は、播種性血管内凝固症候群が存在するため、APL患者のほとんどで確認されていない。COG AAML0631(NCT00866918)試験では、登録された小児101人のうち、診断時にCSF検査を受けた患者28人が特定され、そのうち7人の小児で、無外傷性穿刺で芽球が同定された。[ 28 ]寛解導入期の髄腔内治療および治療中の予防的投与で、CNS再燃を認めた患者はいなかった。
全体として、APL患者で再燃はまれであり、特にWBC数が10×109/L未満の患者で少ない。[ 29 ][ 30 ]1,400人を超えるAPL成人が登録された2件の臨床試験では、CNS予防が実施されておらず、CNS再燃の累積発生率は、WBC数が10×109/L未満の患者で1%未満であったが、WBC数が10×109/L以上の患者では約5%であった。[ 29 ][ 30 ]診断時のWBC数高値に加えて、寛解導入期のCNS出血もCNS再燃の危険因子である。[ 30 ]小児APLで公表された症例のレビューでもCNS再燃率が低いことが認められている。WBC数が10×109/L未満のAPL患児ではCNS再燃の発生率が低いことから、この患者群にCNSサーベイランスおよび予防的なCNS療法は不要な可能性があるものの[ 31 ]、この課題に関してコンセンサスは得られていない。[ 32 ]
APLの治療
現代のAPLに対する治療プログラムは、APL患者から得られた白血病細胞がATRAおよび三酸化ヒ素による分化誘導作用およびアポトーシス促進作用に対して感受性を有することに基づいている。APL療法は、最初に他の非APLのAML亜型の治療から分岐し、化学療法にATRAが追加された。
APLの治療法選択肢には以下を含めてもよい:
- 化学療法。
- ATRA。
- 三酸化ヒ素。
- 支持療法。
APL患児を治療する標準アプローチは、成人を対象とした臨床試験の結果から構築された;このアプローチは、シタラビンの併用または非併用でアントラサイクリン系薬剤とATRAの併用投与を用いた寛解導入療法から始められる。APLに対するATRAの劇的な効果は、ATRAの生理学的濃度でPML-RARA融合蛋白により引き起こされる信号伝達抑制が、ATRAの薬理学的用量で克服できることからもたらされる。信号伝達の回復によりAPL細胞の分化が促進された後、成熟後のアポトーシスに至る。[ 33 ]APL患者の大半はATRAによる治療で完全寛解(CR)を達成するが、ATRA単独では一般に治癒しない。[ 34 ][ 35 ]
一連のランダム化臨床試験では、寛解導入期の化学療法とATRA併用による有益性に加え、維持療法としてATRAを用いる有用性が確立された。[ 36 ][ 37 ][ 38 ]あるレジメンでは、ATRAを標準用量のシタラビンおよびダウノルビシンと併用し[ 14 ][ 39 ]、あるレジメンでは、寛解導入期にシタラビンを併用せずにイダルビシンとATRAを採用している。[ 15 ][ 16 ]これらのアプローチのいずれかで治療を受けたAPL小児のほぼすべてが、凝固障害関連死亡なしにCRを達成する。[ 15 ][ 16 ][ 39 ][ 40 ][ 41 ]
治療開始から1ヵ月後における導入療法に対する反応の評価に形態学的および分子的基準を用いると、最終的にはCRに至る患者に分化が遅れた白血病細胞の残存がみられることがあるため、結果の誤解が生まれる可能性がある。[ 2 ][ 3 ]このような初期の観察に基づいて計画される治療の変更は、ATRA + アントラサイクリン系薬剤を含むレジメンに対するAPLの抵抗性がきわめてまれなため、適切ではない。[ 20 ][ 42 ]
典型的な地固め療法では、シタラビンを併用または非併用のアントラサイクリンと併用投与するATRAが含まれている。地固め療法レジメンにおけるシタラビンの役割には議論の余地がある。低リスクAPL成人を対象にダウノルビシン + ATRAのレジメンに対するシタラビンの貢献度について検討したランダム化研究では、シタラビンの追加で有益性が認められたが[ 43 ]、低リスク患者では、高用量のアントラサイクリン系薬剤を用いたレジメンにより、良好以上の結果が得られると考えられている。[ 44 ]高リスク患者(WBCが10×109/L以上)に関して、Programa para el Tratamiento de Hemopatías Malignas(PETHEMA)のLPA 2005(NCT00408278)試験と先行のLPA 99 (NCT00465933)試験との歴史的比較では、アントラサイクリン系薬剤-ATRA併用療法へシタラビンを追加することで、再燃率が低下する可能性が示唆された。[ 42 ]AIDA 2000(NCT00180128)試験の結果によると、高リスク病態の成人患者における再燃の累積発生率は、ATRA、アントラサイクリン系薬剤、およびシタラビンを含む地固めレジメンにより約10%低下する可能性が確認された。[ 20 ]三酸化ヒ素をベースとした地固め療法を用いた研究では、シタラビンによる地固め療法なしで、優れた生存が実証されている。[ 26 ][ 45 ][ 46 ]
維持療法には、ATRA + メルカプトプリンおよびメトトレキサートが含まれる;この併用療法では相反する有益性が示されており、APL成人を対象としたいくつかのランダム化試験では、ATRA単独を超える優位性が示され[ 36 ][ 47 ]、他の研究では有益性がないことが示された。[ 46 ][ 48 ][ 49 ]しかしながら、APLにおける維持療法の有用性は、多くの因子(例えば、リスク群、寛解導入期に使用したアントラサイクリン系薬剤、三酸化ヒ素の使用、および導入療法および地固め療法の強度)に依存する場合がある。
現時点で、維持療法は依然としてAPL小児に対する標準である。化学療法 + ATRAおよび三酸化ヒ素により良好な転帰(イベントフリー生存[EFS]率が70~80%)が観察されているため、第一CR期に造血幹細胞移植は推奨されない。
三酸化ヒ素は、APLの治療で最も活性のある薬物であり、再燃APLで最初に使用されたが、新規診断患者の治療に組み込まれてきている。三酸化ヒ素の使用を裏付けるデータは、最初に成人患者のみを含む試験から得られたが、最近では、小児および成人患者を両方含む試験および小児患者のみを含む試験で、その有効性が認められている。
証拠(三酸化ヒ素療法):
- 新たにAPLと診断され、CALGB-C9710(NCT00003934)試験で治療を受けた成人で、標準のAPL治療レジメンに三酸化ヒ素による2コースの地固め療法を追加することにより、以下の結果が得られた:[ 45 ]
- 新たにAPLと診断され、COG AAML0631(NCT00866918)試験で治療を受けた小児および青年で、三酸化ヒ素による2コースの地固め療法は、歴史的対照と比較してアントラサイクリン系薬剤の累積用量が低い化学療法レジメンに組み込まれた。[ 28 ]
- 新たにAPLと診断された患者では、三酸化ヒ素をATRAと同時併用することで、高いCR率が得られる。[ 50 ][ 51 ][ 52 ]新たにAPLと診断された小児における初期の経験でも、三酸化ヒ素に対して単剤でもATRAとの併用でも高いCR率が示されている。[ 53 ][証拠レベル:3iiA]
- 三酸化ヒ素は、APML4臨床試験においてイダルビシンとATRAによる導入療法の一部として評価され、小児および成人(評価可能患者 N = 124)が登録された。[ 26 ]患者に対して三酸化ヒ素とATRAによる地固め療法(アントラサイクリン系薬剤は含まない)が2コース施行され、ATRA、メルカプトプリン、およびメトトレキサートによる維持療法が行われた。[ 57 ]
- ドイツとイタリアの第III相臨床試験(APL0406[NCT00482833])では、低リスクから中リスク(WBCが10×109/L以下)に分類されたAPL成人を対象にATRA + 化学療法とATRA + 三酸化ヒ素が比較された。[ 46 ]導入療法および地固め療法でATRA + 三酸化ヒ素を投与する群、または標準のATRA-イダルビシンによる導入療法の後にATRA + 化学療法による3サイクルの地固め療法に加え、低量化学療法とATRAによる維持療法を実施する群のいずれかに患者がランダムに割り付けられた。
APL患児に対して適切な支持療法手段とともにATRAの速やかな開始を規定した治療プログラムを用いることで、現在では80%を超える生存率が達成可能なことが多くの試験により示された[ 2 ][ 14 ][ 15 ][ 16 ][ 19 ][ 20 ][ 40 ][ 41 ];治療レジメンに三酸化ヒ素を追加した1件の試験で、90%を超える生存率が実証された。[ 28 ]完全寛解が5年間以上続いている患者では、再燃はきわめてまれである。[ 59 ][証拠レベル:1iiDi]
臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、全米および/または各医療施設で現在実施されている臨床試験の例である。
- COG AAML1331(NCT02339740)(未治療のAPL患者の治療におけるトレチノインと三酸化ヒ素):これは、リスクに応じて治療を層別化した単群試験で、標準リスク(WBCが10,000/µL未満)の患者に対してATRA + 三酸化ヒ素のみが施行され、高リスク患者(WBCが10,000/µL以上)に対して寛解導入期にイダルビシンの短期追加投与を加えた同じ寛解導入療法が施行された。これは、従来の化学療法を廃し、転帰が悪化しないことを示した成人APL試験を基に構築された。さらに、この試験では、維持療法を廃止しているため、治療期間全体の長さが30ヵ月から8ヵ月に短縮される。この結果がCOG-AAML0631試験を歴史的対照として比較される。
APL療法に特有の合併症
APLを新たに診断された患者では、前述の凝固障害が普遍的に認められることに加えて、APL患者では他にもいくつか臨床医が知っておくべき特有の合併症が発生する。これには、ATRAに関係した2つの病態、偽脳腫瘍および分化症候群(レチノイン酸症候群とも呼ばれる)のほか、三酸化ヒ素関連合併症のQT間隔延長が挙げられる。
微小病変のモニタリング
現在行われている寛解導入および地固め療法によって、ほとんどのAPL患者において、PML-RARAを調べるRT-PCRにより測定される分子的寛解が得られ、地固め療法の終了時点で病変の分子的証拠が認められるのは1%以下である。[ 20 ][ 42 ]治療完了後、2回のRT-PCRアッセイ陰性は長期寛解と関連しているが[ 65 ]、RT-PCR陰性から陽性への転換はその後の血液学的再燃を強く予測する。[ 66 ]
PML-RARAのRT-PCR測定に基づき残存病変または再燃性病変が認められる患者は、再燃治療による介入が有益であろう[ 67 ][ 68 ](詳しい情報に関しては、本要約の再発または難治小児急性骨髄性白血病およびその他の骨髄性悪性疾患のセクションにおける再発急性前骨髄球性白血病[APL]の治療のサブセクションを参照のこと)。
APLにおけるPML-RARA以外の分子的多様体および治療上の影響
APLのまれな分子的多様体は、特異的な遺伝子パートナー(例えば、PLZF、NPM、STAT5B、およびNuMA)がRARAに結合した融合蛋白を生成する。[ 69 ][ 70 ]これらのまれな多様体の認識は、これらのATRAおよび三酸化ヒ素に対する感受性が異なるため、重要である。[ 71 ]
再発APLの治療
歴史的に、APL患者の10~20%で再燃がみられる;しかしながら、三酸化ヒ素療法を取り入れた最近の研究では、再燃の累積発生率が5%未満であることが示された。[ 28 ][ 58 ]
最初に化学療法をベースとした治療を受けた患者で、第一寛解の期間はAPLの予後因子であり、初回診断から12~18ヵ月以内に再燃する患者は転帰不良である。[ 79 ][ 80 ][ 81 ]
再燃する小児における重要な問題は、過去のアントラサイクリン系薬剤への曝露であり、400mg/m2~750mg/m2の範囲に及ぶことがある。[ 2 ]そのため、再燃したAPL患児では、アントラサイクリンを含むレジメンは適切ではない場合が多い。
再発APL患児に対する治療法選択肢には以下を含めてもよい:
- 三酸化ヒ素またはATRA。
- ゲムツズマブオゾガマイシン。
- 造血幹細胞移植(HSCT)。
三酸化ヒ素
再発APLの小児では、第一寛解期に受けた治療に応じて、三酸化ヒ素を単剤として、またはATRAを含むレジメンで使用することを検討すべきである。三酸化ヒ素は、再発APLの患者に活性のある薬物であり、三酸化ヒ素による治療後に約85%の患者が寛解に達する。[ 48 ][ 50 ][ 82 ][ 83 ][ 84 ]三酸化ヒ素では、初回治療で三酸化ヒ素を投与された後に再燃した患者でも再び寛解導入できる。[ 85 ]しかしながら、PML-RARA融合がん遺伝子のPML領域の変異を含む機序を通して、APL細胞が三酸化ヒ素に抵抗性になる可能性がある。[ 86 ]
再燃したAPL成人では、三酸化ヒ素による治療後に約85%が形態学的寛解に達する。[ 83 ][ 84 ][ 87 ]小児における三酸化ヒ素の使用に関するデータは限られているものの、公表された報告によると、再燃したAPL患児は、成人の場合と同様に三酸化ヒ素に反応を示すことが示唆されている。[ 82 ][ 84 ][ 88 ]三酸化ヒ素は再燃したAPL小児において忍容性が良好である。小児における毒性プロファイルおよび奏効率は、成人において観察されているものと同程度である。[ 82 ]
三酸化ヒ素はQT間隔延長を引き起こして、致死的な不整脈につながることがあるため[ 63 ]、三酸化ヒ素を投与する患者では電解質に関する綿密なモニターを行い、カリウム値およびマグネシウム値を正常範囲内に保つことが不可欠である。[ 64 ]
ゲムツズマブオゾガマイシン
抗CD33/カリケアマイシンのモノクローナル抗体であるゲムツズマブオゾガマイシンを単剤として使用することで、分子的寛解が2回の投与後に91%(患者11人中9人)に、また3回の投与後に100%(患者13人中13人)に得られたことから、再燃APLにおけるこの薬剤の優れた効果が実証された。[ 89 ]
HSCT
小児のレトロスペクティブ研究で、自家または同種移植のいずれかのアプローチ後の5年EFS率は同程度で、約70%であったことが報告された。[ 90 ][ 91 ]
証拠(自家HSCT):
- 自家移植を検討する場合、移植前の逆転写ポリメラーゼ連鎖反応により、患者自身および幹細胞産物のいずれでも、前骨髄球性白血病/レチノイン酸受容体α融合転写物が陰性(分子的寛解)であれば、7年EFS(77% vs 50%)が改善することが、成人患者を対象とした研究で実証された。[ 92 ]
- 別の研究で、自家ドナー細胞が微小残存病変(MRD)陽性で自家HSCTを受けた患者7人のうち、すべての患者が移植後9ヵ月に達するまでに再燃したことが明らかになった;しかしながら、自家ドナー細胞がMRD陰性であった患者8人のうち、再燃したのはわずか1人であった。[ 93 ]
- その他の報告により、分子的第二寛解期で自家HSCTを受けた患者の5年EFSは83.3%であったのに対して、維持療法のみを受けた患者では34.5%であったことが明らかにされた。[ 94 ]
このようなデータから、適合同種ドナーが得られそうにない第二CR期のMRD陰性の患者における自家移植の使用が支持される。
小児でAPLがまれであり、この疾患の転帰が良好なため、再燃APLを対象に治療アプローチを比較する臨床試験が実現できる可能性は低い。しかしながら、国際専門家委員会は、小児および成人で報告された経験に基づいて再燃APLの治療に対する推奨を提示した。[ 95 ]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Melnick A, Licht JD: Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93 (10): 3167-215, 1999.[PUBMED Abstract]
- Sanz MA, Grimwade D, Tallman MS, et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113 (9): 1875-91, 2009.[PUBMED Abstract]
- Sanz MA, Lo-Coco F: Modern approaches to treating acute promyelocytic leukemia. J Clin Oncol 29 (5): 495-503, 2011.[PUBMED Abstract]
- Falini B, Flenghi L, Fagioli M, et al.: Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti-PML). Blood 90 (10): 4046-53, 1997.[PUBMED Abstract]
- Gomis F, Sanz J, Sempere A, et al.: Immunofluorescent analysis with the anti-PML monoclonal antibody PG-M3 for rapid and accurate genetic diagnosis of acute promyelocytic leukemia. Ann Hematol 83 (11): 687-90, 2004.[PUBMED Abstract]
- Dimov ND, Medeiros LJ, Kantarjian HM, et al.: Rapid and reliable confirmation of acute promyelocytic leukemia by immunofluorescence staining with an antipromyelocytic leukemia antibody: the M. D. Anderson Cancer Center experience of 349 patients. Cancer 116 (2): 369-76, 2010.[PUBMED Abstract]
- Tallman MS, Hakimian D, Kwaan HC, et al.: New insights into the pathogenesis of coagulation dysfunction in acute promyelocytic leukemia. Leuk Lymphoma 11 (1-2): 27-36, 1993.[PUBMED Abstract]
- Altman JK, Rademaker A, Cull E, et al.: Administration of ATRA to newly diagnosed patients with acute promyelocytic leukemia is delayed contributing to early hemorrhagic death. Leuk Res 37 (9): 1004-9, 2013.[PUBMED Abstract]
- Lehmann S, Ravn A, Carlsson L, et al.: Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia 25 (7): 1128-34, 2011.[PUBMED Abstract]
- Park JH, Qiao B, Panageas KS, et al.: Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood 118 (5): 1248-54, 2011.[PUBMED Abstract]
- Abla O, Ribeiro RC, Testi AM, et al.: Predictors of thrombohemorrhagic early death in children and adolescents with t(15;17)-positive acute promyelocytic leukemia treated with ATRA and chemotherapy. Ann Hematol 96 (9): 1449-1456, 2017.[PUBMED Abstract]
- Breen KA, Grimwade D, Hunt BJ: The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br J Haematol 156 (1): 24-36, 2012.[PUBMED Abstract]
- Visani G, Gugliotta L, Tosi P, et al.: All-trans retinoic acid significantly reduces the incidence of early hemorrhagic death during induction therapy of acute promyelocytic leukemia. Eur J Haematol 64 (3): 139-44, 2000.[PUBMED Abstract]
- de Botton S, Coiteux V, Chevret S, et al.: Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol 22 (8): 1404-12, 2004.[PUBMED Abstract]
- Testi AM, Biondi A, Lo Coco F, et al.: GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood 106 (2): 447-53, 2005.[PUBMED Abstract]
- Ortega JJ, Madero L, Martín G, et al.: Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol 23 (30): 7632-40, 2005.[PUBMED Abstract]
- Guglielmi C, Martelli MP, Diverio D, et al.: Immunophenotype of adult and childhood acute promyelocytic leukaemia: correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br J Haematol 102 (4): 1035-41, 1998.[PUBMED Abstract]
- Sanz MA, Lo Coco F, Martín G, et al.: Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood 96 (4): 1247-53, 2000.[PUBMED Abstract]
- Sanz MA, Martín G, González M, et al.: Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood 103 (4): 1237-43, 2004.[PUBMED Abstract]
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA Group. Blood 116 (17): 3171-9, 2010.[PUBMED Abstract]
- Callens C, Chevret S, Cayuela JM, et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 19 (7): 1153-60, 2005.[PUBMED Abstract]
- Gale RE, Hills R, Pizzey AR, et al.: Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood 106 (12): 3768-76, 2005.[PUBMED Abstract]
- Arrigoni P, Beretta C, Silvestri D, et al.: FLT3 internal tandem duplication in childhood acute myeloid leukaemia: association with hyperleucocytosis in acute promyelocytic leukaemia. Br J Haematol 120 (1): 89-92, 2003.[PUBMED Abstract]
- Noguera NI, Breccia M, Divona M, et al.: Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia 16 (11): 2185-9, 2002.[PUBMED Abstract]
- Tallman MS, Kim HT, Montesinos P, et al.: Does microgranular variant morphology of acute promyelocytic leukemia independently predict a less favorable outcome compared with classical M3 APL? A joint study of the North American Intergroup and the PETHEMA Group. Blood 116 (25): 5650-9, 2010.[PUBMED Abstract]
