

ご利用について
医療専門家向けの本PDQがん情報要約では、小児にはまれながんの治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 小児にはまれながんに関する一般情報
-
序
小児および青年におけるがんはまれであるが、小児がんの全発生率は1975年以降徐々に増加している。[ 1 ]小児および青年のがん患者については、小児期および青年期に発生するがんの治療経験を有するがん専門家から構成される集学的チームのある医療機関への紹介を検討すべきである。この集学的チームのアプローチとは、至適生存期間および至適QOLを得られるような治療、支持療法、およびリハビリテーションを小児が必ず受けられるようにするために、プライマリケア医、小児外科医、放射線腫瘍医、小児内科腫瘍医/血液専門医、リハビリテーション専門家、小児専門看護師、社会福祉士などの技術を集結したものである。(小児および青年のがんの支持療法に関する具体的な情報については、PDQの支持療法および緩和ケアの要約を参照のこと。)
米国小児科学会によって、小児がん施設とそれらが小児がん患者の治療において担う役割に関するガイドラインが概説されている。[ 2 ]このような小児がん施設では、小児および青年に発症するほとんどの種類のがんに関する臨床試験が行われており、大半の患者およびその家族に参加する機会が与えられている。小児および青年のがんに関する臨床試験は一般に、現在標準とされている治療法と、それより効果的であると思われる治療法とを比較するようデザインされる。小児がんの治癒を目指した治療法の進歩の大部分は、このような臨床試験によって達成されたものである。現在実施中の臨床試験に関する情報は、NCIウェブサイトから入手することができる。
小児および青年のがん患者の生存において、劇的な改善が達成されている。1975年から2010年の間に、小児がんの死亡率は50%以上低下した。[ 3 ]小児および青年のがん生存者では、治療から数ヵ月または数年経過後もがん療法の副作用が持続または発現することがあるため、綿密なモニタリングが必要である。(小児および青年のがん生存者における晩期合併症(晩期障害)の発生率、種類、およびモニタリングに関する具体的な情報については、小児がん治療の晩期合併症(晩期障害)に関するPDQ要約を参照のこと。)
小児がんはまれな疾患であり、米国において20歳未満で診断される症例は年間約15,000例である。[ 4 ]米国の2002年希少疾患対策法(Rare Diseases Act of 2002)では、希少疾患を罹患者が20万人未満の疾患と定めている。そのため、小児がんはすべて希少疾患とみなされる。
まれな腫瘍の指定は小児および成人のグループ間で統一されていない。成人のまれながんは、年間発生率が10万人当たり6例未満のがんとして定義され、欧州連合で診断されるすべてのがんの最大24%および米国で診断されるすべてのがんの約20%を占めていると推定される。[ 5 ][ 6 ]また、小児のまれな腫瘍の指定は、以下に示すように国際的グループ間で統一されていない:
このようなまれながんは、個々の診断を受ける患者の発生率が低いこと、青年集団にまれながんが多いこと、および黒色腫などのまれながんの青年についての臨床試験が行われていないことから、研究がきわめて困難である。
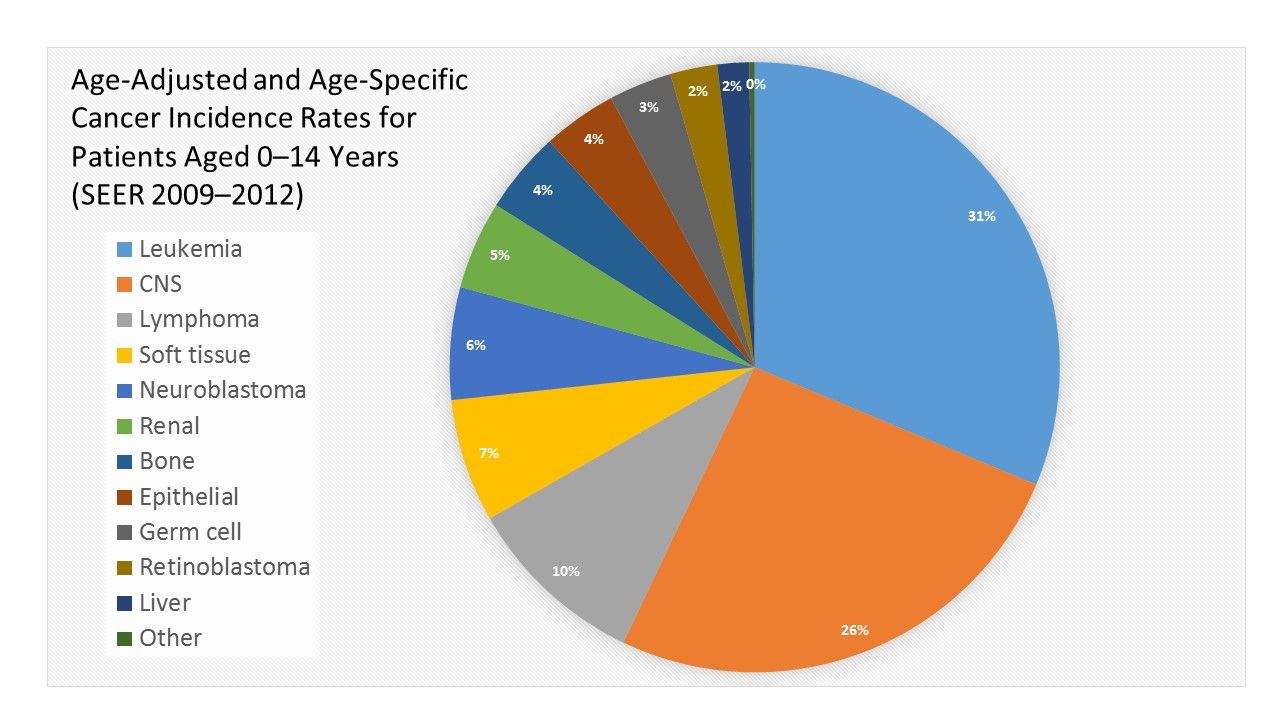
図12009年から2012年の、全人種、男性および女性についての、骨髄異形成症候群およびグループIII良性脳/中枢神経系腫瘍を含む、International Classification of Childhood Cancerのグループおよびサブグループならびに診断時年齢別の、年齢調整および年齢別(0~14歳)のSurveillance, Epidemiology, and End Results(SEER)がん発生率。 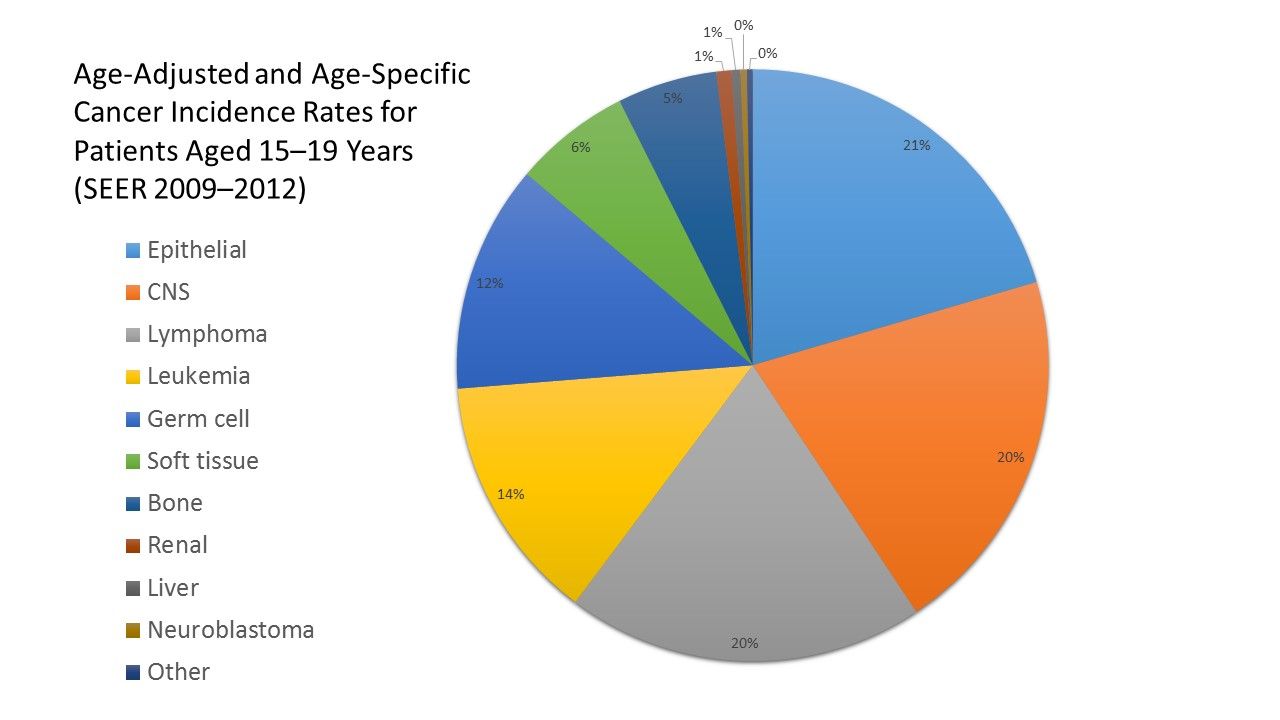
