

ご利用について
医療専門家向けの本PDQがん情報要約では、内分泌および神経内分泌腫瘍の遺伝学について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Cancer Genetics Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
CONTENTS
- 要旨
-
本要旨では、内分泌および神経内分泌腫瘍の遺伝学に関する本PDQ要約で取り扱われている話題について概要を示すとともに、各話題に関するエビデンスを記述した以下の詳細セクションへのハイパーリンクを提示している。
- 序
-
[注: 本要約で用いられている医学および科学用語については、NCI Dictionary of Genetics Termsに解説が用意されている。リンクが張られた用語をクリックすれば、別のウインドウにその定義が表示される。]
[注: 本要約に記載されている多くの遺伝子および病態については、Online Mendelian Inheritance in Man(OMIM)カタログに掲載されている。詳しい情報については、OMIMを参照のこと。]
[注: 現在、遺伝学的多様性を記載するための用語体系を変化させるべく、遺伝学のコミュニティにおいて協調的な取り組みが進められている。その変化とは、研究対象の個人または集団と参照配列との間に存在する遺伝学的な差異、特に生殖細胞系に存在する差異を記述する際に、従来の「mutation(突然変異ないし変異)」ではなく、「variant(多様体ないしバリアント)」という用語を使用するというものである。多様体はさらに、良性(無害)(benign [harmless])、おそらく良性(likely benign)、意義不明(of uncertain significance)、おそらく病原性(likely pathogenic)、病原性(疾患を引き起こす)(pathogenic [disease causing])のいずれかに分類することができる。本要約では、全体を通じて、疾患を引き起こす突然変異に対して病原性多様体(pathogenic variant)という用語を使用する。多様体の分類に関する詳しい情報については、がん遺伝学の概要に関する要約を参照のこと。]
内分泌または神経内分泌腺に関係する遺伝性症候群には、多発性内分泌腫瘍1型(MEN1)、多発性内分泌腫瘍2型(MEN2)、多発性内分泌腫瘍4型(MEN4)、褐色細胞腫(PHEO)、傍神経節腫(PGL)、リー-フラウメニ症候群、家族性大腸腺腫症、フォン・ヒッペル-リンダウ症候群など、いくつかのものがある。本要約では現在、MEN1、MEN2、MEN4、家族性PHEOおよびPGL症候群、カーニー-ストラタキス(CSS)症候群、および家族性非髄様甲状腺がん(FNMTC)について取り上げている。リー-フラウメニ症候群、家族性大腸腺腫症、コーデン症候群、およびフォン・ヒッペル-リンダウ症候群については、乳がんおよび婦人科がんの遺伝学、大腸がんの遺伝学、および腎がんの遺伝学に関するPDQ要約で考察する。
多発性内分泌腫瘍という用語は、内分泌組織の良性または悪性の遺伝性腫瘍のグループを示すために用いられる。一般的にMEN1(ウェルマー症候群としても知られている)およびMEN2という2つの主なカテゴリーに分類される。歴史的に、個人または家系に特定の内分泌腫瘍がみられるかどうかによって、MEN2はさらに次の3つの亜型に分類された:MEN2A、家族性甲状腺髄様がん(FMTC)、およびMEN2B(ときにMEN3と呼ばれる)。現在、FMTCはMEN2Aの亜型とみなされる。[ 1 ]MEN4は、ヒトにおける新たな症候群として2011年に記述され、主要特性として原発性副甲状腺機能亢進症および下垂体腺腫が含まれる。MEN症候群関連腫瘍は通常、ホルモンの過剰産生または腫瘍の増殖、あるいはその両方を発現する。(詳しい情報については、本要約のMEN1、MEN2、およびMEN4のセクションを参照のこと。)
PGLおよびPHEOは、クロム親和性細胞から発生するまれな腫瘍で、カテコールアミンと神経ペプチドを合成・蓄積・分泌する能力がある。[ 2 ]いずれの腫瘍も、遺伝性症候群の顕性化として、つまり家族性のPGLおよびPHEO症候群における単独腫瘍として散発的に発生することがある。(詳しい情報については、本要約の家族性PHEOおよびPGL症候群のセクションを参照のこと。)
CSSを有する罹患者は、多病巣性の局所で侵攻性を示す消化管間質腫瘍および多発性の頸部、胸腔内、および腹腔内PGLを比較的若年で発症する。[ 3 ][ 4 ][ 5 ]類似した名前であるが、この症候群はカーニー複合およびカーニーの三徴と異なる。(詳しい情報については、本要約のCSSのセクションを参照のこと。)
FNMTCは、分化型甲状腺がん全症例の5~10%を占めると考えられている。[ 6 ][ 7 ][ 8 ]少数の例外として非髄様甲状腺がんを伴う数種類のまれな遺伝的症候群も存在するが、FNMTCのほとんどは無症候性であり、基礎にある遺伝的素因ははっきりしない。(詳しい情報については、本要約のFNMTCのセクションを参照のこと。)
参考文献- Wells SA, Asa SL, Dralle H, et al.: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25 (6): 567-610, 2015.[PUBMED Abstract]
- Inherited tumour syndromes. In: Lloyd RV, Osamura RY, Klöppel G, et al.: WHO Classification of Tumours of Endocrine Organs. 4th ed. Lyon, France: International Agency for Research on Cancer, 2017, pp. 262–66.[PUBMED Abstract]
- Carney JA, Stratakis CA: Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 108 (2): 132-9, 2002.[PUBMED Abstract]
- McWhinney SR, Pasini B, Stratakis CA, et al.: Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med 357 (10): 1054-6, 2007.[PUBMED Abstract]
- Pasini B, McWhinney SR, Bei T, et al.: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 16 (1): 79-88, 2008.[PUBMED Abstract]
- Stoffer SS, Van Dyke DL, Bach JV, et al.: Familial papillary carcinoma of the thyroid. Am J Med Genet 25 (4): 775-82, 1986.[PUBMED Abstract]
- Mazeh H, Sippel RS: Familial nonmedullary thyroid carcinoma. Thyroid 23 (9): 1049-56, 2013.[PUBMED Abstract]
- Lupoli G, Vitale G, Caraglia M, et al.: Familial papillary thyroid microcarcinoma: a new clinical entity. Lancet 353 (9153): 637-9, 1999.[PUBMED Abstract]
- 多発性内分泌腫瘍1型
-
臨床記述
多発性内分泌腫瘍1型(MEN1)は常染色体優性症候群で、推定有病率は30,000人当たり約1人である。[ 1 ]MEN1の内分泌に関する主な特徴には以下のものがある:
MEN1の臨床診断は、ある個人にこれら3つの主要な内分泌腫瘍のうち、2つが認められる場合に下される。家族性MEN1は、少なくともMEN1症例が1例いること + 少なくとも1人の第一度近親者(FDR)にこれら3つの腫瘍の1つが認められること、または2人のFDRに生殖細胞病原性多様体が認められることと定義されている。[ 2 ][ 3 ][ 4 ]
最初の臨床症状発現は典型的に20~30歳であるが、MEN1診断はさらに何年も確定されないことがある。MEN1の年齢に関係した浸透度は30歳までは45~73%、50歳までは82%、および70歳までは96%である。[ 2 ][ 5 ][ 6 ]
副甲状腺腫瘍およびPHPT
MEN1に最も一般的な特徴およびしばしば最初に現れる徴候は副甲状腺腫瘍であり、この腫瘍の結果としてPHPTになる。50歳までに80~100%の患者がこれらの腫瘍を発症する。[ 7 ][ 8 ]散発例でみられる孤立性の腺腫とは異なり、MEN1関連副甲状腺腫瘍は典型的に多腺性で、しばしば過形成を示す。[ 9 ]MEN1におけるPHPTの発症時平均年齢は20~25歳であり、一般集団(典型的に50~59歳で発症)とは対照的である。MEN1における副甲状腺がんはまれではあるが、報告されている。[ 10 ][ 11 ][ 12 ]
MEN1関連PHPTの患者では、血中の副甲状腺ホルモン(PTH)およびカルシウム値が高くなる。PHPTの臨床症状は主として高カルシウム血症の結果である。軽度の高カルシウム血症は検出されず、症状がほとんどまたは全く見られないことがある。比較的重度の高カルシウム血症は、以下のような症状を引き起こす:
MEN1関連高カルシウム血症は副甲状腺腫瘍の存在と直接関係しているため、これらの腫瘍を外科的に切除することでカルシウム値およびPTH濃度が正常化し、症状が緩和される;しかしながら、一部のシリーズでは術後の再発率が高いことが報告されている。[ 13 ][ 14 ][ 15 ](詳しい情報については、本要約の介入のセクションを参照のこと。)
膵十二指腸NET
膵十二指腸のNETはMEN1において2番目に一般的な内分泌の症状であり、40歳までに30~80%の患者が発症する。[ 2 ][ 7 ]1件の研究により、MEN1のエクソン2に病原性多様体を有する若年患者(20~40歳)では発生率が2倍も高くなることがあると示されている。こうした個人はまた、より侵攻性の疾患を有し、遠隔転移を来す可能性が高い。[ 16 ]さらに、膵十二指腸NETは外科的切除後でさえも早期死亡に関連している。[ 17 ]
MEN1でみられる膵十二指腸NETには以下のものがある:
表1.MEN1関連膵十二指腸神経内分泌腫瘍 腫瘍の種類 推定浸透度 症状 MEN1 = 多発性内分泌腫瘍1型。 ガストリノーマ ≤70% [ 7 ][ 18 ] 消化性潰瘍疾患および食道炎 下痢 腹痛 体重減少 非機能性 20%–55% [ 7 ][ 19 ] 局所圧迫症状:腹痛、黄疸、食欲不振、体重減少 インスリノーマ 10% [ 7 ] Whippleの三徴:グルコース投与により回復する症候性低血糖および関連するインスリン、Cペプチド、およびプロインスリン値の上昇 血管作動性腸管ペプチド 1% [ 7 ][ 20 ] 水様下痢 低カリウム血症 無酸症 グルカゴノーマ 1% [ 7 ][ 20 ] 糖尿病 下痢 うつ病 壊死性遊走性紅斑 血栓塞栓性疾患 ソマトスタチノーマ <1% [ 20 ] 糖尿病 下痢/脂肪便 胆嚢疾患 低酸症 体重減少 ガストリノーマはMEN1に伴う消化管NETの50%を占めており、MEN1患者における罹病および死亡の主要な原因である。[ 2 ][ 13 ]ガストリノーマは通常、多中心性で、十二指腸全体に小さな(0.5cm未満)病巣が見られる。[ 21 ]ほとんどが消化性潰瘍疾患(ゾリンジャー・エリソン症候群)を引き起こし、半数が診断時に悪性である。[ 13 ][ 21 ][ 22 ]
非機能性膵十二指腸NETは当初、MEN1の個人では比較的まれな腫瘍であると考えられていた。しかしながら、遺伝子検査の出現と画像診断技術の改善によってMEN1における非機能性膵十二指腸NETの有病率の認識が高まっており、1件の研究では、膵臓の超音波内視鏡検査をプロスペクティブに受けたMEN1病原性多様体キャリアにおいて、39歳までに55%という高い頻度が示されている。[ 19 ][ 23 ]これらの腫瘍は転移性の場合がある。非機能性膵十二指腸NETを有するMEN1病原性多様体キャリア108人を対象にした1件の研究により、腫瘍の大きさと転移率および死亡率との間に正の相関があることが示され、2cm超の腫瘍では2cm未満の腫瘍よりも転移率が有意に高かった。[ 24 ](MEN1遺伝子病原性多様体に関する詳しい情報については、本要約のMEN1の分子遺伝学のセクションを参照のこと。)
下垂体腫瘍
MEN1患者の約15~50%が下垂体腫瘍を発症する。[ 2 ][ 7 ]3分の2は微小腺腫(直径1.0cm未満)で、大多数がプロラクチン産生性である。[ 25 ]この他の下垂体腫瘍には、ソマトトロピノーマ(somatotropinoma)およびコルチコトロピノーマ(corticotropinoma)があり、これらは非機能性の場合がある。
この他のMEN1関連腫瘍
MEN1の他の症状発現としては、前腸カルチノイド(MEN1患者の5~10%)がある。これらは典型的に気管支または胸腺に見られ、ときには胃に発生することもある。皮膚病変も一般的に見られ、顔面の血管線維腫(MEN1患者の最大80%)およびコラゲノーマ(collagenoma)(MEN1患者の75%まで)が含まれる。[ 26 ]脂肪腫(MEN1患者の約30%)、および皮質腺腫、びまん性もしくは結節性過形成、またはまれにがんなどの副腎皮質病変(MEN1患者の最大50%)[ 27 ]もよくみられる。[ 28 ][ 29 ][ 30 ]以下の症状発現もまた報告されている:[ 31 ][ 32 ][ 33 ]
MEN1の診断確定
明らかな家族歴が認められないか、MEN1遺伝子の病原性多様体に対する遺伝子検査で陽性の結果が得られない場合、MEN1の診断はしばしば困難である。MEN1の患者560人を対象にした1件の研究により、最初の症状発現時とMEN1診断時間の間に明らかな遅延があることが示された。[ 34 ]この時間の経過は、無月経、消化性潰瘍、低血糖、腎結石など、MEN1関連腫瘍に伴う一部の主症状が、MEN1に特異的ではないためである可能性が高い。
さらに、MEN1関連腫瘍が確認されただけでは、MEN1を臨床的に診断するために十分ではなく、内分泌医に紹介するきっかけとならない場合がある。最初の症状発現からMEN1診断までの期間の中央値は、7.6年から12年に及ぶ。[ 5 ][ 29 ]遺伝子検査により、この遅延がある程度軽減される。数件の研究により、発端者とその家系員との間で、MEN1診断時年齢に統計的有意差が認められることが示されている。1件の研究では、臨床的に症状のある発端者は、平均47.5歳(標準偏差[SD]、±13.5歳)でMEN1と診断されたのに対し、家系員は平均38.5歳(SD、±15.4歳;P < 0.001)で診断された。[ 34 ]MEN1の個人154人を対象にした別の研究では、発端者は平均39.5歳(範囲:18~74歳)で診断されたのに対し、家系員では予測的遺伝子検査によって平均27歳(範囲:14~56歳;P < 0.05)で診断された。[ 35 ]それでも、発端者におけるMEN1診断と家系員におけるMEN1診断間の時間のずれは重要な可能性があり、罹病率および死亡率の増加につながる。[ 36 ]これは、Dutch MEN1 Study Groupの解析で明らかにされたもので、非発端者の10~38%では診断時にMEN1関連症状が既に認められたことが示された;これらの患者の4%は、この遅れ時間にまたはその前に発生したMEN1関連原因により死亡した。家系員では、遅れ時間に関連する罹病率の大半が転移性の膵十二指腸NET、巨大下垂体腫瘍、および複数のMEN1症状によるものであった。[ 36 ]早期の介入は、膵十二指腸NETによる死亡率に関係してくるため、特に重大である。1件の研究では、手術時の年齢が1歳高くなるごとに転移のオッズは6%増加することが示された。[ 17 ]これらの知見は、MEN1関連腫瘍の徴候および症状のほか、MEN1診断を疑う必要のある一連の所見に対する認識を高めることの重要性を強調している。また、遺伝カウンセリングと遺伝子検査、およびMEN1の診断がいったん下された後の家系員間でのコミュニケーションの重要性も強調される。[ 37 ][ 38 ]図1は、ある家系内でのMEN1の特定に関する困難の一部を示している。
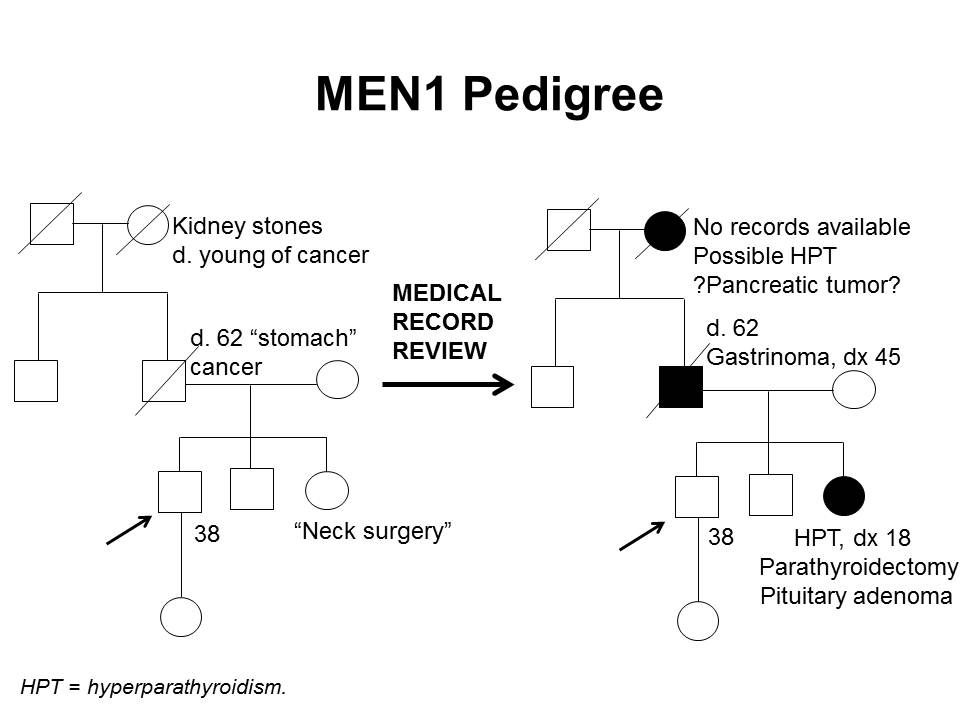
図1.MEN1家系(pedigree) 図。MEN1は単一の家系(pedigree) 内での特定が非常に困難な場合がある。左側の家系(pedigree)図は自己報告に基づき作成された図で、右側の家系(pedigree) 図は同じ家系について利用可能な医療記録を確認して作成された図である。この家系(pedigree) 図は、MEN1病原性多様体を有する家系の特徴の一部を4世代にわたって示し、副甲状腺機能亢進症、下垂体腺腫、ガストリノーマに罹患している家系員と、膵腫瘍の疑いがある家系員を記載している。MEN1に伴う腫瘍は、通常は散発性の腫瘍よりも早い年齢で発生する。MEN1の家系では、これらの特徴の一部またはすべてが認められることがある。常染色体優性症候群と同様に、母系または図のように父系で伝達する場合がある。 MEN1では多くの腫瘍が過小診断または誤診されているため、疾患過程の早期に発端者におけるMEN1遺伝子の病原性多様体を同定することで、腫瘍の早期発見・早期治療が可能となり、リスクのある家系員を早期に特定できる。見たところ散発性のMEN1関連腫瘍を有する患者において、MEN1遺伝子の病原性多様体の保有率を明らかにするために、多くの研究が実施されている。[ 7 ]例えば、ゾリンジャー・エリソン症候群患者の約3分の1は、MEN1病原性多様体を保有している。[ 39 ][ 40 ]見たところ孤立性のPHPTまたは下垂体腺腫の個人における病原性多様体保有率は低く、2~5%程度であるが[ 25 ][ 41 ][ 42 ]、30歳前にこれらの腫瘍を診断された個人における保有率は比較的高い。一部の著者は、以下の条件の1つが認められる場合に、MEN1病原性多様体に対する遺伝相談および/または遺伝子検査への紹介を提唱している:[ 7 ][ 43 ][ 44 ]
MEN1の分子遺伝学
MEN1遺伝子は染色体11q13上に位置し、Menin蛋白をコードする。[ 3 ][ 45 ][ 46 ]MEN1遺伝子における病原性多様体は現在までに1,300以上同定されており、これらの多様体はコード領域全体に散在している。[ 47 ][ 48 ]このうちほとんど(~65%)はナンセンス多様体またはフレームシフト多様体である。残りは、正常と異なる蛋白を発現させるミスセンス多様体(20%)、スプライス部位多様体(9%)、または部分あるいは全遺伝子欠失(1~4%)である。家族間および家族内の変動が一般的である。[ 7 ][ 49 ][ 50 ]1件の大規模研究から下垂体、副腎、および胸腺NETの遺伝率が最も高いことが実証されている。[ 51 ]
遺伝子検査および鑑別診断
MEN1病原性多様体に対する遺伝子検査は、臨床診断基準を満たす個人に推奨され、あまり一般的でない腫瘍のサブセットで検討されうる。(詳しい情報については、本要約のMEN1の診断確定のセクションの・印が付いている一覧を参照のこと。)診断基準を満たす個人では病原性多様体検出率は約75~90%である。[ 49 ][ 52 ]さらに、副甲状腺腫瘍(15.8%)、膵島腫瘍(25.0%)、または下垂体腫瘍(37.5%)の見たところ散発性の症例に対する生殖細胞病原性多様体の割合は16~38%に及んでおり、これらの個人では、MEN1の診断により他のMEN1関連腫瘍に対するスクリーニングが促されるため、遺伝子検査を検討する必要がある。[ 53 ]現在MEN1検査を提供している検査施設は、第一の手法としてDNA塩基配列決定法を使用している。いくつかの施設が、部分または全遺伝子欠失および/または重複に関する追加の解析を提供しているが、このような多様体はまれであり、欠失/重複検査はしばしば、臨床的に非常に強く疑われるが、直接配列決定法では検出可能な病原性多様体が認められない個人または家系においてのみ用いられる。
MEN1および内分泌腫瘍のリスク増加に関連する他の遺伝子を含む多重遺伝子パネルが使用されることもある。このような遺伝子検査は、MEN1と、家族性孤立性副甲状腺機能亢進症(FIHP)、副甲状腺機能亢進症-顎腫瘍症候群(HPT-JT)、家族性低カルシウム尿性高カルシウム血症(FHH)など、他の形態の遺伝性副甲状腺機能亢進症との鑑別に用いることができる。[注: FHHにおける副甲状腺機能亢進症は、MEN1、HPT-JT、およびFIHPで見られるような原発性副甲状腺機能亢進症ではない。]HRPT2遺伝子における生殖細胞病原性多様体によって発生するHPT-JTは、PHPT、上顎骨および下顎骨の骨化病変、ならびに腎病変(通常は両側性腎嚢胞、腎過誤腫、および一部の症例でウィルムス腫瘍)と関連している。[ 54 ][ 55 ]MEN1とは異なり、HPT-JTは副甲状腺がんリスクの増加と関連している。[ 56 ]FIHPは、その名が示す通り、他の内分泌の特徴を示さない孤立性のPHPTを特徴とする;一部の家系では、FIHPは後にMEN1、HPT-JT、またはFHHに発展する疾患の最初の診断となる。[ 57 ][ 58 ][ 59 ]FIHPを臨床的に診断された家系の約20%は、MEN1生殖細胞病原性多様体を保有する。[ 58 ][ 60 ][ 61 ]カルシウム感知受容体(CaSR)遺伝子における病原性多様体は、MEN1における副甲状腺機能亢進症に酷似する場合があるFHHを引き起こす。FHHでは副甲状腺を切除しても患者の副甲状腺機能亢進症は補正されず、症状の軽減を伴わない不必要な手術を行ってしまうため、MEN1とFHHの鑑別は管理において決定的に重要である。[ 62 ]これらの疾患ではリスクおよび管理が異なり、HPT-JTでは副甲状腺がんのリスクが高いため、早期発症型副甲状腺機能亢進症を呈する患者における遺伝子診断は、こうした患者とその家族の管理において重要な役割を果たしうる。[ 63 ]MEN1と他の形態の遺伝性副甲状腺機能亢進症の臨床所見の要約については表3を参照のこと。
表3.MEN1、FIHP、HPT-JT、およびFHHの主要な臨床的特徴 状態 遺伝子 主要な臨床的特徴 CaSR = カルシウム感知受容体遺伝子;FHH = 家族性低カルシウム尿性高カルシウム血症;FIHP = 家族性孤立性副甲状腺機能亢進症;HPT-JT = 副甲状腺機能亢進症-顎腫瘍症候群;HRPT2 = 副甲状腺機能亢進症2遺伝子;MEN1 = 多発性内分泌腫瘍1型(遺伝子は斜体で示される);NET = 神経内分泌腫瘍;PHPT = 原発性副甲状腺機能亢進症。 MEN1 MEN1 PHPT、下垂体腺腫、膵十二指腸NET[ 7 ][ 9 ][ 64 ] FIHP MEN1、HRPT2 PHPT[ 57 ][ 58 ][ 59 ][ 60 ][ 61 ] HPT-JT HRPT2 PHPT;上顎骨および下顎骨の骨腫;腎嚢胞および過誤腫:まれにウィルムス腫瘍および副甲状腺がん[ 54 ][ 55 ][ 56 ] FHH CaSR 副甲状腺機能亢進症(原発性でなはい)[ 62 ][ 65 ] サーベイランス
MEN1に対するスクリーニングおよびサーベイランスでは、生化学検査と画像検査が併用される。利用可能な推奨事項は表4にまとめられている。[ 4 ][ 7 ]