- Iland HJ, Bradstock K, Supple SG, et al.: All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood 120 (8): 1570-80; quiz 1752, 2012.[PUBMED Abstract]
- Kutny MA, Moser BK, Laumann K, et al.: FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 662-7, 2012.[PUBMED Abstract]
- Kutny MA, Alonzo TA, Gerbing RB, et al.: Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients With Acute Promyelocytic Leukemia: Report From the Children's Oncology Group Phase III Historically Controlled Trial AAML0631. J Clin Oncol 35 (26): 3021-3029, 2017.[PUBMED Abstract]
- de Botton S, Sanz MA, Chevret S, et al.: Extramedullary relapse in acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. Leukemia 20 (1): 35-41, 2006.[PUBMED Abstract]
- Montesinos P, Díaz-Mediavilla J, Debén G, et al.: Central nervous system involvement at first relapse in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline monochemotherapy without intrathecal prophylaxis. Haematologica 94 (9): 1242-9, 2009.[PUBMED Abstract]
- Chow J, Feusner J: Isolated central nervous system recurrence of acute promyelocytic leukemia in children. Pediatr Blood Cancer 52 (1): 11-3, 2009.[PUBMED Abstract]
- Kaspers G, Gibson B, Grimwade D, et al.: Central nervous system involvement in relapsed acute promyelocytic leukemia. Pediatr Blood Cancer 53 (2): 235-6; author reply 237, 2009.[PUBMED Abstract]
- Altucci L, Rossin A, Raffelsberger W, et al.: Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med 7 (6): 680-6, 2001.[PUBMED Abstract]
- Huang ME, Ye YC, Chen SR, et al.: Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72 (2): 567-72, 1988.[PUBMED Abstract]
- Castaigne S, Chomienne C, Daniel MT, et al.: All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 76 (9): 1704-9, 1990.[PUBMED Abstract]
- Fenaux P, Chastang C, Chevret S, et al.: A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood 94 (4): 1192-200, 1999.[PUBMED Abstract]
- Fenaux P, Chevret S, Guerci A, et al.: Long-term follow-up confirms the benefit of all-trans retinoic acid in acute promyelocytic leukemia. European APL group. Leukemia 14 (8): 1371-7, 2000.[PUBMED Abstract]
- Tallman MS, Andersen JW, Schiffer CA, et al.: All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med 337 (15): 1021-8, 1997.[PUBMED Abstract]
- Gregory J, Kim H, Alonzo T, et al.: Treatment of children with acute promyelocytic leukemia: results of the first North American Intergroup trial INT0129. Pediatr Blood Cancer 53 (6): 1005-10, 2009.[PUBMED Abstract]
- Imaizumi M, Tawa A, Hanada R, et al.: Prospective study of a therapeutic regimen with all-trans retinoic acid and anthracyclines in combination of cytarabine in children with acute promyelocytic leukaemia: the Japanese childhood acute myeloid leukaemia cooperative study. Br J Haematol 152 (1): 89-98, 2011.[PUBMED Abstract]
- Testi AM, Pession A, Diverio D, et al.: Risk-adapted treatment of acute promyelocytic leukemia: results from the International Consortium for Childhood APL. Blood 132 (4): 405-412, 2018.[PUBMED Abstract]
- Sanz MA, Montesinos P, Rayón C, et al.: Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood 115 (25): 5137-46, 2010.[PUBMED Abstract]
- Adès L, Chevret S, Raffoux E, et al.: Is cytarabine useful in the treatment of acute promyelocytic leukemia? Results of a randomized trial from the European Acute Promyelocytic Leukemia Group. J Clin Oncol 24 (36): 5703-10, 2006.[PUBMED Abstract]
- Adès L, Sanz MA, Chevret S, et al.: Treatment of newly diagnosed acute promyelocytic leukemia (APL): a comparison of French-Belgian-Swiss and PETHEMA results. Blood 111 (3): 1078-84, 2008.[PUBMED Abstract]
- Powell BL, Moser B, Stock W, et al.: Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood 116 (19): 3751-7, 2010.[PUBMED Abstract]
- Lo-Coco F, Avvisati G, Vignetti M, et al.: Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 369 (2): 111-21, 2013.[PUBMED Abstract]
- Sanz M, Martínez JA, Barragán E, et al.: All-trans retinoic acid and low-dose chemotherapy for acute promyelocytic leukaemia. Br J Haematol 109 (4): 896-7, 2000.[PUBMED Abstract]
- Avvisati G, Lo-Coco F, Paoloni FP, et al.: AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood 117 (18): 4716-25, 2011.[PUBMED Abstract]
- Powell BL, Moser BK, Stock W, et al.: Adding mercaptopurine and methotrexate to alternate week ATRA maintenance therapy does not improve the outcome for adults with acute promyelocytic leukemia (APL) in first remission: results from North American Leukemia Intergroup Trial C9710. [Abstract] Blood 118 (21): A-258, 2011. Also available online. Last accessed March 25, 2020.[PUBMED Abstract]
- Shen ZX, Shi ZZ, Fang J, et al.: All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 101 (15): 5328-35, 2004.[PUBMED Abstract]
- Ravandi F, Estey E, Jones D, et al.: Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol 27 (4): 504-10, 2009.[PUBMED Abstract]
- Hu J, Liu YF, Wu CF, et al.: Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A 106 (9): 3342-7, 2009.[PUBMED Abstract]
- Cheng Y, Zhang L, Wu J, et al.: Long-term prognosis of childhood acute promyelocytic leukaemia with arsenic trioxide administration in induction and consolidation chemotherapy phases: a single-centre experience. Eur J Haematol 91 (6): 483-9, 2013.[PUBMED Abstract]
- Wang H, Chen XY, Wang BS, et al.: The efficacy and safety of arsenic trioxide with or without all-trans retinoic acid for the treatment of acute promyelocytic leukemia: a meta-analysis. Leuk Res 35 (9): 1170-7, 2011.[PUBMED Abstract]
- Zhang L, Zhao H, Zhu X, et al.: Retrospective analysis of 65 Chinese children with acute promyelocytic leukemia: a single center experience. Pediatr Blood Cancer 51 (2): 210-5, 2008.[PUBMED Abstract]
- Zhou J, Zhang Y, Li J, et al.: Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood 115 (9): 1697-702, 2010.[PUBMED Abstract]
- Iland HJ, Collins M, Bradstock K, et al.: Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: a non-randomised phase 2 trial. Lancet Haematol 2 (9): e357-66, 2015.[PUBMED Abstract]
- Platzbecker U, Avvisati G, Cicconi L, et al.: Improved Outcomes With Retinoic Acid and Arsenic Trioxide Compared With Retinoic Acid and Chemotherapy in Non-High-Risk Acute Promyelocytic Leukemia: Final Results of the Randomized Italian-German APL0406 Trial. J Clin Oncol 35 (6): 605-612, 2017.[PUBMED Abstract]
- Douer D, Zickl LN, Schiffer CA, et al.: All-trans retinoic acid and late relapses in acute promyelocytic leukemia: very long-term follow-up of the North American Intergroup Study I0129. Leuk Res 37 (7): 795-801, 2013.[PUBMED Abstract]
- Coombs CC, DeAngelis LM, Feusner JH, et al.: Pseudotumor Cerebri in Acute Promyelocytic Leukemia Patients on Intergroup Protocol 0129: Clinical Description and Recommendations for New Diagnostic Criteria. Clin Lymphoma Myeloma Leuk 16 (3): 146-51, 2016.[PUBMED Abstract]
- Sanz MA, Montesinos P: How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood 123 (18): 2777-82, 2014.[PUBMED Abstract]
- Montesinos P, Bergua JM, Vellenga E, et al.: Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood 113 (4): 775-83, 2009.[PUBMED Abstract]
- Unnikrishnan D, Dutcher JP, Varshneya N, et al.: Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood 97 (5): 1514-6, 2001.[PUBMED Abstract]
- Barbey JT: Cardiac toxicity of arsenic trioxide. Blood 98 (5): 1632; discussion 1633-4, 2001.[PUBMED Abstract]
- Jurcic JG, Nimer SD, Scheinberg DA, et al.: Prognostic significance of minimal residual disease detection and PML/RAR-alpha isoform type: long-term follow-up in acute promyelocytic leukemia. Blood 98 (9): 2651-6, 2001.[PUBMED Abstract]
- Diverio D, Rossi V, Avvisati G, et al.: Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter "AIDA" trial. GIMEMA-AIEOP Multicenter "AIDA" Trial. Blood 92 (3): 784-9, 1998.[PUBMED Abstract]
- Lo Coco F, Diverio D, Avvisati G, et al.: Therapy of molecular relapse in acute promyelocytic leukemia. Blood 94 (7): 2225-9, 1999.[PUBMED Abstract]
- Esteve J, Escoda L, Martín G, et al.: Outcome of patients with acute promyelocytic leukemia failing to front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy (PETHEMA protocols LPA96 and LPA99): benefit of an early intervention. Leukemia 21 (3): 446-52, 2007.[PUBMED Abstract]
- Zelent A, Guidez F, Melnick A, et al.: Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 20 (49): 7186-203, 2001.[PUBMED Abstract]
- Yan W, Zhang G: Molecular Characteristics and Clinical Significance of 12 Fusion Genes in Acute Promyelocytic Leukemia: A Systematic Review. Acta Haematol 136 (1): 1-15, 2016.[PUBMED Abstract]
- Rego EM, Ruggero D, Tribioli C, et al.: Leukemia with distinct phenotypes in transgenic mice expressing PML/RAR alpha, PLZF/RAR alpha or NPM/RAR alpha. Oncogene 25 (13): 1974-9, 2006.[PUBMED Abstract]
- Licht JD, Chomienne C, Goy A, et al.: Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85 (4): 1083-94, 1995.[PUBMED Abstract]
- Guidez F, Ivins S, Zhu J, et al.: Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 91 (8): 2634-42, 1998.[PUBMED Abstract]
- Grimwade D, Biondi A, Mozziconacci MJ, et al.: Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood 96 (4): 1297-308, 2000.[PUBMED Abstract]
- Sukhai MA, Wu X, Xuan Y, et al.: Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene 23 (3): 665-78, 2004.[PUBMED Abstract]
- Redner RL, Corey SJ, Rush EA: Differentiation of t(5;17) variant acute promyelocytic leukemic blasts by all-trans retinoic acid. Leukemia 11 (7): 1014-6, 1997.[PUBMED Abstract]
- Wells RA, Catzavelos C, Kamel-Reid S: Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 17 (1): 109-13, 1997.[PUBMED Abstract]
- Wells RA, Hummel JL, De Koven A, et al.: A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia 10 (4): 735-40, 1996.[PUBMED Abstract]
- Marjerrison S, Antillon F, Bonilla M, et al.: Outcome of children treated for relapsed acute myeloid leukemia in Central America. Pediatr Blood Cancer 61 (7): 1222-6, 2014.[PUBMED Abstract]
- Lengfelder E, Lo-Coco F, Ades L, et al.: Arsenic trioxide-based therapy of relapsed acute promyelocytic leukemia: registry results from the European LeukemiaNet. Leukemia 29 (5): 1084-91, 2015.[PUBMED Abstract]
- Holter Chakrabarty JL, Rubinger M, Le-Rademacher J, et al.: Autologous is superior to allogeneic hematopoietic cell transplantation for acute promyelocytic leukemia in second complete remission. Biol Blood Marrow Transplant 20 (7): 1021-5, 2014.[PUBMED Abstract]
- Fox E, Razzouk BI, Widemann BC, et al.: Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood 111 (2): 566-73, 2008.[PUBMED Abstract]
- Niu C, Yan H, Yu T, et al.: Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood 94 (10): 3315-24, 1999.[PUBMED Abstract]
- Shen ZX, Chen GQ, Ni JH, et al.: Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89 (9): 3354-60, 1997.[PUBMED Abstract]
- Lu J, Huang X, Bao L, et al.: Treatment outcomes in relapsed acute promyelocytic leukemia patients initially treated with all-trans retinoic acid and arsenic compound-based combined therapies. Oncol Lett 7 (1): 177-182, 2014.[PUBMED Abstract]