図22009年から2012年の、全人種、男性および女性についての、骨髄異形成症候群およびグループIII良性脳/中枢神経系腫瘍を含む、International Classification of Childhood Cancerのグループおよびサブグループならびに診断時年齢別の、年齢調整および年齢別(15~19歳)のSurveillance, Epidemiology, and End Results(SEER)がん発生率。 一部の研究者は、Surveillance, Epidemiology, and End Results(SEER)およびNational Cancer Databaseなどの大規模なデータベースを用いて、これらのまれな小児がんについて、より多くの見識を得ている。しかしながら、これらのデータベース研究は限られている。COGおよび他のInternational Society of Paediatric Oncology(Société Internationale D'Oncologie Pédiatrique [SIOP])などの国際的なグループにより、まれな小児がんを研究するための新提案がいくつか行われている。Gesellschaft für Pädiatrische Onkologie und Hämatologie(GPOH)のまれな腫瘍プロジェクトは、2006年にドイツで創設された。[ 10 ]TREPは2000年に発足し[ 7 ]、Polish Pediatric Rare Tumor Study Groupは2002年に発足した。[ 11 ]欧州では、フランス、ドイツ、イタリア、ポーランド、および英国のまれな腫瘍研究グループがEuropean Cooperative study Group on Pediatric Rare Tumors (EXPeRT)に参加し、特定のまれな腫瘍についての国際共同研究および分析に主眼を置いている。[ 12 ]COG内では、COGレジストリー(Project Every Child)および腫瘍バンクキングプロトコルへの登録を増進するとともに、単一治療群の臨床試験を開発し、成人の共同研究グループ試験との協力を増やすことに労力を注いでいる。[ 13 ]この構想の成果と課題が詳細に報告されている。[ 8 ][ 14 ]
本要約に記載されている腫瘍は多岐にわたる;これらの腫瘍を解剖学的に上部から下部へと列挙し、頻度の低い頭頸部腫瘍からほとんど発生をみない尿路性器路腫瘍や皮膚がんについて考察した。こうしたがんはすべて発生がまれであり、ほとんどの小児病院の経験でも一部の組織型は数年間で5例に満たない。記載されている組織型の大多数は、成人では比較的頻繁に発生している。これらの腫瘍に関する情報は、成人のがんに関連する情報源でも記載されている場合がある。
小児にはまれながんの治療要約は、各テーマの個別の要約に分割されつつある。個別の要約を探すには、以下のリストまたは次のリンクを使用のこと:https://www.cancer.gov/publications/pdq/information-summaries/pediatric-treatment。
参考文献- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.[PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.[PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.[PUBMED Abstract]
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.[PUBMED Abstract]
- Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017.[PUBMED Abstract]
- DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017.[PUBMED Abstract]
- Ferrari A, Bisogno G, De Salvo GL, et al.: The challenge of very rare tumours in childhood: the Italian TREP project. Eur J Cancer 43 (4): 654-9, 2007.[PUBMED Abstract]
- Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children's Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010.[PUBMED Abstract]
- Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2012. National Cancer Institute, 2015. Also available online. Last accessed June 22, 2021.[PUBMED Abstract]
- Brecht IB, Graf N, Schweinitz D, et al.: Networking for children and adolescents with very rare tumors: foundation of the GPOH Pediatric Rare Tumor Group. Klin Padiatr 221 (3): 181-5, 2009 May-Jun.[PUBMED Abstract]
- Balcerska A, Godziński J, Bień E, et al.: [Rare tumours--are they really rare in the Polish children population?]. Przegl Lek 61 (Suppl 2): 57-61, 2004.[PUBMED Abstract]
- Bisogno G, Ferrari A, Bien E, et al.: Rare cancers in children - The EXPeRT Initiative: a report from the European Cooperative Study Group on Pediatric Rare Tumors. Klin Padiatr 224 (6): 416-20, 2012.[PUBMED Abstract]
- Musselman JR, Spector LG, Krailo MD, et al.: The Children's Oncology Group Childhood Cancer Research Network (CCRN): case catchment in the United States. Cancer 120 (19): 3007-15, 2014.[PUBMED Abstract]
- Pappo AS, Furman WL, Schultz KA, et al.: Rare Tumors in Children: Progress Through Collaboration. J Clin Oncol 33 (27): 3047-54, 2015.[PUBMED Abstract]
- 頭頸部がん
-
15歳未満の患者ではこれらのがんは非常にまれにしかみられず、証拠のほとんどは小規模なケースシリーズまたは小児および成人の患者を合わせたコホートから得られている点を強調する必要がある。
小児肉腫はしばしば頭頸部領域に発生するが、これらは他のセクションで記述する。まれな小児頭頸部がんには以下のものがある:
上咽頭がん
詳しい情報については、小児上咽頭がんの治療に関するPDQ要約を参照のこと。