表4.MEN1のサーベイランスのための実践ガイドラインa 生化学検査または検査法 スクリーニング対象の病態 スクリーニング開始年齢(歳) 頻度 CT = コンピュータ断層撮影法;MEN1 = 多発性内分泌腫瘍1型;MRI = 磁気共鳴画像法;NET = 神経内分泌腫瘍;PHPT = 原発性副甲状腺機能亢進症;PTH = 副甲状腺ホルモン。 a出典:Kloos et al.[ 4 ]、Thakker et al. [ 7 ] b腹部の画像検査に関する推奨は、MEN1の診断および管理のために発表された2つのガイドラインで異なっている。[ 4 ][ 7 ]現時点では、10歳前の年1回の画像検査を支持する証拠は弱い。10歳前に画像検査を行うことで高い割合の患者で疾患を同定できるが、これが予後に影響するかどうかは不明である。[ 19 ][ 66 ] c下垂体腫瘍に対するスクリーニング開始年齢およびスクリーニングの頻度は、小さな非機能性腫瘍の臨床的意義が不明であるため議論の余地がある[ 67 ];さらなる研究が必要であろう。 血清プロラクチンおよび/またはインスリン様成長因子1 下垂体腫瘍 5 年1回 空腹時総血清カルシウムおよび/またはイオン化カルシウムおよびPTH 副甲状腺腫瘍およびPHPT 8 年1回 空腹時血清ガストリン 膵十二指腸ガストリノーマ 20 年1回 クロモグラニンA、膵ポリペプチド、グルカゴン、および血管作用性腸ポリペプチド 膵十二指腸NET <10 年1回 空腹時グルコースおよびインスリン インスリノーマ 5 年1回 脳MRIc 下垂体腫瘍 5 生化学的検査結果に基づき3~5年ごとに1回 腹部CTまたはMRIb[ 4 ] 膵十二指腸NET 20 生化学的検査結果に基づき3~5年ごとに1回 腹部CT、MRI、または超音波内視鏡検査b[ 7 ] 膵十二指腸NET <10 年1回 介入
MEN1は多病巣性で多腺性の性質を有し、手術後も腫瘍再発リスクが高いことを考慮すると、その外科的管理は複雑で議論の余地がある。手術の決定を下す前にMEN1の診断を確定し、MEN1の治療経験を有する外科医に罹患者を紹介することが、不必要な手術または不適切な外科的アプローチを回避する上できわめて重要である。
副甲状腺腫瘍の治療
副甲状腺疾患の証拠が生化学的に確立された時点で推奨される治療コースは、機能が亢進した副甲状腺組織の外科的切除である。しかしながら、手術の時期および範囲については議論の余地が残されている。[ 38 ]MEN1のリスクがある原発性副甲状腺機能亢進症の患者にとって、病原性多様体の術前検出は手術範囲の指針となり、初回手術成功の可能性を高め、疾患再発の可能性を低下させうる。[ 63 ]一部のグループでは、無症状の患者には年1回の生化学的スクリーニングを継続し、外科的介入は明らかに症状がある患者にのみ使用している。手術の継続が決まった場合には、初期治療として、一般的に副甲状腺亜全摘除術(3~3.5腺の切除)が提唱されている。[ 63 ]3.5腺以上切除された場合の持続性疾患の割合は5~6%である。どの腺が機能亢進しているかを決定するための術前画像法の信頼性は片側の検索を正当化するほど十分には高くなく、患者の86%では対側の副甲状腺に肥大した腺が見逃されていた。切除の指針を得るために画像検査を受けた患者の50%で、取り込みが最も多かった対側に、最も肥大した腺が術中に同定された。[ 68 ]切除が不十分であると患者には再手術が必要となる。[ 13 ][ 14 ][ 15 ][ 63 ]前腕などの遠隔部位への副甲状腺組織の自家移植を併用する副甲状腺全摘術もまた、選択肢の1つである。このアプローチの利点は、頸部からよりも前腕からの方が再発病変の切除または減量手術が容易なことである。さらに、左右上肢からの採血の検査結果を比べることで、再発を同定できる利点もある。甲状腺摘出術を実施する場合、再発の可能性は低下するが、これは患者の副甲状腺機能低下または機能欠損(体内でPTHが検出されない)のリスクと比較検討する必要がある。[ 69 ][ 70 ]低カルシウム血症の破壊的な合併が起きると、管理には経口のカルシトリオールおよびカルシウム補給が必要となる。この毎日の薬物依存は患者に対して大きな負担となりうる。同時両側経頸部胸腺摘出により再発率が低下することを示している諸研究により、初回手術時に胸腺を摘出すべきであることが示唆されている。[ 69 ]
膵十二指腸NETの治療
膵十二指腸NETの手術の時期と範囲については見解が一致しておらず、症状の重症度、病変の範囲、機能的要素、位置および単純摘出術、膵亜全摘または膵全摘術、および膵頭十二指腸切除術(Whipple法)の必要性など、多くの因子によって異なる。外科的な摘出術は、再発率が膵体尾部切除術と比較して高く、内分泌機能不全の発生率がWhipple法と比較して低いという関連が認められている。[ 71 ]腫瘍径の増大に伴う転移または再発リスクの増加傾向に基づく外科的切除を提唱するために、腫瘍の大きさが提案されている。[ 72 ][ 73 ]残念ながら、疾患特異的死亡率を予測する特異的腫瘍マーカーまたは腫瘍マーカーの組み合わせは存在しない。[ 74 ]初期のMEN1膵十二指腸NETにおいて長時間作用型ソマトスタチンアナログが役に立つ可能性がある。[ 75 ]薬理学的治療の初期の研究結果から、治療が安全であること、患者の最大10%において腫瘍およびホルモン活性が長期にわたって抑制できること、および患者の80%でホルモンの機能亢進状態が安定することが示唆されている。[ 75 ]手術の主要目的は、ホルモン過剰に伴う症状を減らし、遠隔転移のリスクを低くすることで、長期生存を向上させることである。[ 22 ]遠隔転移の可能性が高いため、ほとんどの機能性腫瘍および非機能性NET(腫瘍が2~3cmを超える場合)に対して一般的に手術が実施される。[ 73 ][ 76 ][ 77 ][ 78 ]膵十二指腸NETの悪性度を予測する上で、構造画像検査法のみでは最適とはいえない。しかしながら、1件の研究で、フッ素18-フルデオキシグルコース・ポジトロン放射断層撮影-コンピュータ断層撮影(18F-FDG PET-CT)を用いたMEN1患者のスクリーニングにより、悪性度の高いこうしたNETが同定されたことが明らかにされた;FDGの結合活性はKi-67指数と相関した。[ 79 ]腫瘍の大きさは確かに患者の生存に影響を与えるとみられ、腫瘍が小さい患者ほど切除後の生存率が増加する。[ 80 ]広範囲の手術アプローチ(例、膵頭十二指腸切除術)を行えば治癒率が高くなり、全生存が改善される一方で[ 81 ][ 82 ][ 83 ]、こうしたアプローチではまた術後の合併症発生率および長期罹病率も高くなる。[ 84 ]したがって、リスクと有益性を注意深く検討すべきであり、手術の決定は症例ごとに下すべきである。開腹アプローチか腹腔鏡によるアプローチかについては、一部の患者では腹腔鏡下膵臓手術が安全で、入院期間の短縮および合併症の少なさと関連しているとみられる。[ 85 ]
NETを診断されたMEN1患者はしばしば、膵および十二指腸全体にさまざまな種類の多発性の腫瘍を有し、これらの一部は磁気共鳴画像法またはコンピュータ断層撮影(CT)を用いて同定可能である。機能的トレーサーの集積と解剖学的画像検査の併用により、腫瘍の部位特定が向上する。ガリウム68-DOTATATEポジトロン放射断層撮影-CTは、膵十二指腸NET病変のマッピングにおいてきわめて優れた感度を実証している。この検査法は最初の精密検査の指針となる可能性があり、特に10mm未満の病変に対して標準のソマトスタチン オクトレオチドよりも優れているようである。[ 86 ][ 87 ]腫瘍の多くが標準の画像技術を用いて発見するにはあまりにも小さく、こうした腫瘍には動脈内セクレチン刺激試験および/または術中超音波検査が有用な場合もある。[ 88 ][ 89 ]さまざまな生化学および画像検査法の組み合わせを用いた術前の評価、腫瘍負荷の術中評価、およびホルモン分泌過多の解決がきわめて重要であり、一部のシリーズによれば、これらが高い治癒率および長期の無病期間と関連している。[ 88 ][ 89 ][ 90 ][ 91 ]
機能性の高いホルモン過剰状態に対して有効な治療がある現代では、MEN1関連死亡のほとんどが膵十二指腸NETの悪性の性質によるものである。頻度は低いが重要な死亡のリスクは、悪性の胸腺カルチノイド腫瘍からもたらされる。MEN1の予後不良指標としては、空腹時血清ガストリン上昇、機能性ホルモン症候群の存在、肝転移または遠隔転移、侵攻性膵十二指腸NET増殖、サイズの大きな膵十二指腸NET、または複数回の副甲状腺切除術の必要性が挙げられる。このコホートにおいてMEN1と無関係な死亡で最も多かったのは、心血管疾患によるものであった。[ 92 ]
他の膵十二指腸NET
グルカゴノーマ、VIP産生腫瘍、およびソマトスタチノーマはまれであるが、しばしば他の膵十二指腸NETよりも悪性腫瘍の割合が高い。[ 20 ]これらの腫瘍はしばしば積極的な手術で治療される。[ 93 ]
インスリノーマ
食事療法および薬物療法を用いたインスリノーマの医学的管理はしばしば成功しない;この腫瘍に対する治療の中心は外科的切除である。[ 7 ]MEN1患者におけるインスリノーマは膵臓のいかなる部位にも位置する可能性があるが、腺遠位部に優勢で[ 94 ][ 95 ][ 96 ]、散発性インスリノーマよりも転移率が高い。[ 93 ]手術の範囲は単一または多発性の大きな腫瘍の核出術から膵部分切除、またはその両方[ 95 ]、膵亜全摘または膵全摘術に及ぶ。[ 94 ][ 95 ]手術アプローチが広範囲になるほど再発率は低くなるが[ 81 ][ 82 ][ 95 ][ 97 ]、術後の罹病率は高くなる。インスリノーマはしばしば非機能性膵腫瘍に伴って発生するため、インスリン過剰の源を明らかにするために選択的動脈内カルシウム注入試験(SAS試験)が必要となることがある。[ 98 ]インスリン/グルコースを手術中にモニタリングすることで、インスリン分泌腫瘍の切除が成功しているかどうかの判定に役立つことがある。[ 89 ][ 99 ]
ガストリノーマ
MEN1関連ガストリノーマのほとんどは十二指腸に発生する。これらの腫瘍は典型的に多病巣性でガストリン分泌過多を引き起こし、その結果、消化性潰瘍疾患を発症する(ゾリンジャー・エリソン症候群)。[ 100 ]多病巣性のため、外科的完全切除は困難である。何らかの外科的介入を検討する前に症状を管理することが不可欠である。[ 101 ]歴史的には、いくつかのグループにより、腫瘍が比較的小さく(画像検査で2.0cm未満)、薬物療法(例、プロトンポンプ阻害薬またはヒスタミン-2拮抗薬)を用いることで症状が緩和している患者では注意深い観察が推奨されている[ 102 ];しかしながら、このアプローチがすべての患者に最適とは限らない。
発表されている数件のシリーズで、原発腫瘍のサイズと遠隔転移率には正の相関があることが示されている。1件のレトロスペクティブ研究により、腫瘍径が3cmを超える患者の61%が肝転移を有したことが示された。[ 22 ]別のシリーズでは、腫瘍径が3cmを超える患者の40%が肝転移を有していた。[ 103 ]対照的に、これらの両シリーズで腫瘍径が3cm未満の患者では肝転移率が有意に低かったことが示された(それぞれ、32%および4.8%)。これらの研究や他のデータに基づいて、多くのグループでは腫瘍径が2cm超の非転移性ガストリノーマの患者で手術が推奨されている。[ 7 ][ 83 ]
ガストリノーマに対する手術の種類は多くの因子によって異なる。Whipple法は、術後の高い合併症発生率および糖尿病や吸収不良などの長期の合併症を考慮すると、初回手術としては一般的に推奨されない。比較的範囲の狭い手術ではさまざまな結果が記述されている。少なくとも、十二指腸腫瘍の位置を突き止めて切除するため術中の触診および/または超音波検査を併用する十二指腸切除術と膵臓周辺リンパ節郭清が実施される。[ 88 ][ 104 ]ガストリノーマ患者のほとんどで膵臓全体にNET(これらの一部は非機能性である)が同時に認められるため、十二指腸腫瘍の切除に加えて膵尾部切除および膵頭部腫瘍の核出術を推奨するグループもある。[ 88 ][ 104 ][ 105 ]
参考文献- Agarwal SK, Ozawa A, Mateo CM, et al.: The MEN1 gene and pituitary tumours. Horm Res 71 (Suppl 2): 131-8, 2009.[PUBMED Abstract]
- Trump D, Farren B, Wooding C, et al.: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM 89 (9): 653-69, 1996.[PUBMED Abstract]
- Chandrasekharappa SC, Guru SC, Manickam P, et al.: Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276 (5311): 404-7, 1997.[PUBMED Abstract]
- Brandi ML, Gagel RF, Angeli A, et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86 (12): 5658-71, 2001.[PUBMED Abstract]
- Carty SE, Helm AK, Amico JA, et al.: The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 124 (6): 1106-13; discussion 1113-4, 1998.[PUBMED Abstract]
- Goudet P, Dalac A, Le Bras M, et al.: MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d'étude des Tumeurs Endocrines. J Clin Endocrinol Metab 100 (4): 1568-77, 2015.[PUBMED Abstract]
- Thakker RV, Newey PJ, Walls GV, et al.: Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97 (9): 2990-3011, 2012.[PUBMED Abstract]
- Goudet P, Murat A, Binquet C, et al.: Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg 34 (2): 249-55, 2010.[PUBMED Abstract]
- Chandrasekharappa SC, Teh BT: Clinical and molecular aspects of multiple endocrine neoplasia type 1. Front Horm Res 28: 50-80, 2001.[PUBMED Abstract]
- del Pozo C, García-Pascual L, Balsells M, et al.: Parathyroid carcinoma in multiple endocrine neoplasia type 1. Case report and review of the literature. Hormones (Athens) 10 (4): 326-31, 2011 Oct-Dec.[PUBMED Abstract]
- Christakis I, Busaidy NL, Cote GJ, et al.: Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature. Int J Surg 31: 10-6, 2016.[PUBMED Abstract]
- Singh Ospina N, Sebo TJ, Thompson GB, et al.: Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 - case report and review of the literature. Clin Endocrinol (Oxf) 84 (2): 244-249, 2016.[PUBMED Abstract]
- Norton JA, Venzon DJ, Berna MJ, et al.: Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg 247 (3): 501-10, 2008.[PUBMED Abstract]
- Hellman P, Skogseid B, Oberg K, et al.: Primary and reoperative parathyroid operations in hyperparathyroidism of multiple endocrine neoplasia type 1. Surgery 124 (6): 993-9, 1998.[PUBMED Abstract]
- Schreinemakers JM, Pieterman CR, Scholten A, et al.: The optimal surgical treatment for primary hyperparathyroidism in MEN1 patients: a systematic review. World J Surg 35 (9): 1993-2005, 2011.[PUBMED Abstract]
- Christakis I, Qiu W, Hyde SM, et al.: Genotype-phenotype pancreatic neuroendocrine tumor relationship in multiple endocrine neoplasia type 1 patients: A 23-year experience at a single institution. Surgery 163 (1): 212-217, 2018.[PUBMED Abstract]
- Donegan D, Singh Ospina N, Rodriguez-Gutierrez R, et al.: Long-term outcomes in patients with multiple endocrine neoplasia type 1 and pancreaticoduodenal neuroendocrine tumours. Clin Endocrinol (Oxf) 86 (2): 199-206, 2017.[PUBMED Abstract]
- Norton JA, Krampitz G, Jensen RT: Multiple Endocrine Neoplasia: Genetics and Clinical Management. Surg Oncol Clin N Am 24 (4): 795-832, 2015.[PUBMED Abstract]
- Thomas-Marques L, Murat A, Delemer B, et al.: Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol 101 (2): 266-73, 2006.[PUBMED Abstract]
- Lévy-Bohbot N, Merle C, Goudet P, et al.: Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol 28 (11): 1075-81, 2004.[PUBMED Abstract]
- Pipeleers-Marichal M, Somers G, Willems G, et al.: Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med 322 (11): 723-7, 1990.[PUBMED Abstract]
- Weber HC, Venzon DJ, Lin JT, et al.: Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology 108 (6): 1637-49, 1995.[PUBMED Abstract]
- Tonelli F, Giudici F, Fratini G, et al.: Pancreatic endocrine tumors in multiple endocrine neoplasia type 1 syndrome: review of literature. Endocr Pract 17 (Suppl 3): 33-40, 2011 Jul-Aug.[PUBMED Abstract]
- Triponez F, Dosseh D, Goudet P, et al.: Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg 243 (2): 265-72, 2006.[PUBMED Abstract]
- Corbetta S, Pizzocaro A, Peracchi M, et al.: Multiple endocrine neoplasia type 1 in patients with recognized pituitary tumours of different types. Clin Endocrinol (Oxf) 47 (5): 507-12, 1997.[PUBMED Abstract]
- Darling TN, Skarulis MC, Steinberg SM, et al.: Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 133 (7): 853-7, 1997.[PUBMED Abstract]
- Ventura M, Melo M, Carrilho F: Outcome and long-term follow-up of adrenal lesions in multiple endocrine neoplasia type 1. Arch Endocrinol Metab 63 (5): 516-523, 2019.[PUBMED Abstract]
- Machens A, Schaaf L, Karges W, et al.: Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 67 (4): 613-22, 2007.[PUBMED Abstract]
- Pieterman CR, Schreinemakers JM, Koppeschaar HP, et al.: Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf) 70 (4): 575-81, 2009.[PUBMED Abstract]
- Waldmann J, Bartsch DK, Kann PH, et al.: Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg 392 (4): 437-43, 2007.[PUBMED Abstract]
- Gibril F, Schumann M, Pace A, et al.: Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 83 (1): 43-83, 2004.[PUBMED Abstract]