- Zhu HH, Qin YZ, Huang XJ: Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med 370 (19): 1864-6, 2014.[PUBMED Abstract]
- Soignet SL, Maslak P, Wang ZG, et al.: Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med 339 (19): 1341-8, 1998.[PUBMED Abstract]
- Zhang P: The use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia. J Biol Regul Homeost Agents 13 (4): 195-200, 1999 Oct-Dec.[PUBMED Abstract]
- Lo-Coco F, Cimino G, Breccia M, et al.: Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood 104 (7): 1995-9, 2004.[PUBMED Abstract]
- Dvorak CC, Agarwal R, Dahl GV, et al.: Hematopoietic stem cell transplant for pediatric acute promyelocytic leukemia. Biol Blood Marrow Transplant 14 (7): 824-30, 2008.[PUBMED Abstract]
- Bourquin JP, Thornley I, Neuberg D, et al.: Favorable outcome of allogeneic hematopoietic stem cell transplantation for relapsed or refractory acute promyelocytic leukemia in childhood. Bone Marrow Transplant 34 (9): 795-8, 2004.[PUBMED Abstract]
- de Botton S, Fawaz A, Chevret S, et al.: Autologous and allogeneic stem-cell transplantation as salvage treatment of acute promyelocytic leukemia initially treated with all-trans-retinoic acid: a retrospective analysis of the European acute promyelocytic leukemia group. J Clin Oncol 23 (1): 120-6, 2005.[PUBMED Abstract]
- Meloni G, Diverio D, Vignetti M, et al.: Autologous bone marrow transplantation for acute promyelocytic leukemia in second remission: prognostic relevance of pretransplant minimal residual disease assessment by reverse-transcription polymerase chain reaction of the PML/RAR alpha fusion gene. Blood 90 (3): 1321-5, 1997.[PUBMED Abstract]
- Thirugnanam R, George B, Chendamarai E, et al.: Comparison of clinical outcomes of patients with relapsed acute promyelocytic leukemia induced with arsenic trioxide and consolidated with either an autologous stem cell transplant or an arsenic trioxide-based regimen. Biol Blood Marrow Transplant 15 (11): 1479-84, 2009.[PUBMED Abstract]
- Abla O, Kutny MA, Testi AM, et al.: Management of relapsed and refractory childhood acute promyelocytic leukaemia: recommendations from an international expert panel. Br J Haematol 175 (4): 588-601, 2016.[PUBMED Abstract]
- 一過性骨髄異常増殖症(TAM)またはダウン症候群でAMLの小児
-
ダウン症候群に伴うTAM
生後3年間でAMLのリスクが高いことに加えて、ダウン症候群の新生児の約10%がTAM(一過性の骨髄増殖性障害[TMD]とも呼ばれる)を発症する。[ 1 ]この疾患は先天性AMLに似ているが、典型的には生後3ヵ月(中央値、49日)以内に自然に改善するものの、TAMは寛解に20ヵ月も要すると報告されている。[ 2 ]この寛解の遅れは、活動性疾患ではなく、TAM関連の肝線維化による肝腫大の持続を反映している可能性が高い。[ 3 ]
TAMは一般に自己消散的な疾患であるが、重大な合併症を伴うことがあり、罹患した乳児の10~17%が死亡する可能性がある。[ 2 ][ 3 ][ 4 ][ 5 ][ 6 ]進行性の臓器肥大、内臓滲出液(visceral effusion)、早産(妊娠37週未満)、出血性素因、自然寛解の失敗、進行性の肝機能障害の臨床検査での証拠(直接ビリルビン上昇)、腎不全、および非常に高い白血球(WBC)数を有する乳児は、早期死亡のリスクが特に高い。[ 3 ][ 4 ][ 6 ]これらの高リスクTAM患者の21%が死亡することが報告されているが、TAMに起因するのは10%のみで、残りの死亡は、ダウン症候群の新生児で多くみられることが知られている共存疾患に起因していた。[ 3 ]
生命を脅かす症状の有無にかかわらず、肝腫大の診断時の臨床像に基づいて、以下の3つのリスク群が特定されている:[ 3 ]
重度の水症または臓器不全が明らかな患者では、治療的介入が正当化される。TAMは、最終的に自然消失するため、治療は短期間で、主に白血病による負担の軽減および差し迫った症状の回復を目的とする。以下を含むいくつかの治療アプローチが用いられている:[ 7 ]
ダウン症候群と関連する骨髄性白血病のその後の発症は、TAMが自然寛解した小児の10~30%で認められ、発症時の平均年齢は生後16ヵ月(範囲、1~30ヵ月)と報告されている。[ 2 ][ 3 ][ 9 ]TAMは一般に21トリソミー以外の細胞遺伝学的異常を伴わない特徴があるが、他の細胞遺伝学的異常所見があると、後にダウン症候群と関連する骨髄性白血病を発症するリスクが高い可能性が暗示される。[ 4 ]2件の研究で報告された追加の危険因子はTAMの回復の遅れで、TAM徴候の完全回復までの期間(診断から47日の中央値を過ぎた回復と定義された)または追跡12週目における末梢血中の微小残存病変(MRD)の存在で測定された。[ 3 ];[ 8 ][証拠レベル:2Di]大規模観察コホート研究で報告されているように、TAM症状またはTAMにおいて持続するMRDに対してシタラビンを使用しても、ダウン症候群と関連するその後の骨髄性白血病の減少を示すことはできていない。[ 3 ][ 6 ]TAMに対するシタラビン治療によりダウン症候群と関連するその後の骨髄性白血病の発症を予防できるかどうかを評価するようにデザインされた1件のプロスペクティブ単一群試験では、歴史的対照と比較して有益性は示されなかった(それぞれ、19% ± 4% vs 22% ± 4%;P = 0.88)。[ 8 ][証拠レベル:2Di]
ダウン症候群と関連する骨髄性白血病
ダウン症候群の小児では、ダウン症候群ではない小児と比較して白血病のリスクが10~20倍と高い;しかしながら、急性骨髄性白血病(AML)に対する急性リンパ芽球性白血病の比率は、小児急性白血病で典型的である。生後3年間に関しては例外で、この期間では、特に巨核芽球性亜型のAMLの比率が高く、GATA1変異およびシタラビンに対する高感受性という特有の生物学的特徴を呈する。[ 10 ][ 11 ][ 12 ][ 13 ][ 14 ][ 15 ][ 16 ][ 17 ][ 18 ]重要な点として、これらのリスクは、小児にダウン症候群表現型の特徴が認められるかどうか、または小児に遺伝的な骨髄モザイクが認められるかどうかにかかわらず、同程度であると考えられている。[ 19 ]
ダウン症候群でAMLの小児の予後および治療
AMLを発症するダウン症候群(世界保健機関分類でダウン症候群関連骨髄性白血病と呼ばれる)の小児では、転帰がおおむね良好である。[ 20 ][ 21 ][ 22 ]
AMLとダウン症候群を伴発している小児の予後因子には以下のものがある:
ダウン症候群患者の約29~47%は、骨髄異形成症候群(MDS)(芽球が20%未満)を呈するが、その転帰はAML患者と同程度である。[ 21 ][ 22 ][ 24 ]
新たにダウン症候群とAMLの併発と診断された小児に対する治療法選択肢には以下のものがある:
- 化学療法。
ダウン症候群とAMLを併発した低年齢小児(4歳以下)に適切な治療法は、現在の標準となっている小児AML向け治療法よりも強度が弱い。第一寛解期で造血幹細胞移植は適応とならない。[ 9 ][ 12 ][ 20 ][ 21 ][ 22 ][ 23 ][ 24 ][ 25 ][ 27 ][ 28 ][ 29 ]
証拠(化学療法):
- 新たにダウン症候群とAMLの併発と診断された小児を対象とした小児腫瘍学グループ(COG)試験(AAML0431[NCT00369317])で、寛解導入の4サイクルの二番目を高用量シタラビンに置き換え、以前のCOG A2971(NCT00003593)試験で使用された強化療法からこのサイクルが廃止されたレジメン(それによりアントラサイクリン系薬剤の累積曝露が320mgから240mgに低減)に小児204人が登録された。[ 21 ][ 22 ]髄腔内投与は、7回から計2回の投与に低減され、強化療法には2サイクルのシタラビン/エトポシドが含められた。
- BFM、Dutch Childhood Oncology Group(DCOG)、およびNordic Society of Pediatric Hematology and Oncology(NOPHO)からの合同試験(ML-DS 2006)で、地固め療法でエトポシドを廃止し、髄腔内投与を11回から4回に減らし、AML-BFM 98のダウン症候群治療低減群から維持療法を廃止することで、治療の低減に焦点を置いた試験にダウン症候群の小児170人が登録された。[
24
]COG試験と同様に、診断時にCNS病変を認める患者はいなかった。
以下の2つの予後因子が特定された:[ 24 ]
21トリソミーでモザイクを認める小児では、臨床的に明らかなダウン症候群の小児と同様な治療を行う。[ 3 ][ 19 ][ 21 ]これらの小児に対する至適治療法は確定していないが、通常はダウン症候群ではない小児に対してデザインされたAMLレジメンによる治療が行われる。
臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、全米および/または各医療施設で現在実施されている臨床試験の例である。
- COG AAML1531(NCT02521493)(ダウン症候群の若年患者で新たに診断されたAMLおよび骨髄異形成症候群の治療における奏効に基づく化学療法):これは、ダウン症候群関連AMLと新たに診断された患者を対象とした第III相単群試験で、寛解導入療法に対する奏効を用いて、寛解導入1サイクルの終了時点でMRDがみられない場合に強度の弱い治療、MRDが認められる場合に強度の強い治療に患者を層別化する。
ダウン症候群の小児における不応性病態または再燃
ダウン症候群で初回療法後に再燃した小児または難治性AMLを合併した小児における転帰について扱った文献は少数である。さまざまな治療アプローチによるこれらのレトロスペクティブ解析のいずれにおいても、再発または不応性の転帰となったこれらの小児では、見通しが不良なことが明らかになった。そのため、これらの小児では、ダウン症候群ではない小児と同様な強化寛解再導入化学療法レジメンによる治療が行われ、寛解に達した場合、その後に造血幹細胞移植(HSCT)による治療が行われる。
ダウン症候群で不応性または再燃AMLの小児に対する治療法選択肢には以下のものがある:
- 化学療法を施行し、その後に同種HSCTを行ってもよい。
証拠(不応性または再燃AMLを伴うダウン症候群の小児の治療):
- 日本の小児白血病/リンパ腫研究グループは、再燃(n = 26)および難治性(n = 3)AMLのダウン症候群患者29人について転帰を報告した。ダウン症候群で予想されたように、このコホートの小児は非常に若い(年齢中央値、2歳);再燃はほぼ全例が早い(中央値、8.6ヵ月;診断から12ヵ月未満が80%);89%がFrench-American-British分類でM7であった。[ 30 ][証拠レベル:3iiA]
- Center for International Blood and Marrow Transplant Researchの研究では、ダウン症候群でAMLの小児がHSCTを受けたが、同様に不良な転帰が報告され、3年OS率は19%であった。[ 31 ][証拠レベル:3iiA]移植後の失敗の主な原因は再燃であり、60%を超えていた;移植関連死亡は約20%であった。
- 日本のレジストリー研究では、ダウン症候群の小児に対して強度縮小前処置レジメンを用いた移植を実施し、骨髄破壊的アプローチと比較して良好な生存が報告されたが、患者数が非常に少なく(n = 5)、ダウン症候群とAMLを合併した小児における強度縮小アプローチの有効性については、さらに研究が必要である。[ 32 ][証拠レベル:3iDi]
参考文献- Gamis AS, Smith FO: Transient myeloproliferative disorder in children with Down syndrome: clarity to this enigmatic disorder. Br J Haematol 159 (3): 277-87, 2012.[PUBMED Abstract]
- Homans AC, Verissimo AM, Vlacha V: Transient abnormal myelopoiesis of infancy associated with trisomy 21. Am J Pediatr Hematol Oncol 15 (4): 392-9, 1993.[PUBMED Abstract]
- Gamis AS, Alonzo TA, Gerbing RB, et al.: Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children's Oncology Group Study A2971. Blood 118 (26): 6752-9; quiz 6996, 2011.[PUBMED Abstract]
- Massey GV, Zipursky A, Chang MN, et al.: A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): Children's Oncology Group (COG) study POG-9481. Blood 107 (12): 4606-13, 2006.[PUBMED Abstract]
- Muramatsu H, Kato K, Watanabe N, et al.: Risk factors for early death in neonates with Down syndrome and transient leukaemia. Br J Haematol 142 (4): 610-5, 2008.[PUBMED Abstract]
- Klusmann JH, Creutzig U, Zimmermann M, et al.: Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood 111 (6): 2991-8, 2008.[PUBMED Abstract]
- Al-Kasim F, Doyle JJ, Massey GV, et al.: Incidence and treatment of potentially lethal diseases in transient leukemia of Down syndrome: Pediatric Oncology Group Study. J Pediatr Hematol Oncol 24 (1): 9-13, 2002.[PUBMED Abstract]
- Flasinski M, Scheibke K, Zimmermann M, et al.: Low-dose cytarabine to prevent myeloid leukemia in children with Down syndrome: TMD Prevention 2007 study. Blood Adv 2 (13): 1532-1540, 2018.[PUBMED Abstract]
- Ravindranath Y, Abella E, Krischer JP, et al.: Acute myeloid leukemia (AML) in Down's syndrome is highly responsive to chemotherapy: experience on Pediatric Oncology Group AML Study 8498. Blood 80 (9): 2210-4, 1992.[PUBMED Abstract]
- Ravindranath Y: Down syndrome and leukemia: new insights into the epidemiology, pathogenesis, and treatment. Pediatr Blood Cancer 44 (1): 1-7, 2005.[PUBMED Abstract]
- Ross JA, Spector LG, Robison LL, et al.: Epidemiology of leukemia in children with Down syndrome. Pediatr Blood Cancer 44 (1): 8-12, 2005.[PUBMED Abstract]
- Gamis AS: Acute myeloid leukemia and Down syndrome evolution of modern therapy--state of the art review. Pediatr Blood Cancer 44 (1): 13-20, 2005.[PUBMED Abstract]
- Bassal M, La MK, Whitlock JA, et al.: Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer 44 (1): 21-8, 2005.[PUBMED Abstract]
- Massey GV: Transient leukemia in newborns with Down syndrome. Pediatr Blood Cancer 44 (1): 29-32, 2005.[PUBMED Abstract]
- Taub JW, Ge Y: Down syndrome, drug metabolism and chromosome 21. Pediatr Blood Cancer 44 (1): 33-9, 2005.[PUBMED Abstract]
- Crispino JD: GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr Blood Cancer 44 (1): 40-4, 2005.[PUBMED Abstract]
- Jubinsky PT: Megakaryopoiesis and thrombocytosis. Pediatr Blood Cancer 44 (1): 45-6, 2005.[PUBMED Abstract]
- Ge Y, Stout ML, Tatman DA, et al.: GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst 97 (3): 226-31, 2005.[PUBMED Abstract]
- Kudo K, Hama A, Kojima S, et al.: Mosaic Down syndrome-associated acute myeloid leukemia does not require high-dose cytarabine treatment for induction and consolidation therapy. Int J Hematol 91 (4): 630-5, 2010.[PUBMED Abstract]
- Lange BJ, Kobrinsky N, Barnard DR, et al.: Distinctive demography, biology, and outcome of acute myeloid leukemia and myelodysplastic syndrome in children with Down syndrome: Children's Cancer Group Studies 2861 and 2891. Blood 91 (2): 608-15, 1998.[PUBMED Abstract]
- Sorrell AD, Alonzo TA, Hilden JM, et al.: Favorable survival maintained in children who have myeloid leukemia associated with Down syndrome using reduced-dose chemotherapy on Children's Oncology Group trial A2971: a report from the Children's Oncology Group. Cancer 118 (19): 4806-14, 2012.[PUBMED Abstract]
- Taub JW, Berman JN, Hitzler JK, et al.: Improved outcomes for myeloid leukemia of Down syndrome: a report from the Children's Oncology Group AAML0431 trial. Blood 129 (25): 3304-3313, 2017.[PUBMED Abstract]
- Creutzig U, Reinhardt D, Diekamp S, et al.: AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia 19 (8): 1355-60, 2005.[PUBMED Abstract]
- Uffmann M, Rasche M, Zimmermann M, et al.: Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood 129 (25): 3314-3321, 2017.[PUBMED Abstract]
- Gamis AS, Woods WG, Alonzo TA, et al.: Increased age at diagnosis has a significantly negative effect on outcome in children with Down syndrome and acute myeloid leukemia: a report from the Children's Cancer Group Study 2891. J Clin Oncol 21 (18): 3415-22, 2003.[PUBMED Abstract]
- Blink M, Zimmermann M, von Neuhoff C, et al.: Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica 99 (2): 299-307, 2014.[PUBMED Abstract]