感覚神経芽腫
詳しい情報については、小児感覚神経芽腫の治療に関するPDQ要約を参照のこと。
甲状腺腫瘍
詳しい情報については、小児甲状腺がんの治療に関するPDQ要約を参照のこと。
口腔がん
詳しい情報については、小児口腔がんの治療に関するPDQ要約を参照のこと。
唾液腺腫瘍
詳しい情報については、小児唾液腺腫瘍の治療に関するPDQ要約を参照のこと。
喉頭がんおよび乳頭腫症
詳しい情報については、小児喉頭腫瘍の治療に関するPDQ要約を参照のこと。
NUT遺伝子が関与する正中線上のがん(NUT正中線がん)
詳しい情報については、NUT遺伝子が関与する小児正中線上のがん(NUT正中線がん)の治療に関するPDQ要約を参照のこと。
- 胸部のがん
-
以下に、こうした胸部のがんの予後、診断、分類、および治療を考察する。15歳未満の患者でこれらのがんがみられることは非常にまれであり、証拠のほとんどはケースシリーズから得られていることを強調する必要がある。[ 1 ]
まれな小児の胸部のがんには以下のものがある:
乳がん
詳しい情報については、小児乳がんの治療に関するPDQ要約を参照のこと。
肺がん
小児における肺の悪性新生物の大半は、転移病変によるもので、原発性悪性腫瘍と転移腫瘍の比率は約1:5である。[ 2 ]
以下は、最も一般的な肺の悪性原発腫瘍である:
気管気管支腫瘍
詳しい情報については、小児気管気管支腫瘍の治療に関するPDQ要約を参照のこと。
胸膜肺芽腫
詳しい情報については、小児胸膜肺芽腫の治療に関するPDQ要約を参照のこと。
食道がん
詳しい情報については、小児食道がんの治療に関するPDQ要約を参照のこと。
胸腺腫および胸腺がん
詳しい情報については、小児胸腺腫および胸腺がんの治療に関するPDQ要約を参照のこと。
心臓腫瘍
詳しい情報については、小児心臓腫瘍の治療に関するPDQ要約を参照のこと。
参考文献- Yu DC, Grabowski MJ, Kozakewich HP, et al.: Primary lung tumors in children and adolescents: a 90-year experience. J Pediatr Surg 45 (6): 1090-5, 2010.[PUBMED Abstract]
- Weldon CB, Shamberger RC: Pediatric pulmonary tumors: primary and metastatic. Semin Pediatr Surg 17 (1): 17-29, 2008.[PUBMED Abstract]
- 腹部のがん
-
以下に、腹部のがんに関する予後、診断、分類、および治療を考察する。15歳未満の患者でこれらのがんがみられることは非常にまれであり、証拠のほとんどがケースシリーズから得られていることを強調する必要がある。(腎腫瘍に関する情報については、ウィルムス腫瘍とその他の小児腎腫瘍の治療に関するPDQ要約を参照のこと。)
まれな小児の腹部のがんには以下のものがある:
副腎皮質がん
詳しい情報については、成人の小児副腎皮質がんの治療に関するPDQ要約を参照のこと。
胃がん
詳しい情報については、小児胃がんの治療に関するPDQ要約を参照のこと。
膵がん
詳しい情報については、小児膵がんの治療に関するPDQ要約を参照のこと。
大腸がん
詳しい情報については、小児大腸がんの治療に関するPDQ要約を参照のこと。
消化管カルチノイド
詳しい情報については、小児消化管カルチノイドの治療に関するPDQ要約を参照のこと。
消化管間質腫瘍(GIST)
詳しい情報については、小児消化管間質腫瘍の治療に関するPDQ要約を参照のこと。
- 生殖器/泌尿器の腫瘍
-
以下に、生殖器/泌尿器の腫瘍の予後、診断、分類、および治療を考察する。15歳未満の患者でこれらの腫瘍がみられることは非常にまれであり、証拠のほとんどがケースシリーズから得られていることを強調する必要がある。
まれな小児の生殖器/泌尿器の腫瘍には以下のものがある:
膀胱がん
詳しい情報については、小児膀胱がんの治療に関するPDQ要約を参照のこと。
精巣腫瘍(非胚細胞)
詳しい情報については、小児精巣腫瘍の治療に関するPDQ要約を参照のこと。
卵巣がん(非胚細胞)
詳しい情報については、小児卵巣がんの治療に関するPDQ要約を参照のこと。
子宮頸がんおよび膣がん
詳しい情報については、小児子宮頸がんおよび膣がんの治療に関するPDQ要約を参照のこと。
- その他の小児にはまれながん
-
以下に、その他の小児にはまれながんの予後、診断、分類、および治療を考察する。15歳未満の患者でこれらのがんがみられることは非常にまれであり、証拠のほとんどがケースシリーズから得られていることを強調する必要がある。
その他の小児にはまれながんには以下のものがある:
中皮腫
詳しい情報については、小児中皮腫の治療に関するPDQ要約を参照のこと。
多発性内分泌腫瘍(MEN)症候群およびカーニー複合
詳しい情報については、小児の多発性内分泌腫瘍(MEN)症候群の治療に関するPDQ要約を参照のこと。
褐色細胞腫と傍神経節腫
詳しい情報については、小児褐色細胞腫と傍神経節腫の治療に関するPDQ要約を参照のこと。
皮膚がん(黒色腫、基底細胞がん[BCC]、および扁平上皮がん[SCC])
(特定の遺伝子変異および関連するがん症候群に関する詳しい情報については、皮膚がんの遺伝学に関するPDQ要約を、小児におけるブドウ膜黒色腫に関する情報については、本要約の眼内[ブドウ膜]黒色腫のセクションを参照のこと。)
黒色腫
詳しい情報については、小児黒色腫の治療に関するPDQ要約を参照のこと。
BCCおよびSCC
詳しい情報については、小児基底細胞がんおよび皮膚扁平上皮がんの治療に関するPDQ要約を参照のこと。
眼内(ブドウ膜)黒色腫
詳しい情報については、小児眼内(ブドウ膜)黒色腫の治療に関するPDQ要約を参照のこと。
脊索腫
詳しい情報については、小児脊索腫の治療に関するPDQ要約を参照のこと。
原発部位不明のがん
詳しい情報については、小児原発不明がんの治療に関するPDQ要約を参照のこと。
- 本要約の変更点 (02/10/2021)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
その他の小児にはまれながん
新規のサブセクションとして 中皮腫が追加された。
本要約はPDQ Pediatric Treatment Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、小児にはまれながんの治療について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Pediatric Treatment Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Pediatric Treatment Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Pediatric Treatment Editorial Board.PDQ Rare Cancers of Childhood Treatment.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/childhood-cancers/hp/rare-childhood-cancers-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389315]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
入手可能な証拠の強さに基づき、治療選択肢は「標準」または「臨床評価段階にある」のいずれかで記載される場合がある。これらの分類は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。