- McKeeby JL, Li X, Zhuang Z, et al.: Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol 159 (3): 1121-7, 2001.[PUBMED Abstract]
- Vortmeyer AO, Lubensky IA, Skarulis M, et al.: Multiple endocrine neoplasia type 1: atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol 12 (9): 919-24, 1999.[PUBMED Abstract]
- Yamazaki M, Suzuki S, Kosugi S, et al.: Delay in the diagnosis of multiple endocrine neoplasia type 1: typical symptoms are frequently overlooked. Endocr J 59 (9): 797-807, 2012.[PUBMED Abstract]
- Lourenço DM, Toledo RA, Coutinho FL, et al.: The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1. Clinics (Sao Paulo) 62 (4): 465-76, 2007.[PUBMED Abstract]
- van Leeuwaarde RS, van Nesselrooij BP, Hermus AR, et al.: Impact of Delay in Diagnosis in Outcomes in MEN1: Results From the Dutch MEN1 Study Group. J Clin Endocrinol Metab 101 (3): 1159-65, 2016.[PUBMED Abstract]
- Strømsvik N, Nordin K, Berglund G, et al.: Living with multiple endocrine neoplasia type 1: decent care-insufficient medical and genetic information: a qualitative study of MEN 1 patients in a Swedish hospital. J Genet Couns 16 (1): 105-17, 2007.[PUBMED Abstract]
- Marini F, Giusti F, Tonelli F, et al.: Management impact: effects on quality of life and prognosis in MEN1. Endocr Relat Cancer 24 (10): T227-T242, 2017.[PUBMED Abstract]
- Roy PK, Venzon DJ, Shojamanesh H, et al.: Zollinger-Ellison syndrome. Clinical presentation in 261 patients. Medicine (Baltimore) 79 (6): 379-411, 2000.[PUBMED Abstract]
- Bardram L, Stage JG: Frequency of endocrine disorders in patients with the Zollinger-Ellison syndrome. Scand J Gastroenterol 20 (2): 233-8, 1985.[PUBMED Abstract]
- Uchino S, Noguchi S, Sato M, et al.: Screening of the Men1 gene and discovery of germ-line and somatic mutations in apparently sporadic parathyroid tumors. Cancer Res 60 (19): 5553-7, 2000.[PUBMED Abstract]
- Scheithauer BW, Laws ER, Kovacs K, et al.: Pituitary adenomas of the multiple endocrine neoplasia type I syndrome. Semin Diagn Pathol 4 (3): 205-11, 1987.[PUBMED Abstract]
- Newey PJ, Thakker RV: Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract 17 (Suppl 3): 8-17, 2011 Jul-Aug.[PUBMED Abstract]
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.[PUBMED Abstract]
- Larsson C, Skogseid B, Oberg K, et al.: Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 332 (6159): 85-7, 1988.[PUBMED Abstract]
- Bassett JH, Forbes SA, Pannett AA, et al.: Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 62 (2): 232-44, 1998.[PUBMED Abstract]
- Lemos MC, Thakker RV: Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29 (1): 22-32, 2008.[PUBMED Abstract]
- Concolino P, Costella A, Capoluongo E: Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet 209 (1-2): 36-41, 2016 Jan-Feb.[PUBMED Abstract]
- Giraud S, Zhang CX, Serova-Sinilnikova O, et al.: Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet 63 (2): 455-67, 1998.[PUBMED Abstract]
- Wautot V, Vercherat C, Lespinasse J, et al.: Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat 20 (1): 35-47, 2002.[PUBMED Abstract]
- Thevenon J, Bourredjem A, Faivre L, et al.: Unraveling the intrafamilial correlations and heritability of tumor types in MEN1: a Groupe d'étude des Tumeurs Endocrines study. Eur J Endocrinol 173 (6): 819-26, 2015.[PUBMED Abstract]
- Agarwal SK, Kester MB, Debelenko LV, et al.: Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 6 (7): 1169-75, 1997.[PUBMED Abstract]
- Klein RD, Salih S, Bessoni J, et al.: Clinical testing for multiple endocrine neoplasia type 1 in a DNA diagnostic laboratory. Genet Med 7 (2): 131-8, 2005.[PUBMED Abstract]
- Teh BT, Farnebo F, Kristoffersson U, et al.: Autosomal dominant primary hyperparathyroidism and jaw tumor syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21-q32 and loss of the wild type allele in renal hamartomas. J Clin Endocrinol Metab 81 (12): 4204-11, 1996.[PUBMED Abstract]
- Carpten JD, Robbins CM, Villablanca A, et al.: HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet 32 (4): 676-80, 2002.[PUBMED Abstract]
- Marx SJ: Multiple endocrine neoplasia type 1. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. New York, NY: McGraw-Hill, 1998, pp 489-506.[PUBMED Abstract]
- Warner J, Epstein M, Sweet A, et al.: Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet 41 (3): 155-60, 2004.[PUBMED Abstract]
- Mizusawa N, Uchino S, Iwata T, et al.: Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clin Endocrinol (Oxf) 65 (1): 9-16, 2006.[PUBMED Abstract]
- Cetani F, Pardi E, Borsari S, et al.: Molecular pathogenesis of primary hyperparathyroidism. J Endocrinol Invest 34 (7 Suppl): 35-9, 2011.[PUBMED Abstract]
- Miedlich S, Lohmann T, Schneyer U, et al.: Familial isolated primary hyperparathyroidism--a multiple endocrine neoplasia type 1 variant? Eur J Endocrinol 145 (2): 155-60, 2001.[PUBMED Abstract]
- Cetani F, Pardi E, Ambrogini E, et al.: Genetic analyses in familial isolated hyperparathyroidism: implication for clinical assessment and surgical management. Clin Endocrinol (Oxf) 64 (2): 146-52, 2006.[PUBMED Abstract]
- Raue F, Frank-Raue K: Primary hyperparathyroidism--what the nephrologist should know--an update. Nephrol Dial Transplant 22 (3): 696-9, 2007.[PUBMED Abstract]
- Romero Arenas MA, Morris LF, Rich TA, et al.: Preoperative multiple endocrine neoplasia type 1 diagnosis improves the surgical outcomes of pediatric patients with primary hyperparathyroidism. J Pediatr Surg 49 (4): 546-50, 2014.[PUBMED Abstract]
- Thakker RV: Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 386 (1-2): 2-15, 2014.[PUBMED Abstract]
- Christensen SE, Nissen PH, Vestergaard P, et al.: Familial hypocalciuric hypercalcaemia: a review. Curr Opin Endocrinol Diabetes Obes 18 (6): 359-70, 2011.[PUBMED Abstract]
- Langer P, Kann PH, Fendrich V, et al.: Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg 28 (12): 1317-22, 2004.[PUBMED Abstract]
- de Laat JM, Dekkers OM, Pieterman CR, et al.: Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab 100 (9): 3288-96, 2015.[PUBMED Abstract]
- Nilubol N, Weinstein LS, Simonds WF, et al.: Limited Parathyroidectomy in Multiple Endocrine Neoplasia Type 1-Associated Primary Hyperparathyroidism: A Setup for Failure. Ann Surg Oncol 23 (2): 416-23, 2016.[PUBMED Abstract]
- Pieterman CR, van Hulsteijn LT, den Heijer M, et al.: Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg 255 (6): 1171-8, 2012.[PUBMED Abstract]
- Lairmore TC, Govednik CM, Quinn CE, et al.: A randomized, prospective trial of operative treatments for hyperparathyroidism in patients with multiple endocrine neoplasia type 1. Surgery 156 (6): 1326-34; discussion 1334-5, 2014.[PUBMED Abstract]
- Ratnayake CBB, Loveday BP, Windsor JA, et al.: Patient characteristics and clinical outcomes following initial surgical intervention for MEN1 associated pancreatic neuroendocrine tumours: A systematic review and exploratory meta-analysis of the literature. Pancreatology 19 (3): 462-471, 2019.[PUBMED Abstract]
- Kishi Y, Shimada K, Nara S, et al.: Basing treatment strategy for non-functional pancreatic neuroendocrine tumors on tumor size. Ann Surg Oncol 21 (9): 2882-8, 2014.[PUBMED Abstract]
- Nell S, Verkooijen HM, Pieterman CRC, et al.: Management of MEN1 Related Nonfunctioning Pancreatic NETs: A Shifting Paradigm: Results From the DutchMEN1 Study Group. Ann Surg 267 (6): 1155-1160, 2018.[PUBMED Abstract]
- Qiu W, Christakis I, Silva A, et al.: Utility of chromogranin A, pancreatic polypeptide, glucagon and gastrin in the diagnosis and follow-up of pancreatic neuroendocrine tumours in multiple endocrine neoplasia type 1 patients. Clin Endocrinol (Oxf) 85 (3): 400-7, 2016.[PUBMED Abstract]
- Ramundo V, Del Prete M, Marotta V, et al.: Impact of long-acting octreotide in patients with early-stage MEN1-related duodeno-pancreatic neuroendocrine tumours. Clin Endocrinol (Oxf) 80 (6): 850-5, 2014.[PUBMED Abstract]
- Triponez F, Goudet P, Dosseh D, et al.: Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg 30 (5): 654-62; discussion 663-4, 2006.[PUBMED Abstract]
- Bettini R, Partelli S, Boninsegna L, et al.: Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery 150 (1): 75-82, 2011.[PUBMED Abstract]
- Triponez F, Sadowski SM, Pattou F, et al.: Long-term Follow-up of MEN1 Patients Who Do Not Have Initial Surgery for Small ≤2 cm Nonfunctioning Pancreatic Neuroendocrine Tumors, an AFCE and GTE Study: Association Francophone de Chirurgie Endocrinienne & Groupe d'Etude des Tumeurs Endocrines. Ann Surg 268 (1): 158-164, 2018.[PUBMED Abstract]
- Kornaczewski Jackson ER, Pointon OP, Bohmer R, et al.: Utility of FDG-PET Imaging for Risk Stratification of Pancreatic Neuroendocrine Tumors in MEN1. J Clin Endocrinol Metab 102 (6): 1926-1933, 2017.[PUBMED Abstract]
- Brunner SM, Weber F, Werner JM, et al.: Neuroendocrine tumors of the pancreas: a retrospective single-center analysis using the ENETS TNM-classification and immunohistochemical markers for risk stratification. BMC Surg 15: 49, 2015.[PUBMED Abstract]
- Bartsch DK, Langer P, Wild A, et al.: Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: surgery or surveillance? Surgery 128 (6): 958-66, 2000.[PUBMED Abstract]
- Bartsch DK, Fendrich V, Langer P, et al.: Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg 242 (6): 757-64, discussion 764-6, 2005.[PUBMED Abstract]
- Norton JA, Jensen RT: Role of surgery in Zollinger-Ellison syndrome. J Am Coll Surg 205 (4 Suppl): S34-7, 2007.[PUBMED Abstract]
- Lopez CL, Waldmann J, Fendrich V, et al.: Long-term results of surgery for pancreatic neuroendocrine neoplasms in patients with MEN1. Langenbecks Arch Surg 396 (8): 1187-96, 2011.[PUBMED Abstract]
- Drymousis P, Raptis DA, Spalding D, et al.: Laparoscopic versus open pancreas resection for pancreatic neuroendocrine tumours: a systematic review and meta-analysis. HPB (Oxford) 16 (5): 397-406, 2014.[PUBMED Abstract]
- Morgat C, Vélayoudom-Céphise FL, Schwartz P, et al.: Evaluation of (68)Ga-DOTA-TOC PET/CT for the detection of duodenopancreatic neuroendocrine tumors in patients with MEN1. Eur J Nucl Med Mol Imaging 43 (7): 1258-66, 2016.[PUBMED Abstract]
- Lastoria S, Marciello F, Faggiano A, et al.: Role of (68)Ga-DOTATATE PET/CT in patients with multiple endocrine neoplasia type 1 (MEN1). Endocrine 52 (3): 488-94, 2016.[PUBMED Abstract]