- Craze JL, Harrison G, Wheatley K, et al.: Improved outcome of acute myeloid leukaemia in Down's syndrome. Arch Dis Child 81 (1): 32-7, 1999.[PUBMED Abstract]
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.[PUBMED Abstract]
- Taga T, Shimomura Y, Horikoshi Y, et al.: Continuous and high-dose cytarabine combined chemotherapy in children with down syndrome and acute myeloid leukemia: Report from the Japanese children's cancer and leukemia study group (JCCLSG) AML 9805 down study. Pediatr Blood Cancer 57 (1): 36-40, 2011.[PUBMED Abstract]
- Taga T, Saito AM, Kudo K, et al.: Clinical characteristics and outcome of refractory/relapsed myeloid leukemia in children with Down syndrome. Blood 120 (9): 1810-5, 2012.[PUBMED Abstract]
- Hitzler JK, He W, Doyle J, et al.: Outcome of transplantation for acute myelogenous leukemia in children with Down syndrome. Biol Blood Marrow Transplant 19 (6): 893-7, 2013.[PUBMED Abstract]
- Muramatsu H, Sakaguchi H, Taga T, et al.: Reduced intensity conditioning in allogeneic stem cell transplantation for AML with Down syndrome. Pediatr Blood Cancer 61 (5): 925-7, 2014.[PUBMED Abstract]
- 骨髄異形成症候群(MDS)
-
骨髄異形成症候群(MDS)および骨髄増殖性症候群(MPS)は、小児におけるすべての骨髄性悪性腫瘍の5~10%を占めている。これらは不均一な疾患群で、MDSは一般に血球減少症を呈し、MPSは末梢血の白血球、赤血球、または血小板の増加を呈する。MDSは造血が非効率的で細胞死が多いという特徴があり、MPSは前期細胞の増殖および生存の増加に関係している。両疾患とも、きわめて初期の多分化能造血幹細胞の疾患であるため、治癒を目的とした治療アプローチには、ほぼすべてで同種造血幹細胞移植(HSCT)が必要である。
危険因子
以下の生殖細胞変異または遺伝性疾患を有する患者は、MDSを発症するリスクが有意に高い:
MDSおよび再生不良性貧血に対して幹細胞移植を受けた患者の末梢血単核細胞検体から抽出したゲノムDNAで、骨髄不全およびMDSの素因であることが知られている変異を標的とする捕捉法(capture assay)を使用したレトロスペクティブ解析が実施された。18歳以下のMDS患児46人のうち、10人(22%)の患者が構成的な素因遺伝子変異(5人がGATA2、各1人がMPL、RTEL1、SBDS、TINF2、およびTP53)を保有しており、そのうち移植前に疑われた患者は2人のみであった。これは、18~40歳の患者でわずか8%(64人中4人)であったことと比較すると、高い発生率の遺伝子変異であると考えられた。[ 11 ]
臨床所見
通常、患者には、蒼白、感染症、または挫傷を含む血球減少症の徴候が認められる。
骨髄では、一般的な特徴的として骨髄系前駆細胞の過形成および異形成の変化が認められる。クローン性増殖が最終的にAMLの発症につながることがある。異常な芽球の割合が20%未満で、一般的なAMLの頻発性細胞遺伝学的異常(t(8;21)、inv(16)、t(15;17)、またはKMT2A[MLL]転座)がみられない。
まれな低形成のMDSは、著しい異形成、クローン性、およびCD34陽性の前駆細胞の割合が高いことによって、ある程度は再生不良性貧血と鑑別できる。[ 12 ][ 13 ]
分子的異常
小児骨髄異形成症候群(MDS)は、成人に発生するMDSと比較して明確に異なった遺伝的変化の集合に関連している。成人におけるMDSは、しばしばクローン性造血から進展し、TET2、DNMT3A、およびTP53における変異を特徴とする。対照的に、これらの遺伝子における変異は小児MDSではまれである一方、GATA2、SAMD9/SAMD9L、SETBP1、ASXL1、およびRas/MAPK経路の遺伝子における変異が小児MDS症例のサブセットで観察されている。[ 14 ][ 15 ]
小児MDSのゲノムの全体像に関する報告で、小児原発性MDS患者32人に対する全エクソーム配列決定法および別の14症例の標的シークエンシングの結果が記述された。[ 14 ]これら46症例が小児不応性血球減少および過剰芽球を伴うMDS(MDS-EB)に等しく分類された。報告の結果は以下の通りである:
2つ目の報告では、MDSの小児患者50人(小児不応性血球減少 = 31およびMDS-EB = 19)への105遺伝子の標的シークエンシングパネルの適用について記述され、7モノソミーの症例(48%)について強化された。[ 14 ][ 15 ]この遺伝子パネルにはSAMD9およびSAMD9Lは含まれなかった。2つ目の報告で以下の結果が記述された:
GATA2の生殖細胞変異を有する患者は、MDSに加えて広範な造血および免疫の欠陥のほか、非造血関係の症状を示す。[ 16 ]前者の欠陥には非定型マイコバクテリア感染症への易感染性を伴う単球減少症およびDCML欠損症(樹状細胞、単球、およびB細胞とナチュラルキラーリンパ球の欠失)が挙げられる。結果として生じる免疫不全により、疣贅、重症ウイルス感染症、マイコバクテリア感染症、真菌感染症、およびヒトパピローマウイルス関連がんへの感受性が増加する。非造血関係の症状には、難聴およびリンパ浮腫がある。GATA2の生殖細胞変異が、European Working Group of MDS in Childhood(EWOG-MDS)の連続した研究に登録された原発性MDSの小児患者426人および二次性MDS症例82例において研究された。[ 17 ]研究で以下の結果が得られた:
SAMD9およびSAMD9L生殖細胞変異はどちらも小児MDS症例に関連し、これらの症例ではさらに7番染色体の全部または一部が欠失している。[ 19 ]2016年に、SAMD9はMIRAGE症候群(骨髄異形成、易感染性、発育障害、副腎低形成症、生殖器症状、および腸疾患)の原因として同定されており、この症候群は7モノソミーを伴う早期発症型MDSに関連している。[ 20 ]その後、SAMD9Lにおける変異は小脳失調-汎血球減少症候群(ataxia pancytopenia syndrome:ATXPC;OMIM 159550)の患者において同定された。SAMD9およびSAMD9L変異はまた、7モノソミーを伴う骨髄異形成および白血病症候群(MLSM7;OMIM 252270[ 21 ];この症候群は小児期に7モノソミーに関連してMDSまたはAMLを発症した表現型が正常な同胞において最初に確認された[ 22 ])の原因として同定された。
(MDSのWHO分類に関する詳しい情報については、本要約の骨髄異形成症候群に関する骨髄および末梢血所見のWHO分類のセクションを参照のこと。)
MDSの分類
フランス-アメリカ-イギリス(FAB)および世界保健機関(WHO)のMDSおよびMPSの分類システムを小児患者に適用するのは困難になっている。小児に対する代替の分類システムがいくつか提案されているが、修正2008年WHO分類システムを除いて、統一的に採用されているものはない。[ 23 ][ 24 ][ 25 ][ 26 ][ 27 ]このWHOシステム[ 28 ]は小児を対象に修正されている。[ 26 ]WHO分類スキームおよび診断基準については、表3および表4を参照のこと。2016年WHO改訂版のMDS分類は、小児における分類に影響しなかった。[ 29 ]
不応性血球減少症の亜型は小児MDSの全症例の約50%を占めている。孤立した7モノソミーの存在は、最も多くみられる細胞遺伝学的異常であるものの、顕性のAMLにおけるその存在と比較して、予後不良を予測するとは考えられていない。しかしながら、他の細胞遺伝学的異常と同時に7モノソミーが存在すると、予後不良との関係が認められる。[ 30 ][ 31 ]成人MDSで比較的多くみられる異常の-Y、20q-、および5q-は、小児MDSでまれである。AMLに認められる細胞遺伝学的異常の存在は、MDSではなくAMLとして治療すべき疾患であることを明確に示している。[ 32 ]
International Prognostic Scoring Systemは、低リスクと高リスクのMDSの区別に有用な可能性があるものの、小児と成人では多くの特徴が異なるため、MDSの小児における有用性は、成人よりも限定される。[ 33 ][ 34 ]高リスクのMDS小児の生存期間中央値は、依然として成人よりかなり良好であり、小児における7モノソミーの存在による予後的に有害な影響は、MDS成人における存在と同じではないとされている。[ 35 ]
小児MDSの治療
MDSの小児に対する治療法選択肢には以下のものがある:
HSCT
MDSおよび関連疾患は、通常未熟な造血幹細胞に関係する。したがって、MDSの小児患者に対する治療には、同種HSCTが至適アプローチであると考えられる。適合同胞移植が望ましいが、十分適合した非血縁者臍帯血およびハプロタイプ一致アプローチにより同程度の生存が認められている。[ 36 ][ 37 ][ 38 ][ 39 ][ 40 ]
治療決定を下す際に、いくつかのデータを考慮すべきである。例えば、初期のMDSで、診断後数ヵ月以内に移植に進んだ患者では、80%と高い生存率が報告されている。さらに、MDS小児では、早期の移植に加え、移植前に化学療法を受けていないことが生存期間延長と関係するとされている。[ 41 ][証拠レベル:3iiA]進行したMDSの小児患者に対して骨髄破壊的移植前処置レジメンを用いた場合、無病生存率(DFS)は50~70%と推定されている。[ 39 ][ 42 ][ 43 ][ 44 ][ 45 ]骨髄非破壊的移植前処置レジメンがMDSおよびAMLの患者で検証されている一方で、このようなレジメンは、これらの疾患の小児に対してまだ研究段階であるが、臨床試験の設定で使用する場合、または患者の臓器機能が損なわれていて骨髄破壊的レジメンに耐えられそうにない場合は妥当であろう。[ 46 ][ 47 ][ 48 ][ 49 ];[ 50 ][証拠レベル:3iiiA]
高リスクMDSでは化学療法を使用すべきかどうかという問題が検討されている。
証拠(HSCT):
- ベルリン-フランクフルト-ミュンスターAMLプロトコル83、87、および93で治療を受けたMDS患児37人の解析では、形質転換して過剰な芽球を伴う不応性貧血の患児で寛解導入療法に対する反応が74%に確認され、移植が有益であることが示唆された。[ 51 ]
- 同じグループによる別の研究では、HSCTの現在のアプローチにより、進行したMDS小児の60%以上で生存が得られ、非血縁ドナー細胞の移植を受けた患者の治療成績は適合家族ドナー(MFD)細胞の移植を受けた患者の治療成績とほぼ同じであったことが示された。[ 52 ]
- Children's Cancer Groupの2891試験では、1989年から1995年にかけてMDSの小児を含めて患者を募集した。[ 42 ]不応性貧血(n = 2)、過剰芽球を伴う不応性貧血(n = 33)、形質転換して過剰な芽球を伴う不応性貧血(n = 26)、またはMDSが先行したAML(n = 16)の患者77人が登録され、標準的な寛解導入療法または投与時期を集中した寛解導入療法にランダムに割り付けられた。その後、適切な家族ドナーが得られた場合、患者は同種HSCTに割り付けられ、そうでない場合は、自家HSCTまたは化学療法にランダムに割り付けられた。
これらの結果を解析すると、形質転換して過剰な芽球を伴う不応性貧血という亜型が顕性のAML患者にみられる可能性が高い一方で、不応性貧血および過剰芽球を伴う不応性貧血がMDSにみられることを考慮することが重要である。WHO分類では現在、移行期芽球増加性不応性貧血は基本的にAMLであるとの結論を下し、このカテゴリーを除外している。
HSCT後の生存は、MDSの初期型(不応性貧血)の患者で改善されているため、後期MDSまたはAMLへの進行前の移植を考慮すべきである。重度の症候性血球減少症患者の症例では通常行われているように、輸血またはその他の治療が必要になった場合は、HSCTを特に検討すべきである。[ 39 ][ 45 ]さまざまな病期のMDS患児における8年無病生存(DFS)率は、HLA適合ドナー移植を受けた場合が65%、HLA非適合非血縁者ドナー移植を受けた場合が40%と報告されている。[ 45 ][証拠レベル:3iiiDii]MDSの小児に対して非血縁臍帯血ドナー移植を用いて2001年以降に移植を実施した場合、3年DFSは50%と報告された。[ 53 ][証拠レベル:3iiiDiii]
小児MDSでは遺伝的素因症候群を伴うことが多いため、これらの疾患の患者の少数で移植の報告がなされている。例えば、ファンコニー貧血とAMLまたは進行性MDSの患者では、5年全生存(OS)率が33~55%と報告されている。[ 54 ][ 55 ][証拠レベル:3iiiA]MDS/MPDの小児患者で、再燃した場合または生着不全であった場合に2回目の移植も用いられている。3年OS率は、再燃後の移植患者で33%、初回の生着不全後の移植患者で57%であった。[ 56 ][証拠レベル:3iiiA]
臨床的に重大な血球減少症の患者では、輸血や抗生物質予防投与などの支持療法が標準医療とみなされる。造血成長因子の使用により、造血状態を改善できるが、このような治療はAMLへの転換を促す恐れがあるという懸念が残されている。[ 57 ]
他の治療法
他に検討されている支持療法には以下のものがある:
臨床評価段階にある治療法の選択肢
さまざまなDNAメチル化阻害剤およびヒストン脱アセチル化酵素阻害剤に加え、分化を誘発するようにデザインされた治療法の使用がMDSの若年から高齢の成人で研究されている。[ 65 ][ 66 ][ 67 ]
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Alter BP, Giri N, Savage SA, et al.: Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150 (2): 179-88, 2010.[PUBMED Abstract]
- Rosenberg PS, Huang Y, Alter BP: Individualized risks of first adverse events in patients with Fanconi anemia. Blood 104 (2): 350-5, 2004.[PUBMED Abstract]
- Ludwig LS, Gazda HT, Eng JC, et al.: Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med 20 (7): 748-53, 2014.[PUBMED Abstract]
- Rosenberg PS, Zeidler C, Bolyard AA, et al.: Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol 150 (2): 196-9, 2010.[PUBMED Abstract]
- Wechsler J, Greene M, McDevitt MA, et al.: Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32 (1): 148-52, 2002.[PUBMED Abstract]
- Liew E, Owen C: Familial myelodysplastic syndromes: a review of the literature. Haematologica 96 (10): 1536-42, 2011.[PUBMED Abstract]
- Owen C, Barnett M, Fitzgibbon J: Familial myelodysplasia and acute myeloid leukaemia--a review. Br J Haematol 140 (2): 123-32, 2008.[PUBMED Abstract]
- Ghauri RI, Naveed M, Mannan J: Congenital amegakaryocytic thrombocytopenic purpura (CAMT). J Coll Physicians Surg Pak 24 (4): 285-7, 2014.[PUBMED Abstract]
- Auer PL, Teumer A, Schick U, et al.: Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet 46 (6): 629-34, 2014.[PUBMED Abstract]
- Vinh DC, Patel SY, Uzel G, et al.: Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 115 (8): 1519-29, 2010.[PUBMED Abstract]
- Keel SB, Scott A, Sanchez-Bonilla M, et al.: Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 101 (11): 1343-1350, 2016.[PUBMED Abstract]
- Kasahara S, Hara T, Itoh H, et al.: Hypoplastic myelodysplastic syndromes can be distinguished from acquired aplastic anaemia by bone marrow stem cell expression of the tumour necrosis factor receptor. Br J Haematol 118 (1): 181-8, 2002.[PUBMED Abstract]
- Orazi A: Histopathology in the diagnosis and classification of acute myeloid leukemia, myelodysplastic syndromes, and myelodysplastic/myeloproliferative diseases. Pathobiology 74 (2): 97-114, 2007.[PUBMED Abstract]
- Schwartz JR, Ma J, Lamprecht T, et al.: The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 8 (1): 1557, 2017.[PUBMED Abstract]
- Pastor V, Hirabayashi S, Karow A, et al.: Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 31 (3): 759-762, 2017.[PUBMED Abstract]
- Collin M, Dickinson R, Bigley V: Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol 169 (2): 173-87, 2015.[PUBMED Abstract]
- Wlodarski MW, Hirabayashi S, Pastor V, et al.: Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 127 (11): 1387-97; quiz 1518, 2016.[PUBMED Abstract]
- Wlodarski MW, Collin M, Horwitz MS: GATA2 deficiency and related myeloid neoplasms. Semin Hematol 54 (2): 81-86, 2017.[PUBMED Abstract]
- Davidsson J, Puschmann A, Tedgård U, et al.: SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32 (5): 1106-1115, 2018.[PUBMED Abstract]
- Narumi S, Amano N, Ishii T, et al.: SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet 48 (7): 792-7, 2016.[PUBMED Abstract]
- Chen DH, Below JE, Shimamura A, et al.: Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 98 (6): 1146-1158, 2016.[PUBMED Abstract]
- Wong JC, Bryant V, Lamprecht T, et al.: Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight 3 (14): , 2018.[PUBMED Abstract]
- Occhipinti E, Correa H, Yu L, et al.: Comparison of two new classifications for pediatric myelodysplastic and myeloproliferative disorders. Pediatr Blood Cancer 44 (3): 240-4, 2005.[PUBMED Abstract]
- Niemeyer CM, Baumann I: Myelodysplastic syndrome in children and adolescents. Semin Hematol 45 (1): 60-70, 2008.[PUBMED Abstract]
- Niemeyer CM, Kratz CP: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol 140 (6): 610-24, 2008.[PUBMED Abstract]
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.[PUBMED Abstract]
- Mandel K, Dror Y, Poon A, et al.: A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol 24 (7): 596-605, 2002.[PUBMED Abstract]
- Nösslinger T, Reisner R, Koller E, et al.: Myelodysplastic syndromes, from French-American-British to World Health Organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98 (10): 2935-41, 2001.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Göhring G, Michalova K, Beverloo HB, et al.: Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 116 (19): 3766-9, 2010.[PUBMED Abstract]
- Haase D, Germing U, Schanz J, et al.: New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood 110 (13): 4385-95, 2007.[PUBMED Abstract]
- Arber DA, Vardiman JW, Brunning RD: Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 110-23.[PUBMED Abstract]
- Cutler CS, Lee SJ, Greenberg P, et al.: A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 104 (2): 579-85, 2004.[PUBMED Abstract]
- Hasle H, Baumann I, Bergsträsser E, et al.: The International Prognostic Scoring System (IPSS) for childhood myelodysplastic syndrome (MDS) and juvenile myelomonocytic leukemia (JMML). Leukemia 18 (12): 2008-14, 2004.[PUBMED Abstract]
- Hasle H, Niemeyer CM: Advances in the prognostication and management of advanced MDS in children. Br J Haematol 154 (2): 185-95, 2011.[PUBMED Abstract]
- Uberti JP, Agovi MA, Tarima S, et al.: Comparative analysis of BU and CY versus CY and TBI in full intensity unrelated marrow donor transplantation for AML, CML and myelodysplasia. Bone Marrow Transplant 46 (1): 34-43, 2011.[PUBMED Abstract]
- Nemecek ER, Guthrie KA, Sorror ML, et al.: Conditioning with treosulfan and fludarabine followed by allogeneic hematopoietic cell transplantation for high-risk hematologic malignancies. Biol Blood Marrow Transplant 17 (3): 341-50, 2011.[PUBMED Abstract]
- Shaw PJ, Kan F, Woo Ahn K, et al.: Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood 116 (19): 4007-15, 2010.[PUBMED Abstract]
- Parikh SH, Mendizabal A, Martin PL, et al.: Unrelated donor umbilical cord blood transplantation in pediatric myelodysplastic syndrome: a single-center experience. Biol Blood Marrow Transplant 15 (8): 948-55, 2009.[PUBMED Abstract]
- Locatelli F, Merli P, Pagliara D, et al.: Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood 130 (5): 677-685, 2017.[PUBMED Abstract]
- Smith AR, Christiansen EC, Wagner JE, et al.: Early hematopoietic stem cell transplant is associated with favorable outcomes in children with MDS. Pediatr Blood Cancer 60 (4): 705-10, 2013.[PUBMED Abstract]
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.[PUBMED Abstract]
- Andolina JR, Kletzel M, Tse WT, et al.: Allogeneic hematopoetic stem cell transplantation in pediatric myelodysplastic syndromes: improved outcomes for de novo disease. Pediatr Transplant 15 (3): 334-43, 2011.[PUBMED Abstract]
- Al-Seraihy A, Ayas M, Al-Nounou R, et al.: Outcome of allogeneic stem cell transplantation with a conditioning regimen of busulfan, cyclophosphamide and low-dose etoposide for children with myelodysplastic syndrome. Hematol Oncol Stem Cell Ther 4 (3): 121-5, 2011.[PUBMED Abstract]
- Woodard P, Carpenter PA, Davies SM, et al.: Unrelated donor bone marrow transplantation for myelodysplastic syndrome in children. Biol Blood Marrow Transplant 17 (5): 723-8, 2011.[PUBMED Abstract]
- Champlin R: Hematopoietic stem cell transplantation for treatment of myleodysplastic syndromes. Biol Blood Marrow Transplant 17 (1 Suppl): S6-8, 2011.[PUBMED Abstract]
- Nelson RP, Yu M, Schwartz JE, et al.: Long-term disease-free survival after nonmyeloablative cyclophosphamide/fludarabine conditioning and related/unrelated allotransplantation for acute myeloid leukemia/myelodysplasia. Bone Marrow Transplant 45 (8): 1300-8, 2010.[PUBMED Abstract]
- Warlick ED: Optimizing stem cell transplantation in myelodysplastic syndromes: unresolved questions. Curr Opin Oncol 22 (2): 150-4, 2010.[PUBMED Abstract]
- Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.[PUBMED Abstract]
- Gao L, Gao L, Gong Y, et al.: Reduced-intensity conditioning therapy with fludarabine, idarubicin, busulfan and cytarabine for allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia and myelodysplastic syndrome. Leuk Res 37 (11): 1482-7, 2013.[PUBMED Abstract]
- Creutzig U, Bender-Götze C, Ritter J, et al.: The role of intensive AML-specific therapy in treatment of children with RAEB and RAEB-t. Leukemia 12 (5): 652-9, 1998.[PUBMED Abstract]
- Strahm B, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia 25 (3): 455-62, 2011.[PUBMED Abstract]
- Madureira AB, Eapen M, Locatelli F, et al.: Analysis of risk factors influencing outcome in children with myelodysplastic syndrome after unrelated cord blood transplantation. Leukemia 25 (3): 449-54, 2011.[PUBMED Abstract]
- Mitchell R, Wagner JE, Hirsch B, et al.: Haematopoietic cell transplantation for acute leukaemia and advanced myelodysplastic syndrome in Fanconi anaemia. Br J Haematol 164 (3): 384-95, 2014.[PUBMED Abstract]
- Ayas M, Saber W, Davies SM, et al.: Allogeneic hematopoietic cell transplantation for fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. J Clin Oncol 31 (13): 1669-76, 2013.[PUBMED Abstract]
- Kato M, Yoshida N, Inagaki J, et al.: Salvage allogeneic stem cell transplantation in patients with pediatric myelodysplastic syndrome and myeloproliferative neoplasms. Pediatr Blood Cancer 61 (10): 1860-6, 2014.[PUBMED Abstract]
- Zwierzina H, Suciu S, Loeffler-Ragg J, et al.: Low-dose cytosine arabinoside (LD-AraC) vs LD-AraC plus granulocyte/macrophage colony stimulating factor vs LD-AraC plus Interleukin-3 for myelodysplastic syndrome patients with a high risk of developing acute leukemia: final results of a randomized phase III study (06903) of the EORTC Leukemia Cooperative Group. Leukemia 19 (11): 1929-33, 2005.[PUBMED Abstract]
- Chan G, DiVenuti G, Miller K: Danazol for the treatment of thrombocytopenia in patients with myelodysplastic syndrome. Am J Hematol 71 (3): 166-71, 2002.[PUBMED Abstract]
- Mathew P, Gerbing R, Alonzo TA, et al.: A phase II study of amifostine in children with myelodysplastic syndrome: a report from the Children's Oncology Group study (AAML0121). Pediatr Blood Cancer 57 (7): 1230-2, 2011.[PUBMED Abstract]
- Schanz J, Jung H, Wörmann B, et al.: Amifostine has the potential to induce haematologic responses and decelerate disease progression in individual patients with low- and intermediate-1-risk myelodysplastic syndromes. Leuk Res 33 (9): 1183-8, 2009.[PUBMED Abstract]
- Sadek I, Zayed E, Hayne O, et al.: Prolonged complete remission of myelodysplastic syndrome treated with danazol, retinoic acid and low-dose prednisone. Am J Hematol 64 (4): 306-10, 2000.[PUBMED Abstract]
- Silverman LR, Demakos EP, Peterson BL, et al.: Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20 (10): 2429-40, 2002.[PUBMED Abstract]
- Yazji S, Giles FJ, Tsimberidou AM, et al.: Antithymocyte globulin (ATG)-based therapy in patients with myelodysplastic syndromes. Leukemia 17 (11): 2101-6, 2003.[PUBMED Abstract]
- Yoshimi A, Baumann I, Führer M, et al.: Immunosuppressive therapy with anti-thymocyte globulin and cyclosporine A in selected children with hypoplastic refractory cytopenia. Haematologica 92 (3): 397-400, 2007.[PUBMED Abstract]
- Mufti G, List AF, Gore SD, et al.: Myelodysplastic syndrome. Hematology (Am Soc Hematol Educ Program) : 176-99, 2003.[PUBMED Abstract]
- Esteller M: DNA methylation and cancer therapy: new developments and expectations. Curr Opin Oncol 17 (1): 55-60, 2005.[PUBMED Abstract]
- Bhalla K, List A: Histone deacetylase inhibitors in myelodysplastic syndrome. Best Pract Res Clin Haematol 17 (4): 595-611, 2004.[PUBMED Abstract]
- 治療関連のAML/骨髄異形成症候群
-
発生機序
電離放射線または化学療法、特にアルキル化剤およびトポイソメラーゼ阻害剤による治療後に急性骨髄性白血病(AML)または骨髄異形成症候群(MDS)が発現した場合は、治療関連AML(t-AML)または治療関連MDS(t-MDS)と呼ばれる。遺伝毒性薬曝露に加えて、遺伝的素因の感受性(薬物の解毒およびDNA修復経路の構成分子における多型など)が二次性AML/MDS発現の一因となっている可能性がある。[ 1 ][ 2 ][ 3 ][ 4 ]
t-AML/t-MDSのリスクはレジメン依存性で、投与した化学療法薬の累積用量のほか、照射した放射線の線量および照射野に関係していることが多い。[ 5 ]過去に使用されていたエピポドフィロトキシン(例えば、エトポシド またはテニポシド)またはアルキル化剤(例えば、メクロレタミン、メルファラン、ブスルファン、およびシクロホスファミド)のいずれかを高い累積用量で採用したレジメンでは、t-AML/t-MDSの発生率が過剰に高く、一部の例で10%を超えていた。[ 5 ][ 6 ]しかしながら、最近の小児がん治療に用いられる化学療法レジメンでは、t-AML/t-MDSの累積発生率が1~2%以下である。
エピポドフィロトキシンおよびその他のトポイソメラーゼII阻害剤(例えば、アントラサイクリン系薬剤)に起因するt-AML/t-MDSは、一般に曝露後2年以内に発現し、染色体11q23異常を伴うことが多いが[ 7 ]、他のAML亜型(例えば、急性前骨髄球性白血病)が報告されている。[ 8 ][ 9 ]アルキル化剤または電離放射線への曝露後に発生するt-AMLは、5~7年後に現れることが多く、一般的に5番および7番染色体のモノソミーまたは欠失を伴っている。[ 1 ][ 7 ]
治療関連AML/MDSの治療
治療関連AML/MDSの治療法選択肢には以下のものがある:
- HSCT。
治療の目的は、AML向けのレジメンを用いて初回の完全寛解(CR)を達成し、その後は通常、利用可能な最適ドナーによる造血幹細胞移植(HSCT)へ直ちに進むことである。しかしながら、以下の理由により治療には困難を伴う:[ 10 ]
- 有害な細胞遺伝学的異常の発生率が高く、その結果、化学療法による寛解が得られない。
- 共存症または以前の悪性腫瘍に対する化学療法に関連する制限。
したがって、t-AMLの患者では、de novoのAML患者と比較してCR率および全生存(OS)率が一般に低い。[ 10 ][ 11 ][ 12 ]また、t-MDSの小児患者の生存は、以前の治療に関連のないMDSの小児患者の生存より不良である。[ 13 ]
t-MDS-不応性貧血の患者では、一般に移植前の導入化学療法は必要ない;過剰芽球を伴う不応性貧血-1の患者における移植前の導入療法の役割については異論がある。
t-AMLに対してHSCTを受けた小児の転帰について示した報告はわずかしかない。
証拠(t-AML/t-MDSに対するHSCT):
- 血縁者および非血縁者ドナーによるHSCTを受けたt-AMLの小児27人の転帰を示した研究が1件ある。[ 14 ]
- 別の研究では、1975年から2007年にt-AML/t-MDSに対して移植を受けた患者14人の二次レトロスペクティブ単一施設の経験が報告された。[ 11 ]
- 多施設共同研究(CCG-2891)では、t-AML/t-MDSの小児24人の転帰について、この研究に登録された他のde novo AML(n = 898)またはMDS(n = 62)の小児と比較する調査が行われた。t-AML/t-MDSの小児は年齢が高く、細胞遺伝学的に低リスクはまれであった。[ 15 ]
- これらの患者における生存に対する寛解の重要性は、1994年から2009年にt-AML/t-MDSに対してHSCTを受けた小児21人についての他の単一施設の報告によってさらに詳しく示されている。小児21人のうち12人がt-AML(移植時点でCRが11人)、7人が不応性貧血(導入療法は実施されなかった)、2人が過剰芽球を伴う不応性貧血であった。[ 16 ]
小児ではt-AMLはまれなため、非血縁者ドナーによるHSCT後の移植関連死亡率が過去数年間にわたって有意な低下を示していることが、この集団における生存の改善につながるかどうかは不明である。初期の治療に起因するHSCT前の罹病状態について患者を注意深く評価すべきであり、移植関連死亡率を最小限に抑えながら、適度の治療強度を与えるアプローチを採用すべきである。
参考文献- Leone G, Fianchi L, Voso MT: Therapy-related myeloid neoplasms. Curr Opin Oncol 23 (6): 672-80, 2011.[PUBMED Abstract]
- Bolufer P, Collado M, Barragan E, et al.: Profile of polymorphisms of drug-metabolising enzymes and the risk of therapy-related leukaemia. Br J Haematol 136 (4): 590-6, 2007.[PUBMED Abstract]
- Ezoe S: Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int J Environ Res Public Health 9 (7): 2444-53, 2012.[PUBMED Abstract]
- Ding Y, Sun CL, Li L, et al.: Genetic susceptibility to therapy-related leukemia after Hodgkin lymphoma or non-Hodgkin lymphoma: role of drug metabolism, apoptosis and DNA repair. Blood Cancer J 2 (3): e58, 2012.[PUBMED Abstract]
- Leone G, Mele L, Pulsoni A, et al.: The incidence of secondary leukemias. Haematologica 84 (10): 937-45, 1999.[PUBMED Abstract]
- Pui CH, Ribeiro RC, Hancock ML, et al.: Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 325 (24): 1682-7, 1991.[PUBMED Abstract]
- Andersen MK, Johansson B, Larsen SO, et al.: Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica 83 (6): 483-8, 1998.[PUBMED Abstract]
- Ogami A, Morimoto A, Hibi S, et al.: Secondary acute promyelocytic leukemia following chemotherapy for non-Hodgkin's lymphoma in a child. J Pediatr Hematol Oncol 26 (7): 427-30, 2004.[PUBMED Abstract]
- Okamoto T, Okada M, Wakae T, et al.: Secondary acute promyelocytic leukemia in a patient with non-Hodgkin's lymphoma treated with VP-16 and MST-16. Int J Hematol 75 (1): 107-8, 2002.[PUBMED Abstract]
- Larson RA: Etiology and management of therapy-related myeloid leukemia. Hematology Am Soc Hematol Educ Program : 453-9, 2007.[PUBMED Abstract]
- Aguilera DG, Vaklavas C, Tsimberidou AM, et al.: Pediatric therapy-related myelodysplastic syndrome/acute myeloid leukemia: the MD Anderson Cancer Center experience. J Pediatr Hematol Oncol 31 (11): 803-11, 2009.[PUBMED Abstract]
- Yokoyama H, Mori S, Kobayashi Y, et al.: Hematopoietic stem cell transplantation for therapy-related myelodysplastic syndrome and acute leukemia: a single-center analysis of 47 patients. Int J Hematol 92 (2): 334-41, 2010.[PUBMED Abstract]