- Imamura M, Komoto I, Ota S, et al.: Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol 17 (10): 1343-53, 2011.[PUBMED Abstract]
- Tonelli F, Fratini G, Nesi G, et al.: Pancreatectomy in multiple endocrine neoplasia type 1-related gastrinomas and pancreatic endocrine neoplasias. Ann Surg 244 (1): 61-70, 2006.[PUBMED Abstract]
- Lewis MA, Thompson GB, Young WF: Preoperative assessment of the pancreas in multiple endocrine neoplasia type 1. World J Surg 36 (6): 1375-81, 2012.[PUBMED Abstract]
- van Asselt SJ, Brouwers AH, van Dullemen HM, et al.: EUS is superior for detection of pancreatic lesions compared with standard imaging in patients with multiple endocrine neoplasia type 1. Gastrointest Endosc 81 (1): 159-167.e2, 2015.[PUBMED Abstract]
- Ito T, Igarashi H, Uehara H, et al.: Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 92 (3): 135-81, 2013.[PUBMED Abstract]
- Akerström G, Stålberg P: Surgical management of MEN-1 and -2: state of the art. Surg Clin North Am 89 (5): 1047-68, 2009.[PUBMED Abstract]
- O'Riordain DS, O'Brien T, van Heerden JA, et al.: Surgical management of insulinoma associated with multiple endocrine neoplasia type I. World J Surg 18 (4): 488-93; discussion 493-4, 1994 Jul-Aug.[PUBMED Abstract]
- Crippa S, Zerbi A, Boninsegna L, et al.: Surgical management of insulinomas: short- and long-term outcomes after enucleations and pancreatic resections. Arch Surg 147 (3): 261-6, 2012.[PUBMED Abstract]
- Sakurai A, Yamazaki M, Suzuki S, et al.: Clinical features of insulinoma in patients with multiple endocrine neoplasia type 1: analysis of the database of the MEN Consortium of Japan. Endocr J 59 (10): 859-66, 2012.[PUBMED Abstract]
- Vezzosi D, Cardot-Bauters C, Bouscaren N, et al.: Long-term results of the surgical management of insulinoma patients with MEN1: a Groupe d'étude des Tumeurs Endocrines (GTE) retrospective study. Eur J Endocrinol 172 (3): 309-19, 2015.[PUBMED Abstract]
- Grant CS: Insulinoma. Best Pract Res Clin Gastroenterol 19 (5): 783-98, 2005.[PUBMED Abstract]
- Giudici F, Nesi G, Brandi ML, et al.: Surgical management of insulinomas in multiple endocrine neoplasia type 1. Pancreas 41 (4): 547-53, 2012.[PUBMED Abstract]
- Plöckinger U: Diagnosis and Treatment of Gastrinomas in Multiple Endocrine Neoplasia Type 1 (MEN-1). Cancers (Basel) 4 (1): 39-54, 2012.[PUBMED Abstract]
- Falconi M, Eriksson B, Kaltsas G, et al.: ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 103 (2): 153-71, 2016.[PUBMED Abstract]
- Mignon M, Cadiot G: Diagnostic and therapeutic criteria in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. J Intern Med 243 (6): 489-94, 1998.[PUBMED Abstract]
- Cadiot G, Vuagnat A, Doukhan I, et al.: Prognostic factors in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. Groupe d'Etude des Néoplasies Endocriniennes Multiples (GENEM and groupe de Recherche et d'Etude du Syndrome de Zollinger-Ellison (GRESZE). Gastroenterology 116 (2): 286-93, 1999.[PUBMED Abstract]
- Dickson PV, Rich TA, Xing Y, et al.: Achieving eugastrinemia in MEN1 patients: both duodenal inspection and formal lymph node dissection are important. Surgery 150 (6): 1143-52, 2011.[PUBMED Abstract]
- Akerström G, Stålberg P, Hellman P: Surgical management of pancreatico-duodenal tumors in multiple endocrine neoplasia syndrome type 1. Clinics (Sao Paulo) 67 (Suppl 1): 173-8, 2012.[PUBMED Abstract]
- Zhang IY, Zhao J, Fernandez-Del Castillo C, et al.: Operative Versus Nonoperative Management of Nonfunctioning Pancreatic Neuroendocrine Tumors. J Gastrointest Surg 20 (2): 277-83, 2016.[PUBMED Abstract]
- Vergès B, Boureille F, Goudet P, et al.: Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 87 (2): 457-65, 2002.[PUBMED Abstract]
- Pieterman CR, Vriens MR, Dreijerink KM, et al.: Care for patients with multiple endocrine neoplasia type 1: the current evidence base. Fam Cancer 10 (1): 157-71, 2011.[PUBMED Abstract]
- 多発性内分泌腫瘍2型
-
臨床記述
多発性内分泌腫瘍2型(MEN2)に観察される内分泌異常は、甲状腺髄様がん(MTC);これに先行するC細胞過形成(CCH)(最近の公表文献でC細胞新生物またはC細胞上皮内(in situ)がんと呼ばれている)[ 1 ];褐色細胞腫(PHEO);ならびに副甲状腺腺腫および/または過形成である。MEN2関連MTCはしばしば両側性および/または多病巣性であり、CCHクローン性C細胞増殖の状況で発生する。対照的に、散発性のMTCは一般に片側性および/または単発性である。散発性の約75~80%の症例はCCHにも関連していることから、この病理組織学的特徴を家族性疾患の予測因子として使用することはできない。[ 2 ]甲状腺に触知可能な腫瘤がある患者または下痢を伴う患者では、所属リンパ節(すなわち、副甲状腺、気管傍、深頸、および上縦隔)、または肝などの遠隔部位にMTCが転移して拡がることが多い。[ 3 ][ 4 ]MEN2で転移性PHEOは報告されていない。[ 5 ]MEN2における副甲状腺異常の範囲は良性副甲状腺腺腫または多腺性の過形成から、高カルシウム血症および腎結石を伴う臨床的に明らかな副甲状腺機能亢進症にわたる。
歴史的に、個人または家族に特定の内分泌腫瘍があるかどうかによって、MEN2を有する個人および家族は以下の3つの臨床的亜型のいずれかに分類されていた:
- MEN2A。
- 家族性甲状腺髄様がん(FMTC)。
- MEN2B(ときにMEN3と呼ばれる)。
現在の層別化は、表現型のみに基づく分類から、遺伝型(すなわち、病原性多様体)と表現型に基づく分類へ移行している。[ 6 ]現在の分類では、2つのMEN2症候群:MEN2AおよびMEN2Bが含まれている。MEN2A症候群は、関連疾患の存在を基にさらに分類される。例えば、古典的なMEN2Aには、MTC、PHEO、および/または副甲状腺機能亢進症を伴うものが含まれる。その他の分類には、皮膚アミロイド苔癬を伴うMEN2A、ヒルシュスプルング病(HSCR)を伴うMEN2A、およびFMTC(RET生殖細胞病原性多様体およびMTCが存在するが、PHEOまたは副甲状腺機能亢進症の家族歴を認めない)がある。[ 1 ]予後および管理方法を決定するためには、MEN2の亜型別に患者または家族を分類するのが有用である。
MEN2の有病率は35,000人に約1人と推定されている。[ 7 ]MEN2症例の大多数がMEN2Aである。
MTCおよびCCH
MTCは甲状腺のカルシトニン分泌細胞(C細胞)に由来する。C細胞の細胞巣が基底膜を越えて拡大し、甲状腺濾胞に浸潤し、これを破壊している場合にMTCであると診断する。CCHは議論のある診断名だが、ほとんどの病理医は、cluster当たりのC細胞が8個以上、C細胞に囲まれた完全な濾胞、正常な解剖学的位置を越えたC細胞の分布としてCCHが定義されることに同意している。[ 1 ][ 8 ][ 9 ][ 10 ]RET病原性多様体およびCCHを認める人は、MTCに進行するリスクが実質的に高い。血漿中カルシトニン濃度が高値を示しているとMTCおよびCCHが疑われる。
10,864人の結節性甲状腺疾患の患者の研究では、カルシトニンによる刺激の後で、MTCが44症例(250例ごとに1例)見つかり、そのうちいずれの症例も臨床的に疑わしいものではなかった。結果的にこれらの患者の半数は、針生検でMTCの証拠を認めず、したがってカルシトニン刺激検査陽性がなければ、手術を受けていなかった可能性がある。[ 11 ]カルシトニン刺激試験陽性と関連するCCHは一般集団の約5%に発生する;それゆえ、血漿中カルシトニンの刺激反応によって必ずしもCCHを小さいMTCと鑑別することはできず、またMEN2家系におけるキャリアと非キャリアを必ずしも識別できない。[ 12 ][ 13 ][ 14 ]
米国において、MTCは毎年診断される甲状腺がん新規症例の1~2%を占める。[ 15 ]米国で診断される甲状腺がん症例の約75%が散発性である(すなわち、MTC、またはMEN2にみられる他の内分泌異常の家族歴がいずれもない場合に発生する)。散発性症例の発生率のピークは40歳代~50歳代にある。[ 3 ][ 16 ]英国の1件の研究では、人口5,500万人のうち年間20~25例が新たにMTCを発生すると推定されている。[ 17 ]
家族歴を有さない場合、MTCが若年で発症するかまたは両側性または多病巣性であればMEN2が疑われよう。散発性MTCと思われる症例の小規模シリーズでは、生殖細胞系のRET病原性多様体の高い保有率が示唆されるが[ 18 ][ 19 ]、より大規模なシリーズでは、保有率において1~7%の幅を示している。[ 20 ][ 21 ]これらのデータに基づき、MTCの全症例に対してRET遺伝子の病原性多様体検査の実施が広く推奨されている。[ 1 ][ 22 ]
証拠レベル(スクリーニング):3
MTCの自然史
米国において、甲状腺がんは毎年新たに発生する悪性腫瘍の約3%を占め、1年当たりのがん診断症例は52,890人、死亡数は2,180人と推定されている。[ 23 ]これらのがん診断症例の1~2%がMTCである。[ 15 ]
MTCは、カルシトニンを分泌する甲状腺傍濾胞細胞に由来する。MTCは散発性および家族性の形で発生し、先にCCHが現れることがあるが、CCHは中年期に比較的よくみられる異常である。[ 8 ][ 9 ]
MTCの平均生存率は、より多くみられる甲状腺がんよりも低い(例えば、甲状腺乳頭がんおよび濾胞がんの5年生存率が94~98%であるのに対して、MTCの5年生存率は86~89%である)。[ 15 ][ 24 ]生存率は診断時の病期に相関し、MTCの生存率が低い原因の1つは後期での診断の割合が高いことにあると説明できる。[ 25 ][ 26 ][ 27 ]
診断時の病期が早期であることに加え、MTCの生存率の向上と関連しているその他の因子には、腫瘍が小さいこと、診断時の年齢が低いこと、症状による診断よりも生化学的または遺伝学的スクリーニング(すなわち、カルシトニン値上昇、RET多様体のスクリーニング)による診断であることなどがある。[ 26 ][ 28 ][ 29 ][ 30 ]
MTC患者1,252人を対象としたSurveillance, Epidemiology, and End Resultsの集団ベースの研究で、局所病変の拡がりによって、生存率が異なることが明らかにされた。例えば、10年生存率では、甲状腺に限局した病変を有する患者に対する95.6%から、遠隔転移を有する患者の40%までの幅があった。[ 28 ]
MEN2関連PHEO
PHEOは、副腎髄質のカテコールアミン産生性のクロム親和性細胞から発生する。褐色細胞腫は比較的まれな腫瘍であるが、難治性高血圧症の患者、または24時間採尿または血漿中のカテコールアミン分泌およびカテコールアミン代謝物(すなわち、ノルエピネフリン、エピネフリン、メタネフリン、およびバニリルマンデル酸)の増加が生化学的スクリーニングによって明らかにされた場合に疑われる。以前は、尿中カテコールアミンの測定が好ましい生化学的スクリーニング方法であると考えられていた。しかしながら、カテコールアミンの放出が間欠性のみであり、副腎髄質でメタネフリンおよびノルメタネフリンに代謝されることから、尿中または血漿分画メタネフリンの測定がゴールドスタンダードとなっている。[ 32 ][ 33 ][ 34 ][ 35 ][ 36 ][ 37 ]MEN2であるか、MEN2のリスクが高い個人における生化学的スクリーニングでPHEOが示唆される場合は、磁気共鳴画像法(MRI)またはコンピュータ断層撮影などの位置確認のための検査を実施できる。[ 38 ]診断の確定は、ヨウ素131-メタヨードベンジルグアニジン・シンチグラフィまたはポジトロン放射断層撮影画像法を用いて行われる。[ 13 ][ 38 ][ 39 ][ 40 ]
PHEOの個人で、MEN2の診断は、両側性PHEOの人、発症年齢が低い(35歳未満)人、ならびにMTCまたは副甲状腺機能亢進症の個人歴および/または家族歴を有する人においてしばしば考慮される。しかしながら、PHEOの素因をもつ遺伝性疾患はMEN2だけではない。他の疾患には、神経線維腫症1型(NF1)、フォン・ヒッペル-リンダウ症候群(VHL)[ 41 ]、および遺伝性パラガングリオーマ症候群がある。[ 42 ](遺伝性のPHEOに関する詳しい情報については、本要約の家族性PHEOおよびPGL症候群のセクションを、またVHLに関する詳しい情報については、腎がんの遺伝学に関するPDQ要約のフォン・ヒッペル-リンダウ症候群のセクションを参照のこと。)
原発性副甲状腺機能亢進症(PHPT)
PHPTは、一般集団において3番目に一般的な内分泌異常である。発生率は年齢とともに増加し、症例の大多数は50歳代を過ぎてから発症している。症例の約80%は1腺にある腺腫の結果である。[ 43 ]PHPTはまた、以下のようないくつかの異なる遺伝性症候群を構成している腫瘍の1つとしてみられる:
遺伝性PHPTは典型的に多腺性で、若年期に発症し、腺腫と腺過形成の両方の組織学的証拠を有することがある。
MEN2亜型の臨床診断
2つのMEN2臨床的亜型の診断は、臨床所見、家族歴、およびRET遺伝子の分子遺伝学的検査を併用して決定される。
MEN2A
古典的MEN2A
1人の人または近い血縁者にMTCに加えて特定の内分泌腫瘍(PHEOおよび/または副甲状腺腺腫および/または過形成)が2つ発生するとMEN2Aであると臨床的に診断される。[ 1 ]
古典的MEN2A亜型は、MEN2症例の約60~90%を占める。MEN2Aは初めシップル症候群と呼ばれていた。[ 47 ]RET病原性多様体の遺伝子検査が可能になったことから、MEN2Aの患者では、約95%がMTCを発症することが明らかになっている。[ 13 ][ 48 ][ 49 ][ 50 ]
一般に、MTCがMEN2Aの最初の症状発現である。症状はないがリスクのある人では、刺激試験によって血漿カルシトニン濃度の増加、およびCCHまたはMTCの存在を明らかにできる場合がある。[ 13 ][ 49 ]MEN2Aの家族では、MTCの生化学的発現時期が一般に5~25歳(平均15歳)のようである。[ 13 ]症状発現前にスクリーニングを実施していなければ、約5~20歳で頸部腫瘤または頸部痛としてMTCが発現するのが一般的である。このような患者の50%以上に頸部リンパ節転移がみられる。[ 3 ]最も好発する全身的症状である下痢は、著しく高い血漿中カルシトニン値または巨大腫瘤および/または肝転移を認める患者にみられ、予後不良を意味する。[ 1 ][ 3 ][ 51 ][ 52 ]MTC患者の最大30%までが下痢および進行疾患を示す。[ 53 ]
MEN2関連PHEOはしばしば両側性、多病巣性であり、腫瘍外の髄質過形成と関連している。[ 54 ][ 55 ][ 56 ]これらはまた発症年齢が早い;散発性の褐色細胞腫よりも悪性である可能性は低い。[ 54 ][ 57 ]通常、MEN2関連PHEOはMTCの後に発現し、概して難治性高血圧症を伴う。[ 58 ]
MEN1にみられるPHPTとは異なり、MEN2患者における副甲状腺機能亢進症は典型的には無症状であるか、カルシウムのわずかな上昇のみと関連している。[ 53 ][ 59 ]MEN2に関連した副甲状腺機能亢進症患者56人の症例集積が、French Calcitonin Tumors Study Groupによって報告されている。[ 59 ]診断時の年齢中央値は38歳であり、この疾患がMEN2の最初の症状発現としては、まれであることが示された。これは、大多数の患者(87~99%)が最初に原発性副甲状腺機能亢進症を発症するMEN1とは著しい対照をなしている。[ 60 ][ 61 ][ 62 ]副甲状腺異常は、甲状腺髄様がんの手術に付随して43人の患者(77%)で認められた。患者の3分の2は無症状であった。手術により副甲状腺を切除した53人の患者のうち、1腺に腺腫が認められたのが24人、2腺に認められたのが4人、また25人に過形成腺が認められた。
皮膚アミロイド苔癬を伴うMEN2A
MEN2Aの少数の家族に、皮膚アミロイド苔癬として知られるそう痒性の皮膚病変がみられる。この苔癬様の皮膚病変は背中の上部にみられ、MTC発症前に現れることもある。[ 63 ][ 64 ]
ヒルシュスプルング病(HSCR)を伴うMEN2A
HSCRは、新生児に腸拡大および便秘または腸の閉塞を典型的に引き起こす結腸の腸壁内神経叢の異常で、MEN2A関連のRET病原性多様体を有する少数の個人に発生する。[ 65 ]エクソン10に位置する特定のシステイン残基(すなわち、コドン609、618、および620)における病原性多様体は、最も多くHSCRと関連しているが、他のエクソンに病原性多様体を有する個人でも罹患することがある。[ 66 ]HSCRは、MEN2A診断以外でも発生することがあり、HSCRの乳児では、HSCRが他の症候群の一部として存在する可能性があるため、MEN2Aの可能性にかかわらず、自身の遺伝子評価から恩恵が得られる場合がある。家族性HSCR例の最高40%および散発性例の3~7%は、RETがん原遺伝子の生殖細胞病原性多様体と関連している。[ 67 ][ 68 ]特定の機能喪失型RET多様体は、孤立性のHSCRと関連していることから[ 69 ]、HSCRおよび生殖細胞RET多様体を有するすべての個人が必ずしもMEN2Aであるとは限らないことが示唆される。
図2は、ある家系内でのMEN2Aの典型的な発現の一部を示している。
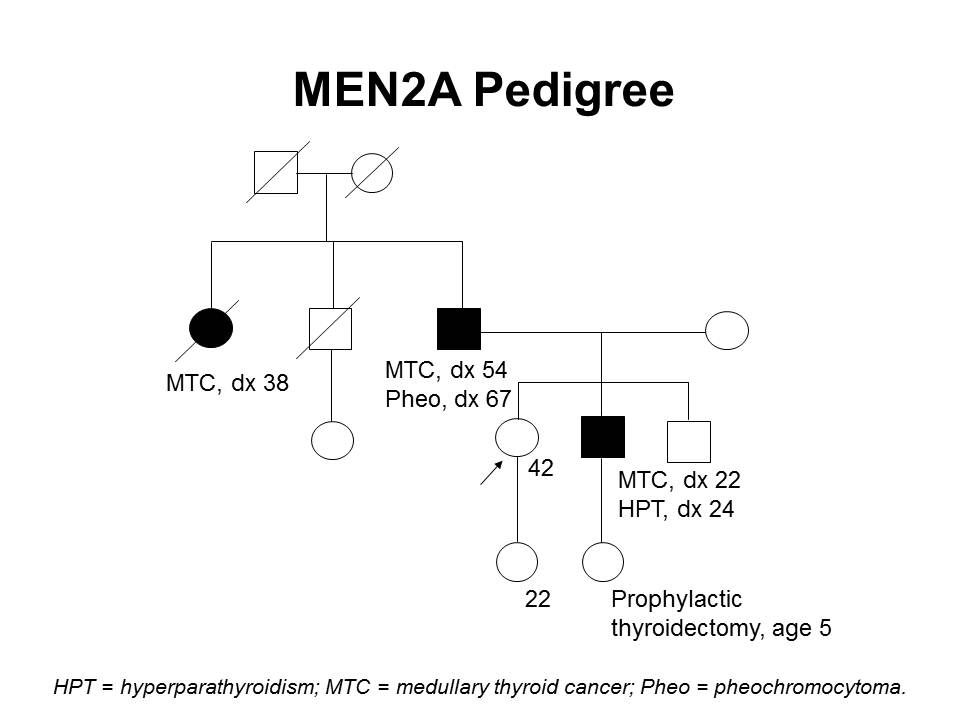
図2.MEN2A家系(pedigree) 図。この家系(pedigree) 図は、RET病原性多様体を有する家系の典型的な特徴の一部を4世代にわたって示し、甲状腺髄様がん、褐色細胞腫、副甲状腺機能亢進症に罹患している家系員を記載している。発症年齢は家系内でさえ、非常に幅広い。MEN2Aの家系では、これらの特徴の一部またはすべてが認められる。常染色体優性症候群と同様に、母系または父系で伝達する場合がある。 家族性甲状腺髄様がん(FMTC)
MEN2A症例の最大50%は、FMTC亜型であり、PHEOまたは副甲状腺腺腫/過形成が認められず、生殖細胞RET病原性多様体およびMTCのみが認められる家族または一人の個人として定義される。[ 1 ]この定義は、単独の診断として以前のFMTCの分類に置き換えられる。[ 1 ]以前に、(家系の人数が少なすぎるため、または他のMEN2A症状の発現が遅いために)家系がFMTCと誤って分類されたことにより、罹病率および死亡率が著しく高い疾患であるPHEOのリスクが見過ごされることがあった。こうした理由から、現在FMTCは、MEN2Aの亜型とみなされ、MEN2A症候群の他の(非甲状腺性)症状が発現しないか、発現が遅れる。[ 70 ]現在の管理ガイドライン[ 1 ]では、純粋なFMTCと考えられる患者も、PHEOおよび副甲状腺機能亢進症についてスクリーニングするよう推奨している。
MEN2B
MEN2Bは、口唇および舌の粘膜神経腫、有髄角膜神経線維、口唇肥大を伴う独特の顔貌、マルファン症候群様の細長い体型、およびMTCの存在によって臨床的に診断される。[ 71 ][ 72 ][ 73 ][ 74 ]de novo病原性多様体の症例におけるMEN2Bの診断はしばしば遅れ、MTCの発生後になる。MTCはしばしば致死的で、特に診断時点で一般的な転移病変が存在すると顕著である。そのため、早期診断により転移が拡がる前に生命を救うMTCの治療を実施できる可能性があるため、小児科医にとって本症候群の内分泌および非内分泌臨床症状を認識することが重要である。[ 75 ]
MEN2BはMEN2症例の約5%を占める。MEN2Bは初め粘膜神経腫症候群またはWagenmann-Froboese症候群と呼ばれていた。[ 76 ]MEN2Bは早い時期にすべての患者で侵攻性のMTCを発生する特徴がある。[ 71 ]幼い時期(約1歳)に甲状腺摘出術を受けていないMEN2Bの患者は、若年齢でMTCが転移する可能性が高い。早期のリスク低減のための甲状腺摘出術による介入の実施前は、MEN2B患者の平均死亡年齢は21歳であった。MEN2B症例の約50%にPHEOが起こる;約半数は多発性で、しばしば両側性である。臨床的に明らかな副甲状腺の病変がみられることはきわめてまれである。[ 48 ][ 71 ]MEN2B患者は、乳児期または幼児期に疾患独特の顔貌、舌の背面前方、口蓋、または咽頭の粘膜神経腫の存在によって同定できる。口唇は徐々に突出していき、朱唇に粘膜下結節を認めることがある。眼瞼神経腫が上部眼瞼縁の肥厚および外反を引き起こすことがある。細隙灯顕微鏡検査で角膜神経の著明な肥厚をみることがある。
MEN2B患者はびまん性消化管神経節神経腫症を有することがあり、その関連症状には腹部膨隆、巨大結腸、便秘、および下痢などがある。[ 77 ]文献のレビューにより、MEN2B患者の72.7%に共通する症状として便秘の存在が報告された。また、消化管症状はMEN2B患者の52.3%で生後1年間に発生した。腸を生検したところ、27.3%の患者で神経節神経腫症が診断された。[ 78 ]
患者の約75%がマルファン症候群様体型であり、しばしば脊柱後側弯症または前側弯症、関節弛緩、および皮下脂肪の減少を伴う。また、近位筋の衰弱および脱力もみられる。[ 73 ][ 74 ]
単一施設で治療を受けたde novo病原性多様体を有するMEN2Bの35症例の臨床所見に関するレトロスペクティブ・レビューでは、本症候群の内分泌症状によって22症例が診断されたことが明らかにされた。[ 75 ]PHEOの診断、頸部腫瘤、および/または骨格異常がMTCの同定につながった。残りの患者13人は、口腔神経腫、角膜神経異常、持続性の下痢、成長障害、または頻繁な転倒を伴う骨格異常などの非内分泌症状を呈していた。全コホートのうち、患者21人は、MEN2B診断の平均5年前にMEN2B関連の特徴の評価のために1人または複数の医師に紹介されていた。
小児科医にとって、MEN2Bに関連する何らかの臨床症状がみられる患者を評価する際には、常に疑念指数を高めておくことがきわめて重要である。小児における口腔および眼球の神経腫の存在および/またはやせてひょろ長い容姿は、追加の調査を要する場合がある。[ 79 ]MTCを有するか、以下の特徴のいずれかを有する個人には、遺伝カウンセリングへの紹介を推奨している著者もいる:[ 71 ][ 79 ]
MEN2の分子遺伝学
MEN2症候群は、染色体領域10q11.2に位置するRET遺伝子の遺伝性病原性多様体の結果である。[ 80 ][ 81 ][ 82 ]RET遺伝子はゲノム55キロベースにわたって存在する21のエクソンで構成されるがん原遺伝子である。[ 83 ][ 84 ]
RETは、細胞外領域、膜貫通領域および細胞内領域をもつ受容体型チロシンキナーゼをコードする。このシグナル伝達経路におけるRET受容体とリガンドとの相互作用の詳細がレビューされている。[ 31 ][ 85 ][ 86 ]簡単にいうと、細胞外領域はカルシウム結合カドヘリン様領域と、今日までに同定されている4つのリガンドのうちの1つと相互作用するシステインに富む領域とで構成される。例えば、グリア細胞株由来神経栄養因子(GDNF)、neurturin、persephin、およびarteminといったこれらのリガンドも、GDNFファミリー受容体-αファミリーの4つの補助受容体のいずれかと相互結合する。[ 85 ]チロシンキナーゼの触媒中心は細胞内領域に位置し、さまざまな第二メッセンジャー分子を介して下流シグナル伝達を引き起こす。
遺伝子検査
MEN2は十分に確定された遺伝性がん症候群であり、遺伝子検査がリスクのある家族員に対する管理の重要な一部とみなされる。これは、米国臨床腫瘍学会の直近の遺伝子検査指針の中で概略が述べられているがん感受性に対する遺伝子検査の適応基準に合致する。[ 87 ]リスクがあるのは、MEN2であると分かっている人の第一度近親者(両親、同胞、および子供)であると定義される。検査によって無症状のMEN2の人々を同定でき、予防処置としてリスク低減のための甲状腺摘出術および生化学的スクリーニングを提供できる。しかしながら、リスクのある近親者における陰性の病原性多様体分析結果は、罹患した近親者に病原性の多様体が同定されて初めて有益となる。(詳しい情報については、がんの遺伝学的リスク評価とカウンセリングに関するPDQ要約を参照のこと。)リスクのある人の早期発見が医学的管理に影響するため、症状がない小児に対する検査が有用と考えられる。[ 88 ][ 89 ](リスクのある人に対する臨床的管理に関する詳しい情報については、本要約の遺伝子型-表現型の相関およびリスク層別化のセクションを参照のこと。)
MEN2を示唆する個人歴または家族歴の有無にかかわらず、MTCの診断を受けたすべての患者に対して、一般的にRET病原性多様体に関する生殖細胞系DNA検査が推奨される。[ 90 ][ 91 ]MEN2AまたはMEN2B患者の約95%が同定可能な生殖細胞系RET病原性多様体を有する。[ 92 ]散発性MTCと思われる個人の1~10%は生殖細胞系RET病原性多様体を保有することから、MTCを診断されたすべての個人を検査する重要性が強調されている。[ 93 ][ 94 ][ 95 ]
これ以外の遺伝子座が関与するという証拠はなく、現在までに分析されている病原性多様体の検出されない家系すべてにおいてRET遺伝子との連鎖が示されている。検出可能な病原性多様体が認められない家系に対する臨床的推奨事項は罹患した患者および家族における臨床的特徴に基づく。
この方法を行うさまざまな検査施設において用いられる手技およびRET病原性多様体検査へのアプローチには、かなりの多様性がある。RETにおける多様体の検出法には、PCR産物の制限酵素消化を伴うポリメラーゼ連鎖反応(PCR)、ヘテロ二本鎖分析、一本鎖高次構造多型(SSCP)分析、変性高速液体クロマトグラフィ、およびDNA塩基配列決定法がある。[ 92 ][ 96 ]ほとんどの検査室では少なくとも、標的エクソンアプローチを用いる検査を提供している;すなわち、検査室では、最も一般的に多様体を保有することが明らかにされているエクソン(エクソン10、11、13、14、15、および16)に多様体がないかが調べられる。他の検査室ではすべてのエクソンについて検査が提供される。MEN2が臨床的に強く疑われる家族における標的エクソン検査が正常である場合は、次に残りのエクソンの塩基配列決定法を実施できる。
多様体発見法および標的 vs 全遺伝子検査におけるこれらの差異は、検査を行う施設選択および検査結果の解釈において重要な考慮事項となる。(臨床的妥当性に関する詳しい情報については、がんの遺伝学的リスク評価とカウンセリングに関するPDQ要約を参照のこと。)
遺伝子型-表現型の相関およびリスク層別化
MEN2における遺伝子型-表現型の相関は十分に確立されており、医学的管理の推奨方針を臨床家に示すために長く用いられている。いくつかのグループにより、臨床的表現型、発症年齢、およびMTCの侵攻性に基づいた病原性多様体-層別化の表が開発されている。[ 1 ][ 90 ][ 97 ]この分類戦略は、2001年のMENに関するSeventh International Workshop後に初めて発表され、遺伝子検査および予防的甲状腺切除の年齢についてのガイドラインを規定した。[ 90 ]この層別化はAmerican Thyroid Association(ATA)により改定されている。[ 1 ][ 98 ][ 99 ]特定の病原性多様体およびそのATA分類は、下の表5に要約する。
ATA最高リスク(HST)(以前にATA-Dレベルとされた)の病原性多様体は侵攻性が最も強く、MTC発症リスクが最も高い。[ 1 ]この分類には、MEN2BおよびRETコドンM918T病原性多様体を伴うものが含まれ、発病時年齢が最も若く、死亡リスクが最も高いという関係がある。ATA高リスク(H)(以前にATA-Cと呼ばれた)の病原性多様体(コドン634およびA883F)はやや低いリスクに関連しているが、これらの病原性多様体がある患者のMTCは、さらに侵攻性が際立っており、若い年齢で発現する場合がある。[ 100 ]以前のATAのレベルAおよびレベルBの病原性多様体は、現在、中リスク(MOD)と呼ばれる単一グループにまとめられており、侵攻性MTCのリスクがATA-HSTおよびATA-Hの病原性多様体キャリアにみられるリスクに比べて低いことと関連している。[ 99 ]しかしながら、MTCのリスクは一般集団のリスクよりまだ相当高く、リスク低減のための甲状腺摘出術を検討する根拠となる。[ 1 ]ATA-MOD分類で一般的な病原性多様体を表5に示す。
コドン883および918の病原性多様体は、MEN2Bにおいてのみ認められており、最も若い発症年齢およびMTCの最も侵攻性の強い型と関連している。[ 100 ][ 101 ][ 102 ][ 103 ][ 104 ][ 105 ]MEN2B患者の約95%はM918T病原性多様体を有する。[ 101 ][ 102 ][ 103 ][ 106 ]上述のように、MEN2B患者の50%はPHEOを発症するが、PHPTはまれである。A883F多様体キャリアの発表されている全症例(N = 13)をレトロスペクティブにレビューしたところ、MTCの疾患経過はM918Tキャリアで観察された疾患経過よりも緩慢であったことが明らかにされた。A883Fキャリアでは疾患の発症が遅く(19歳でのMTCの浸透度は50%)、5年および10年生存率は88%で、63%の患者がMTCの生化学的治癒を達成した。[ 100 ]コドン883および918における多様体に加えて、MEN2B様の表現型を示す患者の一部は、2つの生殖細胞多様体を保有することが明らかにされている。[ 107 ][ 108 ][ 109 ][ 110 ][ 111 ]RETに対する検査が実地臨床でより一般的になるにつれて、さらなる二重多様体の表現型が報告される可能性が高い。
コドン634における病原性多様体(ATA-H)はMEN2Aの家系において頻度が群を抜いて最も高い所見である。RETキャリア477人を対象とした1件の研究により、52.1%がC634R病原性多様体を、26.0%がC634Y病原性多様体を、9.1%がC634G病原性多様体を有することが示された。[ 48 ]一般的に、コドン634における病原性多様体はPHEOおよびPHPTと関連する。[ 48 ][ 112 ]最近まで、皮膚アミロイド苔癬を伴うMEN2Aは、ほぼ例外なくコドン634に病原性多様体を有する患者でみられた。[ 48 ][ 50 ][ 113 ]しかしながら、最近の報告で以前にコドン804病原性多様体のためにFMTCであると考えられたある個人においてMTCと皮膚アミロイド苔癬の併発が記述されている。[ 114 ]コドン634病原性多様体はFMTCにおいても記述されているが、ほぼ全例がC634Yである。[ 48 ]
まとめると、ATA-HSTおよびATA-H(以前はそれぞれレベルDおよびC)の病原性多様体では、MTCのリスクが最も高くなり(約95%の生涯リスク)、より侵攻性の疾患経過をたどる。PHEOのリスクが高い(最大50%)。[ 48 ][ 115 ]コドン634病原性多様体を有する患者(ただし、コドン883または918多様体ではない)はPHPTのリスクも高い。[ 48 ]
中リスクの多様体は、RET遺伝子のエクソン10に位置し、コドン609、611、618、620、および630での多様体を含む。これらの多様体は、RET蛋白の細胞外ドメインにシステイン残基を含んでおり、MEN2Aを有する家系およびMTCのみ(FMTC)を有する家系に認められている。[ 20 ][ 48 ][ 97 ][ 116 ][ 117 ][ 118 ][ 119 ][ 120 ]これらの病原性多様体を有する個人のMTCリスクは約95~100%である;PHEOおよび副甲状腺機能亢進症のリスクは、高リスクの病原性多様体を有する個人でみられるリスクより低い。
以前にATAのレベルA(ATAのレベルBについては現在ATA-MODに分類される、すなわち、コドン321、515、533、600、603、606、531/9塩基対重複、および532重複)として分類された病原性多様体を有する個人は、依然として高いものの、MTCの生涯リスクが低い。これらの病原性多様体に関連するMTCは、より緩慢な経過をたどり、発症年齢が遅い傾向があるものの、これらの病原性多様体を有する個人が20歳前にMTCを発症したという報告がいくつかある。[ 48 ][ 121 ][ 122 ][ 123 ][ 124 ][ 125 ]PHEOおよびPHPTはこれらの病原性多様体とは一般的に関連していないが、これらの疾患も報告されている。[ 125 ]
表5.多発性内分泌腫瘍2型における遺伝子型と表現型の相関a 病原性多様体 エクソン 侵攻性MTCのリスク PHEO概算発生率 HPTH概算発生率 CLAの存在 HSCRの存在 G533C 8 中 10% - No No C609F/G/R/S/Y 10 中 10%–30% 10% No Yes C611F/G/S/Y/W 10 中 10%–30% 10% No Yes C618F/R/S 10 中 10%–30% 10% No Yes C620F/R/S 10 中 10%–30% 10% No Yes C630R/Y 11 中 10%–30% 10% No No D631Y 11 中 50% - No No C634F/G/R/S/W/Y 11 高 50% 20%–30% Yes No K666E 11 中 10% - No No E768D 13 中 - - No No L790F 13 中 10% - No No V804L 14 中 10% 10% No No V804M 14 中 10% 10% Yes No A883F 15 高 50% - No No S891A 15 中 10% 10% No No R912P 16 中 - - No No M918T 16 最高 50% - No No 表5に分類されている病原性多様体に加えて、まれなまたは新たなRET多様体も多数報告されている。これらの一部は、MEN2A表現型に至る病原性多様体である。他に、浸透度の低いアレルまたはMTC発症リスクをわずかに高めるだけの修飾アレルを示すものもある。[ 126 ]1件の多施設研究により、RET K666N多様体を有する8家系が同定された。スクリーニングを受けて病原性多様体を有していることが確認された家系員16人中、MTCを有したのは1人のみであった。[ 126 ]また別のものは、臨床的意義のない良性の多型であろう。例えば、数件の研究から、症例および健康な対照間で頻度が同等であること、ならびに家系内の疾患と共分離する他の病原性多様体と同時発生することに基づいて、RET多様体のY791F(p.Tyr791Phe)およびS649L(p.Ser649Leu)が良性の多型である可能性が高いという有力な証拠が示されている。[ 127 ][ 128 ]デンマークのY791Fキャリア(n = 20)の長期追跡研究で、コホート内にMEN2Aの徴候(MTC、PHPT、PHEO、皮膚アミロイド苔癬、またはHSCR)は認められず、年齢中央値は49.5歳(範囲、7~87歳)であった。[ 129 ]このため、これらの多様体のキャリアはMEN2症候群を有するものとして治療されず、無症候性の家系員についてこれらの多様体の検査は一般に行われない。あらゆる他のRET病原性多様体を除外するためにエクソン8および10~16のすべてのホットスポット多様体の包括的検査が実施されることがあり、またPHEOの診断のために他の疾患関連遺伝子のより広範な検査が必要となる場合がある。(詳しい情報については、本要約の家族性褐色細胞腫および傍神経節腫症候群のセクションを参照のこと。)
MEN2A患者の臨床像を修飾する上で中立的なRET塩基配列多様体の役割を調査する研究が進行中である。特定のRET多型またはハプロタイプの存在は、PHEO、副甲状腺機能亢進症、およびHSCRの発症確率、ならびにMTC転移病変の発現年齢に対する影響について解析されている。[ 130 ][ 131 ][ 132 ][ 133 ]ある遺伝子多様体の機能的および臨床的意義を決定するために、分離比分析、in silico解析、関連解析、機能分析など、さまざまなアプローチを適用することができる。公的に利用できるRET多様体のオンラインデータベースのレポジトリが開発され、これには多様体および関連する病原性、表現型、およびその他の関連する臨床情報と参考文献の完全なリストが含まれている。[ 134 ]
サーベイランス
PHEOのリスクがある人に対するスクリーニング
MEN2AまたはMEN2Bのいずれの患者でも、甲状腺摘出術の前に適切な生化学的スクリーニングを実施して、機能性のPHEOの存在を除外できる。しかしながら、MEN2で小児PHEOはまれである。[ 1 ]ATA-HSTまたはATA-HのRET病原性多様体を有する患者では、11歳までにPHEOの年1回のスクリーニングを検討するよう、ATAは推奨している。[ 1 ]ATA-MODのRET病原性多様体を有する患者では、16歳までにPHEOの定期的なスクリーニングを開始するよう、ATAは推奨している。[ 1 ]生化学検査結果が異常な場合に限り、MRIまたは他の画像検査を指示してもよい。[ 26 ][ 135 ]散発性または遺伝性のPHEO(MEN2の患者を含むが、限定するものではない)の患者を対象とした諸研究では、カテコールアミン排泄が偶発性のために、カテコールアミン代謝物、特に血漿遊離型メタネフリンおよび/または尿分画メタネフリンの測定の方が、尿中カテコールアミンより高い診断的感度が得られることが示唆されている。[ 32 ][ 33 ][ 34 ][ 35 ][ 36 ][ 37 ][ 38 ][ 136 ]数件のレビューでPHEOの生化学的診断、位置確認、および管理に関する簡潔な要約が発表されている。[ 38 ][ 137 ]手術に加えて、カテコールアミン過剰の患者が特に危険となる臨床状況がある。1つの例は、リスクのある健常女性患者が妊娠する場合である。妊娠、分娩または出産により、認識されていなかったPHEOを保有する患者に高血圧の発作が起こることがある。カテコールアミン過剰が認められる妊娠者には、出産前に適切な薬物療法が必要である。
証拠レベル:5
副甲状腺機能亢進症のリスクがある人に対するスクリーニング
一般にMEN2に関係した副甲状腺機能亢進症は、この病気の自然史の初期にみられ、軽度でしばしば無症状であるが、治療を行わないと症状が現れることもある高カルシウム血症と関連している。[ 59 ]MEN2では、小児副甲状腺機能亢進症はまれである。3件の研究では、その診断時年齢の中央値が約38歳であることが明らかになった。[ 59 ][ 138 ][ 139 ]副甲状腺機能亢進症では年1回のスクリーニングをATAは推奨しており[ 1 ]、ATA-HSTおよびATA-Hの病原性多様体キャリアでは11歳までに、ATA-MODのRET病原性多様体キャリアでは16歳までにスクリーニングを開始する。[ 1 ]インタクト副甲状腺ホルモン(PTH)を測定しているかどうかにかかわらず、典型的にはアルブミン補正カルシウムまたはイオン化血清カルシウムを検査に含める。
証拠レベル:5
同定可能なRET病原性多様体をもたない家系(kindred)のリスクがある人に対するスクリーニング
リスクのある個人に対しては、MEN2Aが確認されない限りリスク低減のための甲状腺摘出術はルーチンに実施されない。MEN2Aの患者におけるMTCのスクリーニングプロトコルは年1回のカルシトニン刺激検査である;しかしながら、集団の約5%にMTCの前駆症とはならないCCHがあることから、検査結果の解釈には注意が必要である。[ 12 ][ 13 ][ 140 ]さらに、15歳未満の患者において、偽陰性検査結果のかなりのリスクがある。[ 13 ]PHEOおよび副甲状腺疾患のスクリーニングは上記と同じである。
FMTCのリスクがある患者については、MTCの年1回のスクリーニングはMEN2Aの患者と同じである。
証拠レベル:5
介入
MTCの治療
リスク低減のための甲状腺摘出術
リスク低減のための甲状腺摘出術がMEN2患者に対して選択すべき腫瘍学的治療法となる。M918T RET病原性多様体を有する小児では、生後1年以内、おそらく生後数ヵ月での甲状腺摘出術から恩恵が得られる可能性がある。[ 1 ]同様に、ATA-H分類のRET病原性多様体を有する小児は、血清中カルシトニン値に基づいて5歳以前に甲状腺摘出術を予防的に受けることができる。ATA-MOD分類の小児では、これらの腫瘍が遅れて発生する可能性があるが、これが考慮された時点で同様に侵攻性であるため、身体診察、頸部の超音波検査、および血清中カルシトニン値の測定を5歳程度から開始するよう、ATAは推奨している。[ 1 ][ 99 ]カルシトニン値に異常がみられない場合は、6~12ヵ月ごとに測定を継続するように勧めてもよい。
患者に対応する小児科医、小児内分泌医、および外科医を含む集学的チームは、血清中カルシトニン値の傾向、超音波検査所見、家族の意向、および治療担当医の経験に基づき、小児の親と協力して手術の時期を決定すべきである。[ 1 ]
一部のATA-HまたはATA-MODのRET病原性多様体を有する小児では、甲状腺全摘出術の時期を判断するためにカルシトニンの基礎値およびペンタガストリン刺激後値が使用可能なことが一部の研究で示唆されている。[ 141 ][ 142 ][ 143 ][ 144 ]これらの知見は、RET病原性多様体キャリアにおいて、ルーチンの検査でカルシトニンの基礎値または刺激後値が増加するまで手術を安全に遅らせることができることを示唆している。病原性多様体キャリアの若い世代では、患者が年を経るまで手術を遅らせることで、手術による合併症のリスクが低下する可能性があるため、このアプローチの有益性は特に注目すべきである。16歳以下の小児2,740人を対象とした大規模な研究は、より若い小児におけるカルシトニン値に対する年齢別基準範囲に関するデータを提供しており、意思決定に役立つ可能性がある。[ 145 ]一部のカルシトニン分析法は他の方法より感度が高い可能性があるため[ 143 ]、カルシトニン値と同様に、検査の種類にも注意する考慮が必要になる。しかしながら、術前のカルシトニン基礎値が正常でも、患者がMTCである可能性を否定できない。[ 13 ][ 76 ]