- Xavier AC, Kutny M, Costa LJ: Incidence and outcomes of paediatric myelodysplastic syndrome in the United States. Br J Haematol 180 (6): 898-901, 2018.[PUBMED Abstract]
- Woodard P, Barfield R, Hale G, et al.: Outcome of hematopoietic stem cell transplantation for pediatric patients with therapy-related acute myeloid leukemia or myelodysplastic syndrome. Pediatr Blood Cancer 47 (7): 931-5, 2006.[PUBMED Abstract]
- Barnard DR, Lange B, Alonzo TA, et al.: Acute myeloid leukemia and myelodysplastic syndrome in children treated for cancer: comparison with primary presentation. Blood 100 (2): 427-34, 2002.[PUBMED Abstract]
- Kobos R, Steinherz PG, Kernan NA, et al.: Allogeneic hematopoietic stem cell transplantation for pediatric patients with treatment-related myelodysplastic syndrome or acute myelogenous leukemia. Biol Blood Marrow Transplant 18 (3): 473-80, 2012.[PUBMED Abstract]
- 若年性骨髄単球性白血病(JMML)
-
発生率
若年性骨髄単球性白血病(JMML)はまれな白血病であり、発生頻度は小児急性骨髄性白血病(AML)の約1/10で、年間発生率は100万人当たり約1~2例である。[ 1 ]JMMLは典型的には幼児に認められ(年齢中央値、約1.8歳)、男児に多い(男女比、約2.5:1)。
臨床像および診断基準
診断時によくみられる臨床的特徴には以下のものがある:[ 2 ]
JMMLと疑われる臨床像を呈する小児において、確定診断に現在用いられている基準を表8に示す。[ 3 ]
表8.2016年世界保健機関分類改訂版による若年性骨髄単球性白血病(JMML)の診断基準 カテゴリー1(すべてが必要) カテゴリー2(1つで十分) カテゴリー3(遺伝的特徴を認めない患者は、カテゴリー1に加えて、以下を満たさなければならない 臨床的および血液学的特徴 遺伝子検査 その他の特徴 GM-CSF = 顆粒球マクロファージコロニー刺激因子;NF1 = 神経線維腫症1型。 aカテゴリー2の病変を有することが確認された患者は、カテゴリー1の基準を満たす必要があるが、カテゴリー3の基準を満たす必要はない。カテゴリー2の病変を有することが確認されなかった患者は、カテゴリー1および3の基準を満たさなければならない。 b初発時に脾腫を認めないJMML患者はわずか7%であるが、実質的にすべての患者が数週間ないし数ヵ月以内に脾腫を発症することに注意する。 BCR-ABL1融合遺伝子が認められない KRAS、NRAS、またはPTPN11における体細胞変異 (生殖細胞変異は除外する必要がある) 7モノソミーなどの染色体異常、または以下に掲載する基準の2つ以上: 循環血中単球 >1×109/L NF1の臨床的診断またはNF1遺伝子変異 — 循環血中骨髄系または赤血球系前駆細胞 末梢血中および骨髄中の芽球が20%未満 生殖細胞CBL変異およびCBLのヘテロ接合性消失 — 年齢の割に高いヘモグロビンF値 脾腫 — STAT5の過剰リン酸化 — GM-CSFに対する高感受性 発生機序および関連症候群
JMMLの発生機序は、関連する症候群とともに、RASがん遺伝子経路の活性化に密接に関係している(図1を参照のこと)。[ 4 ][ 5 ]さらに、特有なRNAの発現およびDNAメチル化パターンも報告されている;これらは年齢などの臨床因子との相関がみられ、予後に関係すると考えられている。[ 6 ][ 7 ]
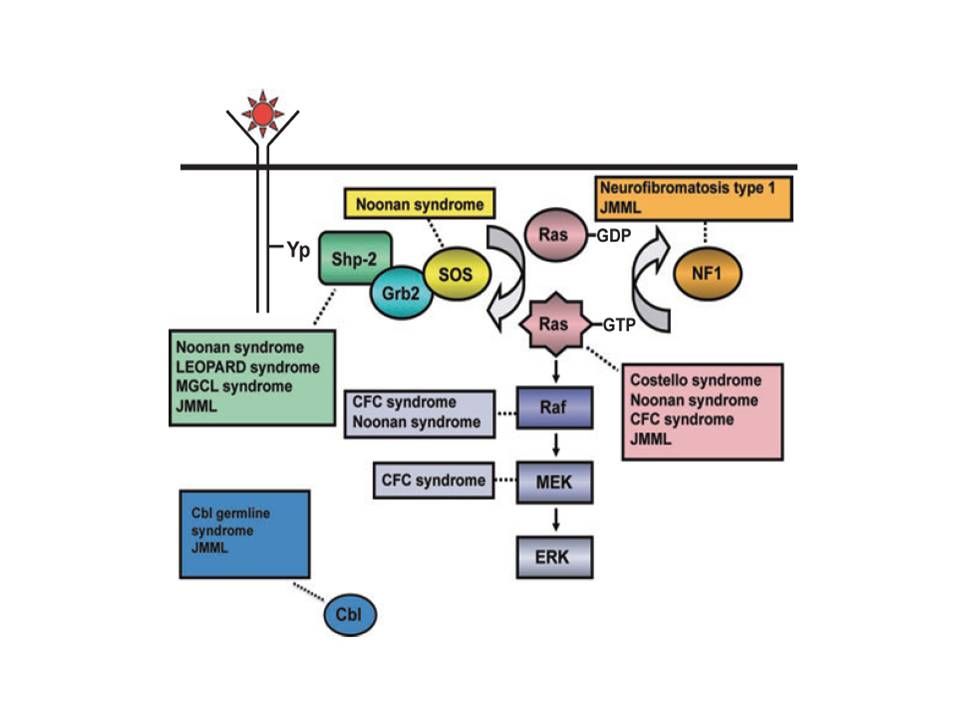
図1.リガンド刺激によるRas活性化、Ras-Erk経路ならびに先天性神経・心・顔面・皮膚障害およびJMMLに寄与する遺伝子変異を示した模式図。NL/MGCL:Noonan-like/multiple giant cell lesion、CFC:心・顔面・皮膚、JMML:若年性骨髄単球性白血病Elsevieから許可を得て転載:Leukemia Research, 33 (3), Rebecca J. Chan, Todd Cooper, Christian P. Kratz, Brian Weiss, Mignon L. Loh, Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium, Pages 355-62, Copyright 2009. 神経線維腫症1型(NF1)およびヌーナン症候群の小児はJMMLを発症するリスクが高い:[ 8 ][ 9 ]
プロテアソームによる分解の標的蛋白、特にチロシンキナーゼに関わるE3ユビキチン蛋白リガーゼであるCBL遺伝子の変異は、JMML症例の10~15%にみられ[ 13 ][ 14 ]、これらの症例の多くが生殖細胞CBL変異を有する小児に発生する。[ 15 ][ 16 ]CBLの生殖細胞変異は、結果的に成長障害、発達遅滞、停留精巣、JMML素因などの特徴を有する常染色体優性の発達障害を引き起こす。[ 15 ]CBLの生殖細胞変異を有する患者の一部は、JMMLの自然消退を経験するが、その後に血管炎を発症する。[ 15 ]CBL変異は、ほとんどの場合、RASおよびPTPN11の変異と相互排他的である。[ 13 ]
JMMLのゲノム情報
JMMLのゲノムの全体像は、次の5つのRas経路遺伝子の1つにおける変異を特徴とする:NF1、NRAS、KRAS、PTPN11、およびCBL。[ 17 ][ 18 ][ 19 ]Ras経路活性化変異を有するJMMLと診断された連続した症例118人のシリーズで、PTPN11が変異を生じた遺伝子として最も多く、症例の51%を占めていた(19%が生殖細胞系および32%が体細胞系)(図2を参照のこと)。[ 17 ]NRAS変異を有する患者が症例の19%を、KRAS変異を有する患者が症例の15%を占めていた。NF1変異は症例の8%を占め、CBL変異は症例の11%を占めていた。これら5つの遺伝子変異は一般的に相互排他的であるが、症例の4~17%はこのようなRas経路の遺伝子の2つに変異を有しており[ 17 ][ 18 ][ 19 ]、これは予後不良と関連する所見である。[ 17 ][ 19 ]
JMML白血病細胞の変異率は非常に低いが、前述の5つのRas経路遺伝子以外のさらなる変異が認められている。[ 17 ][ 18 ][ 19 ]転写抑制因子複合体PRC2の遺伝子に二次的ゲノム変化が認められている(例、ASXL1が症例の7~8%で変異を生じていた)。成人の骨髄増殖性腫瘍に関連するいくつかの遺伝子も、JMMLにおいては低率で変異を生じている(例、SETBP1が症例の6~9%で変異を生じていた)。[ 17 ][ 18 ][ 19 ][ 20 ]JAK3変異もJMML症例のわずかな割合(4~12%)で認められている。[ 17 ][ 18 ][ 19 ][ 20 ]生殖細胞系PTPN11および生殖細胞系CBL変異を有する症例はさらなる変異を低率で示した(図2を参照のこと)。[ 17 ]疾患を定義するRAS経路の変異以外の変異の存在が、より不良な予後に関連している。[ 17 ][ 18 ]
JMMLのゲノムの全体像について記述した報告では、患者150人中16人(11%)で標準のRas経路の変異が認められなかったことが明らかにされた。これら16人の患者のうち、3人は受容体チロシンキナーゼが関与するインフレーム融合(DCTN1-ALK、RANBP2-ALK、およびTBL1XR1-ROS1)を有することが観察された。これらの患者は全員が7モノソミーを有し、生後56ヵ月以上であった。ALK融合が認められた1人の患者は、クリゾチニブ + 従来の化学療法で治療され、分子的完全寛解を達成し、同種骨髄移植に進んだ。[ 19 ]
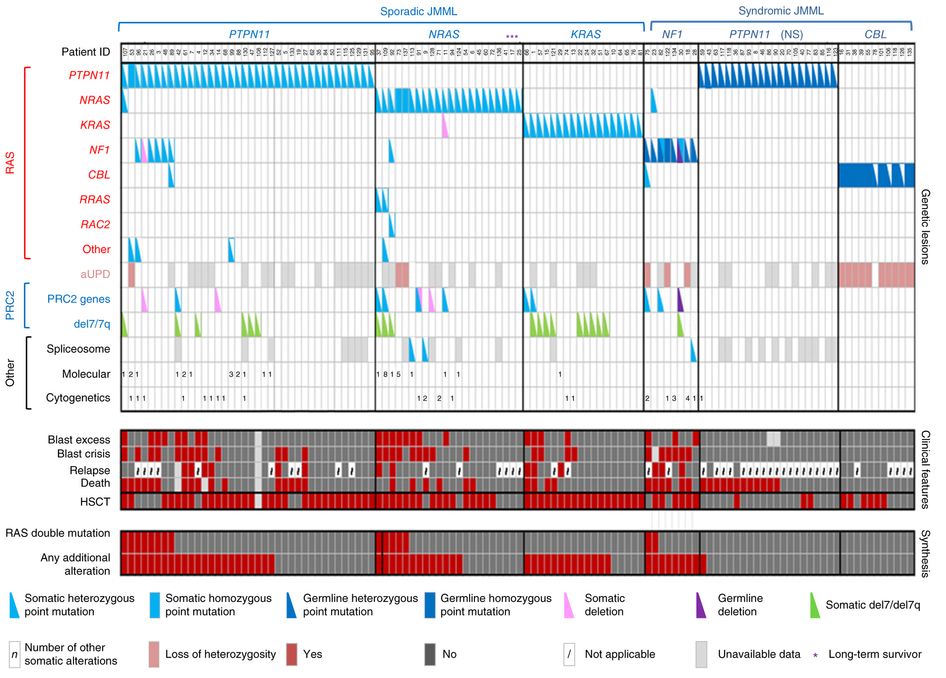
図2.個別のJMML症例における変化プロファイル。JMMLで詳細な遺伝子解析を受けた患者118人について、RAS経路とPRC2ネットワークにおいて反復するヒットにより生じた生殖細胞変異と体細胞変異が示されている。芽球過剰は、診断時の骨髄内における有核細胞のうちの芽球数が10%以上であるが20%未満と定義された。急性転化は骨髄内における有核細胞のうちの芽球数が20%以上と定義された。NS、ヌーナン症候群。Macmillan Publishers Ltdから許諾を得て転載:Nature Genetics (Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network.Nat Genet 47 [11]: 1334-40, 2015), copyright (2015). 予後(ゲノムおよび分子的因子)
以下を含むいくつかのゲノム因子がJMML患者の予後に影響する:
- 非Ras経路の変異数。 JMMLの小児の予後についての予測因子は、疾患を定義するRas経路の変異以外の変異の数である。[ 17 ][ 18 ]
- RAS経路の二重変異。JMMLに関連する標準のRas経路の5つの遺伝子(NF1、NRAS、KRAS、PTPN11、およびCBL)における変異は一般的に相互排他的であるが、症例の4~17%はこのようなRas経路の遺伝子の2つに変異を有しており[ 17 ][ 18 ]、これは予後不良と関連する所見である。[ 17 ][ 18 ]
- DNAメチル化プロファイル。
- LIN28Bの過剰発現。LIN28B過剰発現は、JMML患児の約半数に認められ、生物学的に特徴的なJMMLサブセットが同定される。LIN28Bは幹細胞の再生を調節するRNA結合蛋白の1つである。[ 22 ]
予後(臨床因子)
何らかの治療を行った後の年齢、血小板数、および胎児ヘモグロビン値。過去には、化学療法を実施しても、90%を超えるJMML患者が死亡した[ 23 ];しかしながら、現在では造血幹細胞移植(HSCT)の適用により、約50%の生存率が認められている。[ 24 ]患者は以下の異なる3つの臨床コースに従うと考えられた:
何らかの治療を行った後の生存に関する予後良好因子には、2歳未満、血小板数が33×109/Lを超える、および年齢調整胎児ヘモグロビン低値が含まれる。[ 1 ][ 2 ]対照的に、2歳を過ぎていること、および診断時の血中胎児ヘモグロビン値が高いことは、転帰不良の予測因子である。[ 1 ][ 2 ]
JMMLの治療
JMMLの治療法の選択肢には以下のものが含まれる:
JMMLの治療における従来の抗白血病療法の役割は確定していない。JMMLの奏効判定基準についてはコンセンサスが得られていないことから、JMMLの治療における特定薬物の役割の決定が複雑になっている。[ 25 ]JMMLに対して抗白血病活性が示されている薬物として、エトポシド、シタラビン、チオプリン系薬剤(thioguanineおよびメルカプトプリン)、イソトレチノイン、およびファルネシル阻害剤が挙げられるが、この中で転帰の改善が示されているものはない。[ 25 ][ 26 ][ 27 ][ 28 ][ 29 ];[ 30 ][証拠レベル:2B]
現時点でHSCTは、JMMLで治癒が得られる最も大きなチャンスを提供する。[ 24 ][ 31 ][ 32 ][ 33 ][ 34 ]
証拠(HSCT):
- European Working Group on Childhood Myelodysplastic Syndromesの報告は、抗胸腺細胞グロブリンを併用するまたは併用しない、ブスルファン、シクロホスファミド、およびメルファランの一般的な前処置レジメンによる治療を受けた複数施設の100人の移植レシピエントが対象であった。レシピエントはさまざまな強度の移植前化学療法または分化誘導薬による治療を受けており、中には脾摘出術を受けた患者もいた。[ 24 ]
- 臍帯血移植により44%の5年無病生存率が得られ、診断時に1.4歳未満の小児、7モノソミー以外の核型を有する小児、およびHLAが5/6ないし6/6適合の臍帯血ユニットを移植された小児で転帰が改善した。[ 35 ][証拠レベル:3iiDii]これにより、臍帯血はこの集団の小児に対する追加のドナー供給源になることが示唆されている。
- 一般に骨髄破壊的HSCTに不適格な患者に対して、移植の有害な副作用を減らすための強度縮小前処置レジメンの使用についても少数の患者で報告されている。[
36
][
37
]
COGは、JMMLの小児を対象として標準強度の前処置レジメン(ブスルファン/シクロホスファミド/メルファラン)を強度縮小レジメン(ブスルファン/フルダラビン)と比較するランダム化試験を実施した。[ 38 ]
HSCT後のJMML小児における治療失敗では、疾患再発が主な原因で、症例の30~40%にみられる。[ 24 ][ 31 ][ 32 ]ドナーリンパ球輸注の役割は明らかではないが[ 39 ]、再燃したJMML患者の約50%が2回目のHSCTによる治療を受けて効果が得られることを示す報告がある。[ 40 ]
臨床評価段階にある治療法の選択肢
米国国立がん研究所(NCI)が支援している臨床試験に関する情報は、NCIウェブサイトに掲載されている。他の組織がスポンサーの臨床試験に関する情報については、ClinicalTrials.govウェブサイトを参照のこと。
以下は、全米および/または各医療施設で現在実施されている臨床試験の例である。
参考文献- Passmore SJ, Chessells JM, Kempski H, et al.: Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 121 (5): 758-67, 2003.[PUBMED Abstract]
- Niemeyer CM, Arico M, Basso G, et al.: Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89 (10): 3534-43, 1997.[PUBMED Abstract]
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.[PUBMED Abstract]
- Chan RJ, Cooper T, Kratz CP, et al.: Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res 33 (3): 355-62, 2009.[PUBMED Abstract]
- Loh ML: Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152 (6): 677-87, 2011.[PUBMED Abstract]
- Bresolin S, Zecca M, Flotho C, et al.: Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28 (11): 1919-27, 2010.[PUBMED Abstract]
- Olk-Batz C, Poetsch AR, Nöllke P, et al.: Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 117 (18): 4871-80, 2011.[PUBMED Abstract]
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.[PUBMED Abstract]
- Choong K, Freedman MH, Chitayat D, et al.: Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21 (6): 523-7, 1999 Nov-Dec.[PUBMED Abstract]
- Tartaglia M, Niemeyer CM, Fragale A, et al.: Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34 (2): 148-50, 2003.[PUBMED Abstract]
- Kratz CP, Niemeyer CM, Castleberry RP, et al.: The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106 (6): 2183-5, 2005.[PUBMED Abstract]
- Strullu M, Caye A, Lachenaud J, et al.: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 51 (10): 689-97, 2014.[PUBMED Abstract]
- Loh ML, Sakai DS, Flotho C, et al.: Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114 (9): 1859-63, 2009.[PUBMED Abstract]
- Muramatsu H, Makishima H, Jankowska AM, et al.: Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood 115 (10): 1969-75, 2010.[PUBMED Abstract]
- Niemeyer CM, Kang MW, Shin DH, et al.: Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42 (9): 794-800, 2010.[PUBMED Abstract]
- Pérez B, Mechinaud F, Galambrun C, et al.: Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47 (10): 686-91, 2010.[PUBMED Abstract]
- Caye A, Strullu M, Guidez F, et al.: Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet 47 (11): 1334-40, 2015.[PUBMED Abstract]
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al.: The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47 (11): 1326-33, 2015.[PUBMED Abstract]
- Murakami N, Okuno Y, Yoshida K, et al.: Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 131 (14): 1576-1586, 2018.[PUBMED Abstract]
- Sakaguchi H, Okuno Y, Muramatsu H, et al.: Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet 45 (8): 937-41, 2013.[PUBMED Abstract]
- Stieglitz E, Mazor T, Olshen AB, et al.: Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun 8 (1): 2127, 2017.[PUBMED Abstract]
- Helsmoortel HH, Bresolin S, Lammens T, et al.: LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 127 (9): 1163-72, 2016.[PUBMED Abstract]
- Freedman MH, Estrov Z, Chan HS: Juvenile chronic myelogenous leukemia. Am J Pediatr Hematol Oncol 10 (3): 261-7, 1988 Fall.[PUBMED Abstract]
- Locatelli F, Nöllke P, Zecca M, et al.: Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood 105 (1): 410-9, 2005.[PUBMED Abstract]
- Bergstraesser E, Hasle H, Rogge T, et al.: Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatr Blood Cancer 49 (5): 629-33, 2007.[PUBMED Abstract]
- Castleberry RP, Emanuel PD, Zuckerman KS, et al.: A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N Engl J Med 331 (25): 1680-4, 1994.[PUBMED Abstract]
- Woods WG, Barnard DR, Alonzo TA, et al.: Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol 20 (2): 434-40, 2002.[PUBMED Abstract]
- Loh ML: Childhood myelodysplastic syndrome: focus on the approach to diagnosis and treatment of juvenile myelomonocytic leukemia. Hematology Am Soc Hematol Educ Program 2010: 357-62, 2010.[PUBMED Abstract]
- Hasle H: Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr 19 (1): 1-8, 2007.[PUBMED Abstract]
- Stieglitz E, Ward AF, Gerbing RB, et al.: Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (4): 629-36, 2015.[PUBMED Abstract]
- Smith FO, King R, Nelson G, et al.: Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol 116 (3): 716-24, 2002.[PUBMED Abstract]
- Yusuf U, Frangoul HA, Gooley TA, et al.: Allogeneic bone marrow transplantation in children with myelodysplastic syndrome or juvenile myelomonocytic leukemia: the Seattle experience. Bone Marrow Transplant 33 (8): 805-14, 2004.[PUBMED Abstract]
- Baker D, Cole C, Price J, et al.: Allogeneic bone marrow transplantation in juvenile myelomonocytic leukemia without total body irradiation. J Pediatr Hematol Oncol 26 (3): 200-3, 2004.[PUBMED Abstract]
- Locatelli F, Niemeyer CM: How I treat juvenile myelomonocytic leukemia. Blood 125 (7): 1083-90, 2015.[PUBMED Abstract]
- Locatelli F, Crotta A, Ruggeri A, et al.: Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: a EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood 122 (12): 2135-41, 2013.[PUBMED Abstract]
- Yabe M, Sako M, Yabe H, et al.: A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr Transplant 12 (8): 862-7, 2008.[PUBMED Abstract]
- Koyama M, Nakano T, Takeshita Y, et al.: Successful treatment of JMML with related bone marrow transplantation after reduced-intensity conditioning. Bone Marrow Transplant 36 (5): 453-4; author reply 454, 2005.[PUBMED Abstract]
- Dvorak CC, Satwani P, Stieglitz E, et al.: Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children's Oncology Group study. Pediatr Blood Cancer 65 (7): e27034, 2018.[PUBMED Abstract]
- Yoshimi A, Bader P, Matthes-Martin S, et al.: Donor leukocyte infusion after hematopoietic stem cell transplantation in patients with juvenile myelomonocytic leukemia. Leukemia 19 (6): 971-7, 2005.[PUBMED Abstract]
- Yoshimi A, Mohamed M, Bierings M, et al.: Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia 21 (3): 556-60, 2007.[PUBMED Abstract]
- 慢性骨髄性白血病(CML)
-
発生率
慢性骨髄性白血病(CML)は、小児白血病全体に占める割合が5%未満で、小児期では年齢の高い青年に最も多くみられる。[ 1 ]
分子的異常
CMLで最も特徴的な細胞遺伝学的異常は、9番染色体と22番染色体の転座(t(9;22))で、BCR-ABL1融合蛋白を生じるフィラデルフィア染色体(Ph)である。[ 2 ]
臨床所見
CMLの特徴として著しい白血球増加がみられ、多くは血小板増加症で、ときに血小板の機能異常を伴う。骨髄穿刺および生検により、比較的正常な顆粒球の成熟を伴う過形成が明らかになり、白血病性芽球数の明らかな増加はみられない。CMLでは白血球アルカリホスファターゼ活性の低下がみられるが、これは特異的な所見ではない。
CMLには、以下の3つの臨床段階がある:
CMLの治療:歴史的視点
チロシンキナーゼ阻害剤(TKI)が現れる前は、同種造血幹細胞移植(HSCT)がCMLの小児に対する一次治療であった。この期間に発表されている報告では、HLA適合家族ドナー(MFD)を慢性期初期の小児の移植治療に用いた場合の生存率が70~80%で、HLA適合非血縁ドナーを用いた場合の生存率はこれより低いとされている。[ 4 ][ 5 ][ 6 ]
慢性期に移植を実施した場合の再燃率は低かった(20%未満)。[ 4 ][ 5 ]死亡の主な原因は治療関連死であり、ほとんどの報告でHLA-MFDと比較してHLA適合非血縁ドナーを用いた場合が多かった。[ 4 ][ 5 ]HLAアレルに対する高分解能DNAマッチングにより治療関連死亡の割合が減少し、非血縁ドナーを用いるHSCTの治療成績改善につながると考えられた。[ 7 ]
慢性期の移植と比較した場合、加速期または急性転化期における移植および二次慢性期における移植では、生存率に有意な低下がみられた。[ 4 ][ 5 ][ 6 ]移植片対宿主病を回避するためにTリンパ球除去を用いた場合は、再燃率が高く、全生存(OS)期間が短くなることから[ 8 ]、同種HSCT後の良好な転帰に移植片対白血病効果が寄与していることが裏付けられた。
BCR-ABL融合キナーゼの阻害を目指した治療薬として、TKIのイマチニブが導入されたことで、小児および成人のいずれにおいてもCML患者の治療に大きな変革がもたらされた。[ 9 ]CMLに対してTKIを用いた場合のデータのほとんどは、成人の臨床試験から得られたものであるため、成人での経験が最初に報告され、その後にかなり限定された数の小児に関する経験が報告された。
TKIによる成人CMLの治療
イマチニブは、ABLチロシンキナーゼの強力な阻害剤で、血小板由来増殖因子(PDGF)受容体(αおよびβ)およびKITの阻害剤でもある。イマチニブ治療により慢性期に治療を受けたCML患者では、臨床的、細胞遺伝学的、および分子的な寛解(BCR-ABL融合転写産物が認められないことで定義)が高い割合で達成される。[ 10 ]
証拠(成人に対するイマチニブ):
- イマチニブをインターフェロン + シタラビンと比較した大規模な第III相試験(IRIS)の結果に基づいて、CMLの初期治療で、イマチニブが遺伝子組換えインターフェロンαの使用に置き換えられた。[ 11 ][ 12 ]
治療に対する患者の反応に基づいたCMLの成人向けのイマチニブ治療に関するガイドラインが策定されており、その中には、血液学的完全奏効、細胞遺伝学的完全奏効、および分子的大奏効(BCR-ABL1/対照遺伝子比率における3logの低下として定義)を達成する時期が含まれている。[ 13 ][ 14 ][ 15 ][ 16 ]
長期治療を受けている成人CML患者で、細胞遺伝学的完全奏効が得られないことおよびイマチニブ治療失敗の主な理由は、服薬遵守不良である。[ 17 ]イマチニブ治療に対して失敗または効果不十分であった時点で、BCR-ABL1キナーゼドメインの変異を同定することも臨床的に重要であり[ 18 ]、これは、イマチニブに対する抵抗性の原因となる一部の(すべてではない)変異に対して活性を維持する代替のBCR-ABLキナーゼ阻害剤(例えば、ダサチニブおよびニロチニブ)があるためである。[ 13 ][ 19 ][ 20 ]
2つのTKI、ダサチニブおよびニロチニブは、イマチニブに対する反応が不十分な患者に有効であることが示されているが、T315I変異を有する患者には無効である。またダサチニブおよびニロチニブはどちらも、以下の研究に基づいて、成人において新たに診断された慢性期CMLの治療を適応として規制当局の承認を得ている:
ダサチニブおよびニロチニブはいずれも、細胞遺伝学的完全奏効率および分子的大奏効率に関してイマチニブより優れているため、CMLの成人における第一選択治療として広く用いられている。しかしながら、第一選択療法として用いた場合、ダサチニブおよびニロチニブによる効果はイマチニブより早く現れるにもかかわらず、PFSおよびOSは、3つのすべての薬剤で同程度であると考えられている。[ 23 ][ 24 ]長期のPFSおよびOSに対するこれらの薬剤の効果をさらによく確定するには、追加の追跡が必要である。
ボスチニブは、BCR-ABL融合を標的とした別のTKIで、すべての相のCMLの成人に対して、別のTKIによる前治療に対して不耐性または病変が抵抗性を示す場合の治療法として米国食品医薬品局(FDA)により承認されている。小児集団でボスチニブは研究されていない。
ポナチニブは、BCR-ABL阻害剤の1つで、T315I変異に対して有効である。[ 25 ]ポナチニブにより、慢性期CMLで多くの前治療歴がある成人患者の約70%に客観的奏効が得られ、ベースライン時のBCR-ABLキナーゼドメインの変異とは無関係に奏効が観察された。[ 26 ]ポナチニブの開発は、この薬剤を投与された患者の高い割合に血管閉塞が観察されたことにより困難になっており、動脈および静脈血栓症および閉塞(心筋梗塞や脳卒中を含む)が治療を受けた20%を超える患者に発生している。[ 27 ]小児集団でポナチニブは研究されていない。
同種HSCTに進む成人のCML患者について、移植前のイマチニブが転帰に悪影響を及ぼすという証拠は示されていない。
証拠(成人におけるイマチニブとその後のHSCT):
- 移植前にイマチニブを使用した患者145人と歴史的コホートの患者231人を比較したレトロスペクティブ研究では、早期の肝毒性作用または生着の遅延に差がみられないことが示された。[ 28 ]
- 移植前のイマチニブが移植後の転帰に影響しないことに関するさらなる証拠は、Center for International Blood and Marrow Transplant Researchによる報告からもたらされた;この報告では、第一慢性期でHSCT前にイマチニブによる治療を受けた小児および成人のCML被験者181人に対して、HSCT前にイマチニブを受けなかった被験者657人との転帰が比較された。[ 29 ]
- イマチニブとその後の同種HSCTの第三の報告では、第一慢性期でイマチニブ治療に失敗した患者におけるこの移植戦略の有効性が裏付けられる。[ 13 ]
(HSCTを併用しない)TKI単独で治療された成人患者について、至適治療期間は依然として不明であり、ほとんどの患者がTKI治療を無期限に継続している。
証拠(成人におけるイマチニブ治療の長さ):
- 治療の長さに関する疑問に答えようと、イマチニブによる治療が2年を超え、細胞遺伝学的大奏効が2年を超えている成人69人に関して、プロスペクティブ研究により報告された。患者は毎月1回のモニタリングを受け、分子的再燃の証拠があればイマチニブが再開された。[ 30 ]
- 別の研究では、ポリメラーゼ連鎖反応(PCR)により微小残存病変(MRD)が少なくとも2年以上継続して検出できなかったため、イマチニブによる治療を中止した慢性期CML患者40人について報告された。[ 31 ]
分子的寛解状態にある選択されたCML患者に対して、イマチニブまたは他のBCR-ABL標的薬の中止が標準的臨床医療として推奨できるようになるには、研究がさらに必要である。
小児CMLの治療
小児CMLに対する治療法選択肢には以下を含めてもよい:
- イマチニブなどのチロシンキナーゼ阻害剤。
CMLの小児で、イマチニブは成人に観察されている活性と同程度の高レベルの活性を示している。[ 32 ][ 33 ][ 34 ][ 35 ][ 36 ]
証拠(小児におけるイマチニブ):
- プロスペクティブ試験において、新たにCMLと診断された小児患者44人がイマチニブ(260mg/日)による治療を受けた。[ 36 ]
この高レベルの活性の結果として、CML患児では、直ちに同種幹細胞移植へ進むのではなく、イマチニブによる治療を開始することが一般的である。[ 37 ]小児におけるイマチニブの薬物動態は、成人における以前の結果と一致していると考えられる。[ 38 ]
CMLの小児を対象とした第II相試験で用いられたイマチニブの用量は、260mg/m2~340mg/m2の範囲で、成人における400~600mgの一定用量に匹敵する薬物曝露が得られる。[ 34 ][ 35 ][ 36 ]
証拠(小児におけるイマチニブの用量):
- イマチニブ340mg/m2/日による治療を受けた47人の慢性期CMLの小児患者を対象としたイタリアの研究では、中央値6ヵ月で細胞遺伝学的完全奏効が91.5%の患者で得られ、12ヵ月後の分子的大奏効率は66.6%であった。[
36
]
したがって、340mg/m2という比較的高用量で開始することでより優れた有効性が得られ、毒性に対し必要に応じた用量調節を行うことで、一般的に忍容性があると考えられる。[ 35 ][ 36 ]
- 治療から3ヵ月経過時のPCRに基づいたMRDの測定でBCR-ABL1/ABLが最大10%といった早期の分子的奏効は、成人における早期の分子的奏効データに類似したPFSの改善と関係することが報告されている。[ 39 ]
前述の成人CMLに対して記述されたモニタリングガイドラインは、小児に使用することが妥当である。
イマチニブは、小児における忍容性がおおむね良好であり、有害作用は一般に軽度から中等度で、治療中止または用量減量により改善可能である。[ 34 ][ 35 ]イマチニブを服用している思春期前のほとんどの小児に発育遅延がみられる。[ 40 ]イマチニブを服用し、成長障害がみられる小児では、思春期成長スパート中に、ある程度の遅れを取り戻す成長を示すことがあるが、ほとんどの患者が親の中間身長に達しないことから、予想より低い成人身長となるリスクがある。[ 40 ][ 41 ]
CMLの小児における使用がFDAにより承認された他の2つTKI-ダサチニブおよびニロチニブの効力および毒性作用に関するデータはほとんど発表されていない。
証拠(小児におけるダサチニブ):
- 小児を対象としたダサチニブの第I相試験では、この薬物の体内動態、忍容性、および有効性が成人で観察されたものとほぼ同じであることが示された。[ 42 ][ 43 ]
- 新たに慢性期CMLと診断された小児84人を含むダサチニブの第II相試験では、60mg/m2(錠剤)または72mg/m2(経口溶液)を用いて患者に1日1回投与された。[ 44 ]
証拠(小児におけるニロチニブ):
- 2018年3月のCMLの小児の治療に対するFDAによるニロチニブの承認は、2件の民間主導の試験に基づいていた。[ 45 ][ 46 ]患者11人を対象とした1つ目の試験(NCT01077544 [CAMN107A2120])では、薬物動態、安全性、および予備的な有効性データが評価され、患者58人を対象とした2つ目の試験(NCT01844765 [CAMN107A2203;AAML1321])では、有効性および安全性が評価された。両研究からのデータは、患者69人の併合データ解析で統合され、新規診断CML患者25人および耐性または不耐容のCML患者44人が含まれていた。いずれの研究でも230mg/m2(最も近い50mg単位で端数処理;最大用量400mg)の1日2回投与が使用された。[ 46 ]
他のTKI(ボスチニブおよびポナチニブ)で、小児での安全用量はまだ確立されていない。
再発または不応性小児CMLの治療
再発または不応性CMLに対する治療法選択肢には以下を含めてもよい:
- ダサチニブまたはニロチニブのような代替のキナーゼ阻害剤。
- 同種HSCT。
イマチニブによる治療中に血液学的または細胞遺伝学的な再燃を起こした小児、またはイマチニブに対する初期反応が不十分であった小児では、今後の治療方針に役立てるため、BCR-ABLキナーゼドメインの変異状態の検査を検討すべきである。患者の変異状態に応じて、ダサチニブまたはニロチニブのような代替のキナーゼ阻害剤がこれらの薬剤の成人および小児における経験に基づいて検討可能である。[ 21 ][ 22 ][ 44 ][ 47 ][ 48 ][ 49 ]
証拠(耐性または不耐容のCML患児におけるダサチニブ):
- 耐性または不耐容のCML患児14人で、ダサチニブ治療の12ヵ月後に76%の患者が細胞遺伝学的完全寛解に達し、41%の患者が分子的大奏効に達した。48ヵ月でのPFSは78%であった。[ 44 ]
証拠(耐性または不耐容のCML患児におけるニロチニブ):
- イマチニブまたはダサチニブに耐性または不耐容のCML患児44人で、ニロチニブ治療の12ヵ月後に40.7%の患者が分子的大奏効に達した。中央値で11.3ヵ月後、疾患進行が認められた患者はいなかった。[ 45 ]
ダサチニブおよびニロチニブは、イマチニブに対する抵抗性の原因となる多くのBCR-ABL変異に対して活性を示すものの、T315I変異を有する患者には無効である。FDA承認を受けたすべてのキナーゼ阻害剤に対して抵抗性を示すT315I変異が存在する場合は、同種移植を考慮すべきである。
複数のTKIが利用できる場合に、CMLの小児患者が同種移植を受けるべきかどうかという問題は依然として解明されていない;しかしながら、HSCTを使用した場合、イマチニブの持続的投与の場合との比較で、PFSが改善しないことがいくつかの報告により示唆される。[ 36 ]考えられる長所と短所について、患者および家族と話し合う必要がある。HSCTは現在のところCMLに対して治癒が得られることが知られている唯一の根治治療であるが、持続的な分子的寛解後にTKIによる治療を終了し、分子的寛解を維持した患者が報告されている。[ 31 ]
最新の臨床試験
NCIが支援しているがん臨床試験で現在患者登録中の試験を検索するには、臨床試験アドバンスト・サーチを使用のこと(なお、このサイトは日本語検索に対応していない。)。このサーチでは、試験の場所、治療の種類、薬物名やその他の基準による絞り込みが可能である。臨床試験に関する一般情報も入手することができる。
参考文献- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed January 31, 2020.[PUBMED Abstract]
- Quintás-Cardama A, Cortes J: Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113 (8): 1619-30, 2009.[PUBMED Abstract]
- O'Dwyer ME, Mauro MJ, Kurilik G, et al.: The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood 100 (5): 1628-33, 2002.[PUBMED Abstract]
- Millot F, Esperou H, Bordigoni P, et al.: Allogeneic bone marrow transplantation for chronic myeloid leukemia in childhood: a report from the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC). Bone Marrow Transplant 32 (10): 993-9, 2003.[PUBMED Abstract]
- Cwynarski K, Roberts IA, Iacobelli S, et al.: Stem cell transplantation for chronic myeloid leukemia in children. Blood 102 (4): 1224-31, 2003.[PUBMED Abstract]
- Weisdorf DJ, Anasetti C, Antin JH, et al.: Allogeneic bone marrow transplantation for chronic myelogenous leukemia: comparative analysis of unrelated versus matched sibling donor transplantation. Blood 99 (6): 1971-7, 2002.[PUBMED Abstract]
- Lee SJ, Klein J, Haagenson M, et al.: High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 110 (13): 4576-83, 2007.[PUBMED Abstract]
- Horowitz MM, Gale RP, Sondel PM, et al.: Graft-versus-leukemia reactions after bone marrow transplantation. Blood 75 (3): 555-62, 1990.[PUBMED Abstract]
- Druker BJ: Translation of the Philadelphia chromosome into therapy for CML. Blood 112 (13): 4808-17, 2008.[PUBMED Abstract]
- Kantarjian H, Sawyers C, Hochhaus A, et al.: Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 346 (9): 645-52, 2002.[PUBMED Abstract]
- O'Brien SG, Guilhot F, Larson RA, et al.: Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 348 (11): 994-1004, 2003.[PUBMED Abstract]
- Druker BJ, Guilhot F, O'Brien SG, et al.: Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 355 (23): 2408-17, 2006.[PUBMED Abstract]
- Saussele S, Lauseker M, Gratwohl A, et al.: Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood 115 (10): 1880-5, 2010.[PUBMED Abstract]
- Hughes TP, Hochhaus A, Branford S, et al.: Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood 116 (19): 3758-65, 2010.[PUBMED Abstract]
- Kantarjian H, Cortes J: Considerations in the management of patients with Philadelphia chromosome-positive chronic myeloid leukemia receiving tyrosine kinase inhibitor therapy. J Clin Oncol 29 (12): 1512-6, 2011.[PUBMED Abstract]
- Bisen A, Claxton DF: Tyrosine kinase targeted treatment of chronic myelogenous leukemia and other myeloproliferative neoplasms. Adv Exp Med Biol 779: 179-96, 2013.[PUBMED Abstract]
- Ibrahim AR, Eliasson L, Apperley JF, et al.: Poor adherence is the main reason for loss of CCyR and imatinib failure for chronic myeloid leukemia patients on long-term therapy. Blood 117 (14): 3733-6, 2011.[PUBMED Abstract]
- Soverini S, Hochhaus A, Nicolini FE, et al.: BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 118 (5): 1208-15, 2011.[PUBMED Abstract]
- Hazarika M, Jiang X, Liu Q, et al.: Tasigna for chronic and accelerated phase Philadelphia chromosome--positive chronic myelogenous leukemia resistant to or intolerant of imatinib. Clin Cancer Res 14 (17): 5325-31, 2008.[PUBMED Abstract]
- Brave M, Goodman V, Kaminskas E, et al.: Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res 14 (2): 352-9, 2008.[PUBMED Abstract]
- Kantarjian H, Shah NP, Hochhaus A, et al.: Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 362 (24): 2260-70, 2010.[PUBMED Abstract]
- Saglio G, Kim DW, Issaragrisil S, et al.: Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 362 (24): 2251-9, 2010.[PUBMED Abstract]
- Jabbour E, Kantarjian HM, Saglio G, et al.: Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 123 (4): 494-500, 2014.[PUBMED Abstract]