RET病原性多様体を有する患者では、予防的甲状腺切除時の年齢が高いほど、術後の病変の持続または再発のリスクが高いという有意な関係があるとされている。[ 146 ]これと一致して、MEN2Aを有する若い臨床的に無症状の個人を対象とした研究で、早期(8歳未満と定義)に甲状腺摘出術を受け、リンパ節転移の証拠が認められない患者では病変の持続または再発の発生率がより低いことが示された。[ 147 ]いくつかの研究で、早期の年齢で手術を受けた患者では、晩期の年齢で手術を受けた患者よりも浸潤性または転移性MTCの発生率が有意に低いことが示されている。[ 148 ]最も侵攻性のM918T RET多様体を有する患者では、手術を4歳後に実施した場合、治癒は非常にまれである。[ 141 ][ 149 ]これらの知見を合わせると、早期にリスク低減のための手術を受けた患者の方が転帰が良好なことと一致している。[ 148 ][ 150 ][ 151 ]
疾患の生化学的証拠が得られる前(術前のカルシトニンが正常)に甲状腺切除を行うことで、疾患再発のリスクが低下しうるが、術後の生涯にわたるサーベイランスに対する選択的な戦略は、がんが存在するか否か、および微小髄様(micromedullary)がんまたは巨大髄様(macromedullary)がんのいずれであるかの病理学的な最終判定に依存する可能性がある。[ 1 ][ 152 ]甲状腺切除を受けたMEN2A患者で、最初は検知できなかったカルシトニンの基礎値および刺激後値(2pg/mL未満)が術後5~10年で陽性になったことを基に、10%に疾患の再発が認められたことを明らかにした研究が1件ある。[ 147 ]予防的手術後にカルシトニンの基礎値および刺激後値の継続的な上昇で評価される残存病変が認められた患者はわずか2%であった。[ 147 ]
MEN2の自然史に関する疑問は残る。多くの情報が得られるにつれて、甲状腺摘出術に最適な年齢に関する推奨内容や、遺伝的および生化学的スクリーニングの潜在的役割が変わる可能性がある。MTCの最も早い報告は、MEN2Bで3歳前、ATA-HまたはATA-MODのRET多様体を有するMEN2A症例で6歳前とされている。[ 141 ][ 147 ][ 149 ][ 153 ]これに対して、別の症例報告では、MEN2AのFMTCサブタイプを有する一部の家系における中年期以降でのがんの発生、およびがんを発症していないもののRET多様体遺伝子型を保有している高齢の血縁者でのがんの発生が報告された。[ 154 ]その後のデータによると、以前により緩慢性であると考えられていた一部のATA-MOD RET多様体は、ATA-H多様体と同様に侵攻性の可能性があるが、疾患の遅発と関連していることが示唆されている。[ 99 ]このような臨床的観察から、MEN2症候群の自然史が多岐にわたり、RET病原性多様体の種類、RET以外の遺伝子、行動因子、または環境曝露に関連する修飾作用に左右される可能性が示唆されている。
証拠レベル:4b
治療としての甲状腺切除
MTCの成人に対する標準的治療法は、後部カプセルを含めた甲状腺全体の外科的切除および中央区域リンパ節郭清である。[ 1 ]治療としてのcentral neck dissectionは、典型的にリンパ節転移の証拠が画像検査で得られている場合または血清中カルシトニン値が40pg/mLを上回る場合に実施される。[ 1 ]予防的なcentral neck dissectionの実施決定は、一般に患者の年齢、病原性多様体、同時性PHPTの存在、および副甲状腺の血管分布などの多くの因子に基づいてなされる。[ 1 ]予防的甲状腺摘除術および/またはcentral neck clearance時に脈管切除された副甲状腺の選択的自家移植が推奨される。選択的アプローチによっても副甲状腺機能低下症の有害な転帰が有意に減少する。[ 155 ]
MEN2BのRET多様体M918Tは、生後1年以内での約100%のMTC発生率と関連しており[ 149 ]、最も侵攻性のMEN2表現型と考えられる。MEN2Aの患者で、ATA-H高リスクのコドン634の病原性多様体は、コドン804、618、または620における病原性多様体よりも、浸潤性または転移性MTCおよび病変の持続または再発と関連する傾向がはるかに高い。[ 148 ]ATA-MOD分類の病原性多様体(コドン533、609、611、618、620、791、および804を含む)を有するリスクのある個人503人を対象とした1件のシリーズで、累積浸透率、MTC発症までの期間中央値、および術前カルシトニンの陽性適中率が報告された。[ 142 ]50歳までにMTCを発症するリスクは18~95%で、コドンによって異なり、コドン620で浸透率が最も高かった。MTCのほとんどの患者はリンパ節転移陰性であったことから、これらの病原性多様体で過去に報告されている比較的緩慢な疾患経過が確認された。術前のカルシトニン高値によりMTCの存在が強く予測されるが、多くのコドンで偽陰性率が比較的高い(MTCでは通常のカルシトニン値が低い)ことが注目された。この情報は、外科的切除の範囲について病原性多様体キャリアにカウンセリングを行う場合に有用である。
ATAは、局所または領域病変(遠隔転移の証拠がない場合)に対するコンパートメント方向のリンパ節郭清を以下の状況で推奨している:[ 1 ]
カルシトニン基礎値では術前にMTC患者をすべて特定できない可能性があるが、この検査は術後の寛解、リンパ節転移、および遠隔転移の予測因子として有用である。[ 156 ]単一施設の患者224人を対象としたある研究では、術前のカルシトニンの基礎値が500pg/mLを超えていれば、生化学的寛解を達成できないことが予測された。[ 156 ]この研究の著者らは、カルシトニン基礎値が40pg/mL(正常値は10pg/mL未満)でリンパ節転移が現れ始めることを明らかにした。リンパ節陽性の患者では、カルシトニン基礎値が150pg/mL~400pg/mLで遠隔転移が明らかになった。RET病原性多様体キャリア308人を対象とした別の研究では、術前のカルシトニン基礎値が正常であれば、リンパ節転移の存在が否定される(陰性適中率、100%)ことが判明した。[ 144 ]したがって、術前のカルシトニンの基礎値は有用な予後予測因子であり、外科的アプローチの指針に役立つ可能性がある。
甲状腺全摘術を受けた患者は、生涯にわたって甲状腺ホルモン補充療法が必要となる。薬の処方は年齢に依存し、治療は理想体重に基づいて開始してもよい。心疾患がない60歳以下の健康な成人では、妥当な開始用量として1.6~1.8μg/kgを1日1回投与する。[ 157 ]これ以上の年齢の患者では、甲状腺ホルモンの20~30%減量が必要な場合がある。[ 158 ]小児では成人より速くT4が代謝されるため、体重に応じて比較的多い補充が必要となる。小児の年齢に応じて、補充量は典型的に2~6μg/kgである。[ 159 ]しかしながら、抑制的治療よりも補充療法が好ましいことに注意することが重要である。C細胞腫瘍の増殖は甲状腺刺激ホルモン(TSH)に依存しないことから、MTC患者に対するT4補充療法では、TSHを正常基準範囲内に維持するよう調節してもよい。TSH値を正常基準範囲内に調整・維持し、残存する甲状腺組織のさらなる再増殖を避けるためには、サイログロブリンの測定も有用な場合がある。[ 160 ]
証拠レベル(central neck dissection):5
証拠レベル(ホルモン補充療法):3c
証拠レベル(治療としての甲状腺摘出術):4
補助療法
MTCに対して、化学療法および放射線療法は有効ではない。[ 4 ][ 161 ][ 162 ]RETの機能を遮断するためにマルチキナーゼ阻害薬を用いる分子標的療法がMTCの管理に使用されている。バンデタニブおよびカボザンチニブの使用は、手術に適さない進行した転移性MTCの成人患者に対して米国食品医薬品局により承認されている。第III相研究では、バンデタニブを投与された成人患者の方がプラセボを投与された成人患者より無増悪生存期間(PFS)が長かったことが明らかになった。[ 163 ]MEN2Bの小児を対象にした第I相/第II相研究では、バンデタニブにより47%の客観的部分奏効率が明らかにされた。[ 164 ]このコホートに関するその後のフォローアップ解析で、患者17人中10人に部分奏効がみられ;別の6人に安定がみられたことが明らかになった。PFS中央値は6.7年であった。[ 165 ]進行性MTC患者330人を対象にしてカボザンチニブとプラセボを比較した第III相二重盲検試験により、すべてのサブグループでPFS期間中央値の改善が示された。[ 166 ][ 167 ]この試験では、RETまたはRASなどの病原性変異をもつ患者は、両方の病原性多様体をもたない患者と比較して、PFSが長くなる可能性が高かった。[ 168 ]プロスペクティブ研究により、特定の病原性多様体を治療法の指針とすることができるか否かが、さらに明確になる可能性がある。カボザンチニブもバンデタニブも全生存を改善することは実証されていない。[ 163 ][ 166 ][ 167 ]重要なこととして、これらの薬物は一部のMEN2 RET多様体、特にコドン804における多様体[ 169 ]の阻害に有効ではないことから、RET阻害薬を用いる治療では遺伝子型が考慮すべき重要事項となっている。さらに、2018年の研究で、RETにおけるV804M変異の体細胞性獲得を介してこれらの薬物に対する抵抗性の発生が実証された。[ 170 ]最後に、マルチキナーゼ阻害薬は、おそらく他のキナーゼに対するオフターゲット作用のために、重大な毒性と関連している。[ 171 ]複数の臨床試験で、RETを標的にする他のマルチキナーゼ阻害薬が研究されている;しかしながら、こうした薬物がバンデタニブおよびカボザンチニブより優位である可能性はごくわずかであろう。こうした理由から、進行中のMTCにおける研究では、すべてのRET多様体の活性を阻害することが可能でオフターゲット作用の少ない選択的なRET阻害薬の開発および併用療法の使用に焦点が当てられている。今後の研究では、MTCを対象とした新しい標的療法の開発および併用療法の使用を中心に検討される可能性が高い。[ 172 ][ 173 ]RET転座を来したMTCおよび分化型甲状腺がん患者に対して、複数の臨床試験(ARROW[NCT03037385]およびLIBRETTO-001[NCT03157128])で新規のRET阻害薬が研究されている。
証拠レベル(バンデタニブ):2
証拠レベル(カボザンチニブ):1
(甲状腺がんの治療に関する詳しい情報については、甲状腺がんの治療[成人]に関するPDQ要約を参照のこと。)
MEN2関連PHEOの治療
MEN2患者におけるPHEOは、片側性の場合もあれば両側性の場合もある。腹腔鏡下副腎摘出術(腹部または背部)は、片側性PHEOの治療に対して適切な術前の内科的ブロック後の推奨されているアプローチである。[ 1 ][ 90 ][ 98 ][ 174 ]このリスク、有益性、および生命を脅かす副腎機能障害の可能性は、最初の手術計画時点で考慮すべきである。疾患が片側性とみられる場合は、17~72%の患者で対側副腎に異時性疾患が現れることがある。[ 5 ][ 175 ]1件のシリーズにおいて、片側性PHEOおよび肉眼的に正常な対側副腎をもつ23人の患者が最初に片側副腎摘出術による治療を受けた。[ 176 ]これらの患者の12人(52%)にPHEOが遺残腺内に発現したが、これは最初の手術後平均11.9年経過した後のことであった。追跡中に、高血圧クリーゼまたは診断未確定のPHEOによるその他の問題を経験した患者はなかった。これに対して、両側副腎摘出術を受けた43人の患者のうち10人(23%)が急性副腎皮質機能不全のエピソードを1つ以上経験した。したがって、片側副腎摘出術は、MEN2患者における片側性PHEOに対して妥当な管理戦略であるとみられている。[ 1 ][ 177 ][ 178 ]一見して片側性疾患に対する最初の手術でも皮質を残す手技を考慮することを提案している専門家が多い。[ 1 ][ 179 ](詳しい情報については、本要約の家族性PHEOおよび傍神経節腫症候群セクションの介入セクションを参照のこと。)対側副腎疾患のリスクのため、対側副腎における疾患の発現について定期的サーベイランス(血清中または尿中カテコールアミン測定)が推奨される。[ 1 ]
外科的アプローチに関して、背面からの後腹膜鏡下副腎摘出術の有用性を検討し、この手術が安全かつ有効なことを明らかにしたいくつか研究では、死亡率が非常に低く、軽度の合併症発生率が低い上に、開腹手術への変更が必要になることはまれであった。[ 175 ][ 180 ][ 181 ][ 182 ][ 183 ][ 184 ][ 185 ][ 186 ]
証拠レベル:4
副甲状腺機能亢進症の治療
MEN2に関連した副甲状腺疾患の患者のほとんどは、無症候性であるか、または甲状腺摘出術の術前の計画中あるいは甲状腺摘出術時に偶発的に診断される。典型的に、高カルシウム血症(存在する場合)は、カルシウムの尿への排泄増加および腎石症が随伴することがあるが、軽度である。結果として、外科的介入の適応は一般に散発性のPHPT患者に推奨されるそれと類似している。[ 90 ]一般に、カルシウム代謝における異常が見つかるときには4腺未満の副甲状腺が関連している。[ 1 ]
副甲状腺機能亢進症の治療では、典型的に異常な腺の外科的切除がある程度まで用いられる。1件の大規模シリーズの患者のうち89%で、手術により副甲状腺機能亢進症の治癒が達成された[ 59 ];しかしながら、この研究で切除術を受けた患者の22%に、術後、副甲状腺機能低下症が発現した。5人の患者(9%)が再発副甲状腺機能亢進症を引き起こした。このシリーズでは、利き腕ではない方の前腕への自己移植を伴う副甲状腺全摘術(術後に副甲状腺機能低下症を発症した患者11人中4人[36%])、甲状腺亜全摘術(副甲状腺機能低下症を発症した患者12人中6人[50%])、および肉眼的に肥大した腺のみの切除(副甲状腺機能低下症を発症した患者29人中3人[10%])といったさまざまな外科手術が使用された。これらのデータは、ほとんどの症例で肥大した副甲状腺のみの切除で十分であろうことを示している。
一部の研究者は、甲状腺手術時に同定される副甲状腺を配置すべき位置を決定するのにMEN2亜型を用いることを提案している。副甲状腺疾患のリスクが非常に低いMEN2B患者に関しては、副甲状腺を頸部にそのまま残すことができる。MTCに対する初回の外科治療中に副甲状腺が不注意に脈管切除されたMEN2Aの成人患者について、再移植を要する副甲状腺は利き腕ではない方の前腕に移植すべきであると提唱されている。このアプローチは、副甲状腺機能亢進症が発症または再発した場合に、頸部における追加の外科的介入の必要性を最小限に抑える。[ 1 ][ 155 ][ 187 ][ 188 ]小児については、過剰治療とその後の上皮小体欠損を回避するため、リスク/便益比を慎重に比較検討する必要がある。[ 189 ]残されたまたは自家移植された副甲状腺組織が機能していることを確認することが重要である。[ 1 ][ 98 ][ 190 ][ 191 ]
循環血中のカルシウム濃度に対する副甲状腺上のカルシウム感覚性受容体の感度を高めることで、循環血中のPTH濃度を減少させる薬剤であるカルシウム擬態薬の出現により、副甲状腺機能亢進症の薬物療法が人気を得ている。ランダム化二重盲検プラセボ対照試験では、PHPT患者において、シナカルセト塩酸の投与によりカルシウムおよびPTHの循環血中濃度に持続性の減少が認められたことが示された。[ 192 ]高リスクで手術の候補となる患者、再発性甲状腺機能亢進症の患者、または余命が限られている患者では、薬物療法が手術アプローチに対する実行可能な代替案となる可能性がある。[ 1 ]シナカルセトによる長期治療の結果は不明である。
証拠レベル:5
遺伝カウンセリング
遺伝様式
MEN2亜型はすべて常染色体優性で遺伝する。MEN2を有する人の子供がMEN2病原性多様体を受け継ぐリスクは50%である。しかし、MEN2を有する個人の中にはde novo病原性多様体を保有している人もいる;つまり彼らは前の世代までその家系に存在しなかった新たな病原性多様体を有しており、このため罹患者である親がいない。親が罹患者であるMEN2を有する人の割合は、それぞれの亜型によって異なる。
MEN2A:親が罹患者であるのは約95%である。MEN2Aの人の両親が、この疾患を発現していないかどうかを評価することは適切である。家族性ではない症例の5%では、de novoの病原性多様体または変異アレルの不完全浸透が考えられる。[ 193 ]
FMTC:多数の家族員が罹患する;そのため、罹患したすべての患者がその変異遺伝子を親から受け継いでいた。
MEN2B:罹患者の約50%にde novoのRET遺伝子の病原性多様体が認められ、50%には親から受け継いだMEN2B病原性多様体が認められる。[ 194 ][ 195 ]de novo病原性多様体の大部分は父方由来であるが、母方に由来するケースも報告されている。[ 196 ]
発端者の同胞:同胞へのリスクは親の遺伝的状態に依存し、これは家系解析および/またはDNAベースの検査によって明らかにできる。de novo病原性多様体が明らかな場合でも、見かけ上罹患していない親が生殖細胞にモザイクを有する可能性を検討する必要があるが、そのようなケースはこれまでに報告されていない。
着床前遺伝子検査に対する意識
1件の研究により、MEN1およびMEN2を有する個人の着床前遺伝子検査(PGT)に対する意識が調査された。[ 197 ]MEN1でMEN1病原性多様体を有するか、MEN2でRET病原性多様体を有する米国の単一施設からのクリニックベースの患者91人が調査された。この研究により、MEN1患者の30%(33人中10人)およびMEN2患者の37%(57人中21人)がPGTを認識していた;MEN1患者の82%(33人中27人)およびMEN2患者の61%(56人中34人)がPGTを提案されるべきであると考えていた;MEN1患者の61%(31人中19人)およびMEN2患者の43%(54人中23人)がPGTを検討することが明らかにされた。
心理社会的問題
RET病原性多様体に対する遺伝子検査が及ぼす心理社会的影響は広範囲にわたっては研究されていない。サンプルサイズが小さかったり、集団が不均一であったりするなど、公表済みの研究は限界がある;したがって、これらの知見の臨床的妥当性の解釈は慎重に行うべきである。病原性多様体のキャリアと同定されると、自尊心、家族関係、生活の質に影響が出る可能性がある。[ 198 ]加えて、遺伝性疾患についての誤った考えは、家族内に非難および罪悪感をもたらすことがある。[ 199 ][ 200 ]数件の総説に、特に小児の遺伝子検査に関係する医学的および心理学的問題がまとめられている。[ 201 ][ 202 ][ 203 ][ 204 ]早期スクリーニングおよびリスク低減のための治療のもつ医学的意義と、その個人の意思決定における自律性の喪失が対比されている。遺伝子検査および外科手術の重要性およびタイミングについて両親の意見が一致していないと、家族内に情緒的問題が発生するきっかけになりかねない。
ある研究では、血液サンプルを提出してから遺伝子検査の結果を受け取るまでの期間における心理的苦悩のレベルを調べた。最大の苦悩レベルを経験した集団は、25歳未満の独身で、不安を伴う辛い状況に対処した経験がある人たちであった。[ 205 ]子供が陰性の検査結果を受け取った病原性多様体陽性の両親は、安心した様子ではなく、DNA検査の信頼性に疑問をもち、非キャリアである子供のスクリーニングの継続を強く希望した。[ 206 ]
ある小規模の定性研究(N = 21)では、MEN2Aを有する患者と家族が、生涯にわたる高リスクサーベイランスへの参加をどのように概念化するのかを評価した。[ 207 ]進行中のサーベイランスは、健康への脅威を思い出させるものとしてみることができる。生涯にわたるサーベイランスを受け入れルーチンの健康管理へ組み入れることは、この状況のもつ裏の意味とうまく共生していくことにおいて、必要不可欠なことであった。がんの遺伝的素因についての懸念は、サーベイランスについての懸念の周辺的なものである。インターネット討論などの支持的介入は、生涯にわたるサーベイランスを受けるMTC患者に対する情報提供とサポートの必要性に対処するための持続的な手段として機能しうる。[ 208 ]
参考文献- Wells SA, Asa SL, Dralle H, et al.: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25 (6): 567-610, 2015.[PUBMED Abstract]
- Kaserer K, Scheuba C, Neuhold N, et al.: Sporadic versus familial medullary thyroid microcarcinoma: a histopathologic study of 50 consecutive patients. Am J Surg Pathol 25 (10): 1245-51, 2001.[PUBMED Abstract]
- Robbins J, Merino MJ, Boice JD, et al.: Thyroid cancer: a lethal endocrine neoplasm. Ann Intern Med 115 (2): 133-47, 1991.[PUBMED Abstract]
- Moley JF, Debenedetti MK, Dilley WG, et al.: Surgical management of patients with persistent or recurrent medullary thyroid cancer. J Intern Med 243 (6): 521-6, 1998.[PUBMED Abstract]
- Thosani S, Ayala-Ramirez M, Palmer L, et al.: The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab 98 (11): E1813-9, 2013.[PUBMED Abstract]
- Machens A, Lorenz K, Dralle H: Constitutive RET tyrosine kinase activation in hereditary medullary thyroid cancer: clinical opportunities. J Intern Med 266 (1): 114-25, 2009.[PUBMED Abstract]
- DeLellis RA, Lloyd RV, Heitz PU, et al., eds.: Pathology and Genetics of Tumours of Endocrine Organs. Lyon, France: IARC Press, 2004. World Health Organization classification of tumours, vol. 8.[PUBMED Abstract]
- Guyétant S, Rousselet MC, Durigon M, et al.: Sex-related C cell hyperplasia in the normal human thyroid: a quantitative autopsy study. J Clin Endocrinol Metab 82 (1): 42-7, 1997.[PUBMED Abstract]
- LiVolsi VA: C cell hyperplasia/neoplasia. J Clin Endocrinol Metab 82 (1): 39-41, 1997.[PUBMED Abstract]