- Hochhaus A, Saglio G, Hughes TP, et al.: Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 30 (5): 1044-54, 2016.[PUBMED Abstract]
- O'Hare T, Shakespeare WC, Zhu X, et al.: AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16 (5): 401-12, 2009.[PUBMED Abstract]
- Cortes JE, Kim DW, Pinilla-Ibarz J, et al.: A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med 369 (19): 1783-96, 2013.[PUBMED Abstract]
- Prasad V, Mailankody S: The accelerated approval of oncologic drugs: lessons from ponatinib. JAMA 311 (4): 353-4, 2014 Jan 22-29.[PUBMED Abstract]
- Oehler VG, Gooley T, Snyder DS, et al.: The effects of imatinib mesylate treatment before allogeneic transplantation for chronic myeloid leukemia. Blood 109 (4): 1782-9, 2007.[PUBMED Abstract]
- Lee SJ, Kukreja M, Wang T, et al.: Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood 112 (8): 3500-7, 2008.[PUBMED Abstract]
- Mahon FX, Réa D, Guilhot J, et al.: Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 11 (11): 1029-35, 2010.[PUBMED Abstract]
- Ross DM, Branford S, Seymour JF, et al.: Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood 122 (4): 515-22, 2013.[PUBMED Abstract]
- Champagne MA, Capdeville R, Krailo M, et al.: Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104 (9): 2655-60, 2004.[PUBMED Abstract]
- Millot F, Guilhot J, Nelken B, et al.: Imatinib mesylate is effective in children with chronic myelogenous leukemia in late chronic and advanced phase and in relapse after stem cell transplantation. Leukemia 20 (2): 187-92, 2006.[PUBMED Abstract]
- Millot F, Baruchel A, Guilhot J, et al.: Imatinib is effective in children with previously untreated chronic myelogenous leukemia in early chronic phase: results of the French national phase IV trial. J Clin Oncol 29 (20): 2827-32, 2011.[PUBMED Abstract]
- Champagne MA, Fu CH, Chang M, et al.: Higher dose imatinib for children with de novo chronic phase chronic myelogenous leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer 57 (1): 56-62, 2011.[PUBMED Abstract]
- Giona F, Putti MC, Micalizzi C, et al.: Long-term results of high-dose imatinib in children and adolescents with chronic myeloid leukaemia in chronic phase: the Italian experience. Br J Haematol 170 (3): 398-407, 2015.[PUBMED Abstract]
- Andolina JR, Neudorf SM, Corey SJ: How I treat childhood CML. Blood 119 (8): 1821-30, 2012.[PUBMED Abstract]
- Menon-Andersen D, Mondick JT, Jayaraman B, et al.: Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother Pharmacol 63 (2): 229-38, 2009.[PUBMED Abstract]
- Millot F, Guilhot J, Baruchel A, et al.: Impact of early molecular response in children with chronic myeloid leukemia treated in the French Glivec phase 4 study. Blood 124 (15): 2408-10, 2014.[PUBMED Abstract]
- Shima H, Tokuyama M, Tanizawa A, et al.: Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia. J Pediatr 159 (4): 676-81, 2011.[PUBMED Abstract]
- Millot F, Guilhot J, Baruchel A, et al.: Growth deceleration in children treated with imatinib for chronic myeloid leukaemia. Eur J Cancer 50 (18): 3206-11, 2014.[PUBMED Abstract]
- Aplenc R, Blaney SM, Strauss LC, et al.: Pediatric phase I trial and pharmacokinetic study of dasatinib: a report from the children's oncology group phase I consortium. J Clin Oncol 29 (7): 839-44, 2011.[PUBMED Abstract]
- Zwaan CM, Rizzari C, Mechinaud F, et al.: Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31 (19): 2460-8, 2013.[PUBMED Abstract]
- Gore L, Kearns PR, de Martino ML, et al.: Dasatinib in Pediatric Patients With Chronic Myeloid Leukemia in Chronic Phase: Results From a Phase II Trial. J Clin Oncol 36 (13): 1330-1338, 2018.[PUBMED Abstract]
- Novartis Pharmaceuticals Corporation: TASIGNA (nilotinib): Prescribing Information. East Hanover, NJ: Novartis, 2018. Available online. Last accessed March 25, 2020.[PUBMED Abstract]
- Hijiya N, Maschan A, Rizzari C, et al.: Phase 2 study of nilotinib in pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia. Blood 134 (23): 2036-2045, 2019.[PUBMED Abstract]
- Hochhaus A, Baccarani M, Deininger M, et al.: Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia 22 (6): 1200-6, 2008.[PUBMED Abstract]
- le Coutre P, Ottmann OG, Giles F, et al.: Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood 111 (4): 1834-9, 2008.[PUBMED Abstract]
- Kantarjian H, O'Brien S, Talpaz M, et al.: Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Cancer 109 (8): 1556-60, 2007.[PUBMED Abstract]
- がんの小児の治療に関する特別な考慮事項
-
小児および青年におけるがんはまれであるが、小児がんの全発生率は、1975年以降徐々に増加している。[ 1 ]小児および青年のがん患者は、小児期および青年期に発生するがんの治療経験を有する専門家から構成される集学的チームのある医療機関に紹介すべきである。[ 2 ]この集学的チームのアプローチでは、至適な生存期間および生活の質が得られる治療、支持療法、およびリハビリテーションを小児が必ず受けられるように、以下の小児およびその他の専門家の技能を取り入れている。
(小児および青年のがんの支持療法に関する具体的な情報については、PDQの支持療法および緩和ケアの要約を参照のこと。)
小児がん施設のためのガイドラインと、それらの施設が小児がん患者の治療において担う役割は、米国小児科学会(American Academy of Pediatrics)によって概説されている。[ 3 ]このような小児がん施設では、小児および青年にみられるほとんどの種類のがんに関して、臨床試験が利用可能であり、これらの試験へ参加する機会がほとんどの患者およびその家族に提供される。小児および青年のがんに関する臨床試験は一般に、現在標準とされている治療法と、それより効果的であると思われる治療法とを比較するようデザインされる。小児がんの治癒を目指した治療法の進歩の大部分は、このような臨床試験によって達成されたものである。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
参考文献- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Wolfson J, Sun CL, Wyatt L, et al.: Adolescents and Young Adults with Acute Lymphoblastic Leukemia and Acute Myeloid Leukemia: Impact of Care at Specialized Cancer Centers on Survival Outcome. Cancer Epidemiol Biomarkers Prev 26 (3): 312-320, 2017.[PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.[PUBMED Abstract]
- 生存者と有害な晩期続発症
-
がんおよびその治療による長期合併症の問題は多くの疾患カテゴリーに及ぶが、骨髄性悪性疾患の治療に関連して、強調に値する重大な問題がいくつかある。(詳しい情報については、PDQの小児がん治療の晩期合併症(晩期障害)の要約を参照のこと。)
造血幹細胞移植(HSCT)を受けていない成人生存者を対象としたAML治療による晩期合併症(晩期障害)に関して選択した研究には以下のものがある:
- 心臓。
- Children's Cancer Survivor Studyでは、小児急性骨髄性白血病(AML)でHSCTを受けなかった生存者272人について調査された。[ 1 ]
- 英国医学研究審議会ベースのレジメンで治療を受けた小児の心機能を評価したレトロスペクティブ研究では、治療後中央値13ヵ月での左室の1回拍出量において、ベースライン値との比較で平均8.4%の有害な変化が報告された。[ 3 ]
- 小児患者では、早期毒性が発現するリスクが13.7%で、晩期心毒性(第一選択治療終了後1年として定義)が発現するリスクが17.4%であった。早期心毒性作用は、晩期心毒性作用のほか、長期的な治療を要する臨床的な心筋症の発生を示す有意な予測因子であった。[ 4 ]
- 単一研究のレトロスペクティブ分析では、ダウン症候群の小児において心疾患リスクが高い可能性が示唆されるが[ 5 ]、この結果を確認するにはプロスペクティブ研究が必要である。
- 心理社会的問題。
- Nordic Society for Pediatric Hematology and Oncologyによるレトロスペクティブ試験では、化学療法のみを受けたAML患者について、医療機関を受診した自己報告を基に中央値で11年追跡し、医療機関の利用状況および婚姻状況が各同胞とほぼ同じであることが明らかにされた。[ 6 ]
- 小児AMLでHSCTを受けなかった生存者を対象とした集団ベース研究では、同胞と比較した学業成績、雇用、および配偶者の有無の割合が同程度であったことが報告された。しかしながら、AML生存者は処方薬を使用している傾向が同胞と比較して有意に高く(23% vs 9%;P = 0.03)、特に喘息に対する薬が多かった。小児AMLの生存者では、慢性疲労も他の悪性腫瘍の生存者より有意に多くみられる有害な晩期合併症(晩期障害)であることが明らかになっている。[ 7 ]
AMLの治療として化学療法のみを受けた小児では、腎臓、消化管、および肝臓への有害な晩期合併症(晩期障害)はまれであると報告されている。[ 8 ]
HSCTによる治療を受けた成人生存者を対象としたAML治療による晩期合併症(晩期障害)に関して選択した研究には以下のものがある:
- ある施設からのレビューによると、AMLの治療を受けた小児に最も高頻度にみられた有害な長期続発症として、以下の発生率のものがあった:成長異常(51%)、神経認知的異常(30%)、輸血で罹患した肝炎(28%)、不妊症(25%)、内分泌疾患(16%)、拘束性肺疾患(20%)、慢性移植片対宿主病(20%)、二次悪性腫瘍(14%)、および白内障(12%)。[ 9 ]
- 別の研究では、AMLまたは急性リンパ芽球性白血病(ALL)でHSCTを受けた3歳未満の小児の転帰が調査された。[ 10 ]
- 対照的に、Bone Marrow Transplant Survivor Studyでは、自己報告式質問票を用いて、小児AMLまたはALLの生存者を同胞と比較した。[ 11 ]追跡期間中央値は8.4年で、患者の86%が移植前処置の一環として全身放射線照射(TBI)を受けていた。
- 小児腫瘍学グループ(COG)研究では、健康関連の生活の質について比較され、重度または命を脅かす慢性的な健康障害を有する5年生存者が全体で21%と報告された;治療法の種類ごとに比較した場合の割合は、化学療法のみの治療群が16%、自家HSCTによる治療群が21%、同種HSCTによる治療群が33%であった。[ 12 ]
有害な長期的続発症を低減するためには、特に骨髄破壊的HSCTに関連した晩期続発症を低減する新しい治療アプローチが必要である。
がん生存者の経過観察およびリスクに関する詳細についての重要な情報源として、COGのLong-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers、およびNational Comprehensive Cancer NetworkのGuidelines for Acute Myeloid Leukemiaが公開されている。さらに、がん生存者にとって、治療を終えた後の医療提供者と共有できる過去の医療経過へのアクセスが重要であることが、ますます認識されるようになってきている。
参考文献- Mulrooney DA, Dover DC, Li S, et al.: Twenty years of follow-up among survivors of childhood and young adult acute myeloid leukemia: a report from the Childhood Cancer Survivor Study. Cancer 112 (9): 2071-9, 2008.[PUBMED Abstract]
- Creutzig U, Diekamp S, Zimmermann M, et al.: Longitudinal evaluation of early and late anthracycline cardiotoxicity in children with AML. Pediatr Blood Cancer 48 (7): 651-62, 2007.[PUBMED Abstract]
- Orgel E, Zung L, Ji L, et al.: Early cardiac outcomes following contemporary treatment for childhood acute myeloid leukemia: a North American perspective. Pediatr Blood Cancer 60 (9): 1528-33, 2013.[PUBMED Abstract]
- Temming P, Qureshi A, Hardt J, et al.: Prevalence and predictors of anthracycline cardiotoxicity in children treated for acute myeloid leukaemia: retrospective cohort study in a single centre in the United Kingdom. Pediatr Blood Cancer 56 (4): 625-30, 2011.[PUBMED Abstract]
- O'Brien MM, Taub JW, Chang MN, et al.: Cardiomyopathy in children with Down syndrome treated for acute myeloid leukemia: a report from the Children's Oncology Group Study POG 9421. J Clin Oncol 26 (3): 414-20, 2008.[PUBMED Abstract]
- Molgaard-Hansen L, Glosli H, Jahnukainen K, et al.: Quality of health in survivors of childhood acute myeloid leukemia treated with chemotherapy only: a NOPHO-AML study. Pediatr Blood Cancer 57 (7): 1222-9, 2011.[PUBMED Abstract]
- Jóhannsdóttir IM, Hjermstad MJ, Moum T, et al.: Increased prevalence of chronic fatigue among survivors of childhood cancers: a population-based study. Pediatr Blood Cancer 58 (3): 415-20, 2012.[PUBMED Abstract]
- Skou AS, Glosli H, Jahnukainen K, et al.: Renal, gastrointestinal, and hepatic late effects in survivors of childhood acute myeloid leukemia treated with chemotherapy only--a NOPHO-AML study. Pediatr Blood Cancer 61 (9): 1638-43, 2014.[PUBMED Abstract]
- Leung W, Hudson MM, Strickland DK, et al.: Late effects of treatment in survivors of childhood acute myeloid leukemia. J Clin Oncol 18 (18): 3273-9, 2000.[PUBMED Abstract]
- Perkins JL, Kunin-Batson AS, Youngren NM, et al.: Long-term follow-up of children who underwent hematopoeitic cell transplant (HCT) for AML or ALL at less than 3 years of age. Pediatr Blood Cancer 49 (7): 958-63, 2007.[PUBMED Abstract]
- Baker KS, Ness KK, Weisdorf D, et al.: Late effects in survivors of acute leukemia treated with hematopoietic cell transplantation: a report from the Bone Marrow Transplant Survivor Study. Leukemia 24 (12): 2039-47, 2010.[PUBMED Abstract]
- Schultz KA, Chen L, Chen Z, et al.: Health conditions and quality of life in survivors of childhood acute myeloid leukemia comparing post remission chemotherapy to BMT: a report from the children's oncology group. Pediatr Blood Cancer 61 (4): 729-36, 2014.[PUBMED Abstract]
- 心臓。
- 本要約の変更点(03/25/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
小児骨髄性悪性疾患の分類
参考文献20としてOrgel et al.および証拠レベル:3iiAを追加。
小児急性骨髄性白血病(AML)の組織化学的、免疫表現型の、および分子的評価
本文に以下の記述が追加された; 難治性AMLの小児に関する研究で、寛解に達したコホートと比較してNUP98の発現が過剰であった(引用、参考文献153として、McNeer et al.)。
本文に以下の記述が追加された;難治性AMLの小児に関する研究で、寛解に達したコホートと比較してWT1の発現が過剰であった。
慢性骨髄性白血病(CML)
参考文献46としてHijiya et al.が追加された。
本文でCML患者の2つの研究の併合データ解析について、第II相試験で新規診断CML患者の64%が1年で分子的大奏効を示したと改訂された。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児急性骨髄性白血病とその他の骨髄性悪性疾患の治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Childhood Acute Myeloid Leukemia/Other Myeloid Malignancies Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/leukemia/hp/child-aml-treatment-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389454]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。