- Mete O, Asa SL: Precursor lesions of endocrine system neoplasms. Pathology 45 (3): 316-30, 2013.[PUBMED Abstract]
- Elisei R, Bottici V, Luchetti F, et al.: Impact of routine measurement of serum calcitonin on the diagnosis and outcome of medullary thyroid cancer: experience in 10,864 patients with nodular thyroid disorders. J Clin Endocrinol Metab 89 (1): 163-8, 2004.[PUBMED Abstract]
- Landsvater RM, Rombouts AG, te Meerman GJ, et al.: The clinical implications of a positive calcitonin test for C-cell hyperplasia in genetically unaffected members of an MEN2A kindred. Am J Hum Genet 52 (2): 335-42, 1993.[PUBMED Abstract]
- Lips CJ, Landsvater RM, Höppener JW, et al.: Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med 331 (13): 828-35, 1994.[PUBMED Abstract]
- Kudo T, Miyauchi A, Ito Y, et al.: Serum calcitonin levels with calcium loading tests before and after total thyroidectomy in patients with thyroid diseases other than medullary thyroid carcinoma. Endocr J 58 (3): 217-21, 2011.[PUBMED Abstract]
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2017. Bethesda, Md: National Cancer Institute, 2020. Available online. Last accessed May 21, 2020.[PUBMED Abstract]
- Gharib H, McConahey WM, Tiegs RD, et al.: Medullary thyroid carcinoma: clinicopathologic features and long-term follow-up of 65 patients treated during 1946 through 1970. Mayo Clin Proc 67 (10): 934-40, 1992.[PUBMED Abstract]
- Ponder BA: Multiple endocrine neoplasia type 2. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. New York, NY: McGraw-Hill, 2002, pp 501-513.[PUBMED Abstract]
- Decker RA, Peacock ML, Borst MJ, et al.: Progress in genetic screening of multiple endocrine neoplasia type 2A: is calcitonin testing obsolete? Surgery 118 (2): 257-63; discussion 263-4, 1995.[PUBMED Abstract]
- Kitamura Y, Goodfellow PJ, Shimizu K, et al.: Novel germline RET proto-oncogene mutations associated with medullary thyroid carcinoma (MTC): mutation analysis in Japanese patients with MTC. Oncogene 14 (25): 3103-6, 1997.[PUBMED Abstract]
- Eng C, Mulligan LM, Smith DP, et al.: Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf) 43 (1): 123-7, 1995.[PUBMED Abstract]
- Wohllk N, Cote GJ, Bugalho MM, et al.: Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 81 (10): 3740-5, 1996.[PUBMED Abstract]
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Thyroid Carcinoma. Version 2.2019. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2019. Available online with free subscription. Last accessed June 08, 2020.[PUBMED Abstract]
- American Cancer Society: Cancer Facts and Figures 2020. Atlanta, Ga: American Cancer Society, 2020. Available online. Last accessed May 12, 2020.[PUBMED Abstract]
- Randle RW, Balentine CJ, Leverson GE, et al.: Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 161 (1): 137-146, 2017.[PUBMED Abstract]
- Hundahl SA, Fleming ID, Fremgen AM, et al.: A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see comments] Cancer 83 (12): 2638-48, 1998.[PUBMED Abstract]
- Modigliani E, Vasen HM, Raue K, et al.: Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The Euromen Study Group. J Intern Med 238 (4): 363-7, 1995.[PUBMED Abstract]
- Bhattacharyya N: A population-based analysis of survival factors in differentiated and medullary thyroid carcinoma. Otolaryngol Head Neck Surg 128 (1): 115-23, 2003.[PUBMED Abstract]
- Roman S, Lin R, Sosa JA: Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 107 (9): 2134-42, 2006.[PUBMED Abstract]
- Bergholm U, Bergström R, Ekbom A: Long-term follow-up of patients with medullary carcinoma of the thyroid. Cancer 79 (1): 132-8, 1997.[PUBMED Abstract]
- Kebebew E, Ituarte PH, Siperstein AE, et al.: Medullary thyroid carcinoma: clinical characteristics, treatment, prognostic factors, and a comparison of staging systems. Cancer 88 (5): 1139-48, 2000.[PUBMED Abstract]
- Romei C, Ciampi R, Elisei R: A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol 12 (4): 192-202, 2016.[PUBMED Abstract]
- Lenders JW, Pacak K, Walther MM, et al.: Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287 (11): 1427-34, 2002.[PUBMED Abstract]
- Gerlo EA, Sevens C: Urinary and plasma catecholamines and urinary catecholamine metabolites in pheochromocytoma: diagnostic value in 19 cases. Clin Chem 40 (2): 250-6, 1994.[PUBMED Abstract]
- Guller U, Turek J, Eubanks S, et al.: Detecting pheochromocytoma: defining the most sensitive test. Ann Surg 243 (1): 102-7, 2006.[PUBMED Abstract]
- Raber W, Raffesberg W, Bischof M, et al.: Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med 160 (19): 2957-63, 2000.[PUBMED Abstract]
- Sawka AM, Jaeschke R, Singh RJ, et al.: A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 88 (2): 553-8, 2003.[PUBMED Abstract]
- Unger N, Pitt C, Schmidt IL, et al.: Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol 154 (3): 409-17, 2006.[PUBMED Abstract]
- Pacak K, Eisenhofer G, Ahlman H, et al.: Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 3 (2): 92-102, 2007.[PUBMED Abstract]
- van der Harst E, de Herder WW, Bruining HA, et al.: [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. J Clin Endocrinol Metab 86 (2): 685-93, 2001.[PUBMED Abstract]
- Pacak K, Linehan WM, Eisenhofer G, et al.: Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 134 (4): 315-29, 2001.[PUBMED Abstract]
- Kaelin WG: Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2 (9): 673-82, 2002.[PUBMED Abstract]
- Maher ER, Eng C: The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet 11 (20): 2347-54, 2002.[PUBMED Abstract]
- Fraser WD: Hyperparathyroidism. Lancet 374 (9684): 145-58, 2009.[PUBMED Abstract]
- Tonelli F, Marcucci T, Giudici F, et al.: Surgical approach in hereditary hyperparathyroidism. Endocr J 56 (7): 827-41, 2009.[PUBMED Abstract]
- Villablanca A, Calender A, Forsberg L, et al.: Germline and de novo mutations in the HRPT2 tumour suppressor gene in familial isolated hyperparathyroidism (FIHP). J Med Genet 41 (3): e32, 2004.[PUBMED Abstract]
- Marx SJ, Simonds WF, Agarwal SK, et al.: Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res 17 (Suppl 2): N37-43, 2002.[PUBMED Abstract]
- Sipple JH: The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med 31 (1): 163-166, 1961.[PUBMED Abstract]
- Eng C, Clayton D, Schuffenecker I, et al.: The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276 (19): 1575-9, 1996.[PUBMED Abstract]
- Sanso GE, Domene HM, Garcia R, et al.: Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer 94 (2): 323-30, 2002.[PUBMED Abstract]
- Yip L, Cote GJ, Shapiro SE, et al.: Multiple endocrine neoplasia type 2: evaluation of the genotype-phenotype relationship. Arch Surg 138 (4): 409-16; discussion 416, 2003.[PUBMED Abstract]
- Rambaud JC, Jian R, Flourié B, et al.: Pathophysiological study of diarrhoea in a patient with medullary thyroid carcinoma. Evidence against a secretory mechanism and for the role of shortened colonic transit time. Gut 29 (4): 537-43, 1988.[PUBMED Abstract]
- Cox TM, Fagan EA, Hillyard CJ, et al.: Rôle of calcitonin in diarrhoea associated with medullary carcinoma of the thyroid. Gut 20 (7): 629-33, 1979.[PUBMED Abstract]
- Raue F, Frank-Raue K, Grauer A: Multiple endocrine neoplasia type 2. Clinical features and screening. Endocrinol Metab Clin North Am 23 (1): 137-56, 1994.[PUBMED Abstract]
- Perren A, Komminoth P: Familial pheochromocytomas and paragangliomas: stories from the sign-out room. Endocr Pathol 17 (4): 337-44, 2006.[PUBMED Abstract]
- Webb TA, Sheps SG, Carney JA: Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2. Am J Surg Pathol 4 (2): 121-6, 1980.[PUBMED Abstract]
- Lips KJ, Van der Sluys Veer J, Struyvenberg A, et al.: Bilateral occurrence of pheochromocytoma in patients with the multiple endocrine neoplasia syndrome type 2A (Sipple's syndrome). Am J Med 70 (5): 1051-60, 1981.[PUBMED Abstract]
- Neumann HP, Berger DP, Sigmund G, et al.: Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med 329 (21): 1531-8, 1993.[PUBMED Abstract]
- Conte-Devolx B, Schuffenecker I, Niccoli P, et al.: Multiple endocrine neoplasia type 2: management of patients and subjects at risk. French Study Group on Calcitonin-Secreting Tumors (GETC). Horm Res 47 (4-6): 221-6, 1997.[PUBMED Abstract]
- Kraimps JL, Denizot A, Carnaille B, et al.: Primary hyperparathyroidism in multiple endocrine neoplasia type IIa: retrospective French multicentric study. Groupe d'Etude des Tumeurs á Calcitonine (GETC, French Calcitonin Tumors Study Group), French Association of Endocrine Surgeons. World J Surg 20 (7): 808-12; discussion 812-3, 1996.[PUBMED Abstract]
- Benson L, Ljunghall S, Akerström G, et al.: Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. Am J Med 82 (4): 731-7, 1987.[PUBMED Abstract]
- Trump D, Farren B, Wooding C, et al.: Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM 89 (9): 653-69, 1996.[PUBMED Abstract]
- Vasen HF, Lamers CB, Lips CJ: Screening for the multiple endocrine neoplasia syndrome type I. A study of 11 kindreds in The Netherlands. Arch Intern Med 149 (12): 2717-22, 1989.[PUBMED Abstract]
- Bugalho MJ, Limbert E, Sobrinho LG, et al.: A kindred with multiple endocrine neoplasia type 2A associated with pruritic skin lesions. Cancer 70 (11): 2664-7, 1992.[PUBMED Abstract]
- Robinson MF, Furst EJ, Nunziata V, et al.: Characterization of the clinical features of five families with hereditary primary cutaneous lichen amyloidosis and multiple endocrine neoplasia type 2. Henry Ford Hosp Med J 40 (3-4): 249-52, 1992.[PUBMED Abstract]
- Romeo G, Ceccherini I, Celli J, et al.: Association of multiple endocrine neoplasia type 2 and Hirschsprung disease. J Intern Med 243 (6): 515-20, 1998.[PUBMED Abstract]
- Mulligan LM, Eng C, Attié T, et al.: Diverse phenotypes associated with exon 10 mutations of the RET proto-oncogene. Hum Mol Genet 3 (12): 2163-7, 1994.[PUBMED Abstract]
- Decker RA, Peacock ML, Watson P: Hirschsprung disease in MEN 2A: increased spectrum of RET exon 10 genotypes and strong genotype-phenotype correlation. Hum Mol Genet 7 (1): 129-34, 1998.[PUBMED Abstract]
- Carrasquillo MM, McCallion AS, Puffenberger EG, et al.: Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet 32 (2): 237-44, 2002.[PUBMED Abstract]
- Fewtrell MS, Tam PK, Thomson AH, et al.: Hirschsprung's disease associated with a deletion of chromosome 10 (q11.2q21.2): a further link with the neurocristopathies? J Med Genet 31 (4): 325-7, 1994.[PUBMED Abstract]
- Pacini F, Castagna MG, Cipri C, et al.: Medullary thyroid carcinoma. Clin Oncol (R Coll Radiol) 22 (6): 475-85, 2010.[PUBMED Abstract]
- Brauckhoff M, Machens A, Hess S, et al.: Premonitory symptoms preceding metastatic medullary thyroid cancer in MEN 2B: An exploratory analysis. Surgery 144 (6): 1044-50; discussion 1050-3, 2008.[PUBMED Abstract]
- Castinetti F, Moley J, Mulligan L, et al.: A comprehensive review on MEN2B. Endocr Relat Cancer 25 (2): T29-T39, 2018.[PUBMED Abstract]
- Gorlin RJ, Sedano HO, Vickers RA, et al.: Multiple mucosal neuromas, pheochromocytoma and medullary carcinoma of the thyroid--a syndrome. Cancer 22 (2): 293-9 passim, 1968.[PUBMED Abstract]
- Gorlin RJ, Vickers RA: Multiple mucosal neuromas, pheochromocytoma, medullary carcinoma of the thyroid and marfanoid body build with muscle wasting: reexamination of a syndrome of neural crest malmigration. Birth Defects Orig Artic Ser 7 (6): 69-72, 1971.[PUBMED Abstract]
- Makri A, Akshintala S, Derse-Anthony C, et al.: Multiple Endocrine Neoplasia Type 2B Presents Early in Childhood but Often Is Undiagnosed for Years. J Pediatr 203: 447-449, 2018.[PUBMED Abstract]
- Skinner MA, DeBenedetti MK, Moley JF, et al.: Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg 31 (1): 177-81; discussion 181-2, 1996.[PUBMED Abstract]
- Wells SA, Pacini F, Robinson BG, et al.: Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab 98 (8): 3149-64, 2013.[PUBMED Abstract]
- Gfroerer S, Theilen TM, Fiegel H, et al.: Identification of intestinal ganglioneuromatosis leads to early diagnosis of MEN2B: role of rectal biopsy. J Pediatr Surg 52 (7): 1161-1165, 2017.[PUBMED Abstract]
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.[PUBMED Abstract]
- Gardner E, Papi L, Easton DF, et al.: Genetic linkage studies map the multiple endocrine neoplasia type 2 loci to a small interval on chromosome 10q11.2. Hum Mol Genet 2 (3): 241-6, 1993.[PUBMED Abstract]
- Mole SE, Mulligan LM, Healey CS, et al.: Localisation of the gene for multiple endocrine neoplasia type 2A to a 480 kb region in chromosome band 10q11.2. Hum Mol Genet 2 (3): 247-52, 1993.[PUBMED Abstract]
- Takahashi M, Ritz J, Cooper GM: Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42 (2): 581-8, 1985.[PUBMED Abstract]
- Kwok JB, Gardner E, Warner JP, et al.: Structural analysis of the human ret proto-oncogene using exon trapping. Oncogene 8 (9): 2575-82, 1993.[PUBMED Abstract]
- Myers SM, Eng C, Ponder BA, et al.: Characterization of RET proto-oncogene 3' splicing variants and polyadenylation sites: a novel C-terminus for RET. Oncogene 11 (10): 2039-45, 1995.[PUBMED Abstract]
- Airaksinen MS, Saarma M: The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3 (5): 383-94, 2002.[PUBMED Abstract]
- Mulligan LM: RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 14 (3): 173-86, 2014.[PUBMED Abstract]
- Robson ME, Bradbury AR, Arun B, et al.: American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J Clin Oncol 33 (31): 3660-7, 2015.[PUBMED Abstract]
- Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.[PUBMED Abstract]
- Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. American Society of Human Genetics Board of Directors, American College of Medical Genetics Board of Directors. Am J Hum Genet 57 (5): 1233-41, 1995.[PUBMED Abstract]
- Brandi ML, Gagel RF, Angeli A, et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86 (12): 5658-71, 2001.[PUBMED Abstract]
- Cooper DS, Doherty GM, Haugen BR, et al.: Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 19 (11): 1167-214, 2009.[PUBMED Abstract]
- Wells SA: Advances in the management of MEN2: from improved surgical and medical treatment to novel kinase inhibitors. Endocr Relat Cancer 25 (2): T1-T13, 2018.[PUBMED Abstract]
- Romei C, Cosci B, Renzini G, et al.: RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin Endocrinol (Oxf) 74 (2): 241-7, 2011.[PUBMED Abstract]
- Elisei R, Cosci B, Romei C, et al.: Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab 93 (3): 682-7, 2008.[PUBMED Abstract]
- Sarika HL, Papathoma A, Garofalaki M, et al.: Genetic screening of patients with medullary thyroid cancer in a referral center in Greece during the past two decades. Eur J Endocrinol 172 (4): 501-9, 2015.[PUBMED Abstract]
- Mathiesen JS, Kroustrup JP, Vestergaard P, et al.: Distribution of RET Mutations in Multiple Endocrine Neoplasia 2 in Denmark 1994-2014: A Nationwide Study. Thyroid 27 (2): 215-223, 2017.[PUBMED Abstract]
- Kouvaraki MA, Shapiro SE, Perrier ND, et al.: RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 15 (6): 531-44, 2005.[PUBMED Abstract]
- Kloos RT, Eng C, Evans DB, et al.: Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19 (6): 565-612, 2009.[PUBMED Abstract]
- Voss RK, Feng L, Lee JE, et al.: Medullary Thyroid Carcinoma in MEN2A: ATA Moderate- or High-Risk RET Mutations Do Not Predict Disease Aggressiveness. J Clin Endocrinol Metab 102 (8): 2807-2813, 2017.[PUBMED Abstract]
- Mathiesen JS, Habra MA, Bassett JHD, et al.: Risk Profile of the RET A883F Germline Mutation: An International Collaborative Study. J Clin Endocrinol Metab 102 (6): 2069-2074, 2017.[PUBMED Abstract]
- Eng C, Smith DP, Mulligan LM, et al.: Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 3 (2): 237-41, 1994.[PUBMED Abstract]
- Hofstra RM, Landsvater RM, Ceccherini I, et al.: A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 367 (6461): 375-6, 1994.[PUBMED Abstract]
- Carlson KM, Dou S, Chi D, et al.: Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci U S A 91 (4): 1579-83, 1994.[PUBMED Abstract]
- Gimm O, Marsh DJ, Andrew SD, et al.: Germline dinucleotide mutation in codon 883 of the RET proto-oncogene in multiple endocrine neoplasia type 2B without codon 918 mutation. J Clin Endocrinol Metab 82 (11): 3902-4, 1997.[PUBMED Abstract]
- Smith DP, Houghton C, Ponder BA: Germline mutation of RET codon 883 in two cases of de novo MEN 2B. Oncogene 15 (10): 1213-7, 1997.[PUBMED Abstract]
- Eng C, Mulligan LM, Healey CS, et al.: Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Res 56 (9): 2167-70, 1996.[PUBMED Abstract]
- Cranston AN, Carniti C, Oakhill K, et al.: RET is constitutively activated by novel tandem mutations that alter the active site resulting in multiple endocrine neoplasia type 2B. Cancer Res 66 (20): 10179-87, 2006.[PUBMED Abstract]
- Miyauchi A, Futami H, Hai N, et al.: Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn J Cancer Res 90 (1): 1-5, 1999.[PUBMED Abstract]
- Kameyama K, Okinaga H, Takami H: RET oncogene mutations in 75 cases of familial medullary thyroid carcinoma in Japan. Biomed Pharmacother 58 (6-7): 345-7, 2004 Jul-Aug.[PUBMED Abstract]
- Iwashita T, Murakami H, Kurokawa K, et al.: A two-hit model for development of multiple endocrine neoplasia type 2B by RET mutations. Biochem Biophys Res Commun 268 (3): 804-8, 2000.[PUBMED Abstract]
- Menko FH, van der Luijt RB, de Valk IA, et al.: Atypical MEN type 2B associated with two germline RET mutations on the same allele not involving codon 918. J Clin Endocrinol Metab 87 (1): 393-7, 2002.[PUBMED Abstract]
- Mulligan LM, Eng C, Healey CS, et al.: Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 6 (1): 70-4, 1994.[PUBMED Abstract]
- Seri M, Celli I, Betsos N, et al.: A Cys634Gly substitution of the RET proto-oncogene in a family with recurrence of multiple endocrine neoplasia type 2A and cutaneous lichen amyloidosis. Clin Genet 51 (2): 86-90, 1997.[PUBMED Abstract]
- Rothberg AE, Raymond VM, Gruber SB, et al.: Familial medullary thyroid carcinoma associated with cutaneous lichen amyloidosis. Thyroid 19 (6): 651-5, 2009.[PUBMED Abstract]
- Quayle FJ, Fialkowski EA, Benveniste R, et al.: Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery 142 (6): 800-5; discussion 805.e1, 2007.[PUBMED Abstract]
- Bolino A, Schuffenecker I, Luo Y, et al.: RET mutations in exons 13 and 14 of FMTC patients. Oncogene 10 (12): 2415-9, 1995.[PUBMED Abstract]
- Boccia LM, Green JS, Joyce C, et al.: Mutation of RET codon 768 is associated with the FMTC phenotype. Clin Genet 51 (2): 81-5, 1997.[PUBMED Abstract]
- Lesueur F, Cebrian A, Cranston A, et al.: Germline homozygous mutations at codon 804 in the RET protooncogene in medullary thyroid carcinoma/multiple endocrine neoplasia type 2A patients. J Clin Endocrinol Metab 90 (6): 3454-7, 2005.[PUBMED Abstract]
- Shannon KE, Gimm O, Hinze R: Germline V804M mutation in the RET protooncogene in 2 apparently sporadic cases of MTC presenting in the 7th decade of life. The Journal of Endocrine Genetics 1 (1): 39-46, 1999.[PUBMED Abstract]
- Raue F, Frank-Raue K: Genotype-phenotype relationship in multiple endocrine neoplasia type 2. Implications for clinical management. Hormones (Athens) 8 (1): 23-8, 2009 Jan-Mar.[PUBMED Abstract]
- Mulligan LM, Marsh DJ, Robinson BG, et al.: Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. J Intern Med 238 (4): 343-6, 1995.[PUBMED Abstract]
- Moers AM, Landsvater RM, Schaap C, et al.: Familial medullary thyroid carcinoma: not a distinct entity? Genotype-phenotype correlation in a large family. Am J Med 101 (6): 635-41, 1996.[PUBMED Abstract]
- Niccoli-Sire P, Murat A, Rohmer V, et al.: Familial medullary thyroid carcinoma with noncysteine ret mutations: phenotype-genotype relationship in a large series of patients. J Clin Endocrinol Metab 86 (8): 3746-53, 2001.[PUBMED Abstract]
- Machens A, Ukkat J, Brauckhoff M, et al.: Advances in the management of hereditary medullary thyroid cancer. J Intern Med 257 (1): 50-9, 2005.[PUBMED Abstract]
- Mukherjee S, Zakalik D: RET codon 804 mutations in multiple endocrine neoplasia 2: genotype-phenotype correlations and implications in clinical management. Clin Genet 79 (1): 1-16, 2011.[PUBMED Abstract]
- Xu JY, Grubbs EG, Waguespack SG, et al.: Medullary Thyroid Carcinoma Associated with Germline RETK666N Mutation. Thyroid 26 (12): 1744-1751, 2016.[PUBMED Abstract]
- Erlic Z, Hoffmann MM, Sullivan M, et al.: Pathogenicity of DNA variants and double mutations in multiple endocrine neoplasia type 2 and von Hippel-Lindau syndrome. J Clin Endocrinol Metab 95 (1): 308-13, 2010.[PUBMED Abstract]
- Toledo RA, Hatakana R, Lourenço DM, et al.: Comprehensive assessment of the disputed RET Y791F variant shows no association with medullary thyroid carcinoma susceptibility. Endocr Relat Cancer 22 (1): 65-76, 2015.[PUBMED Abstract]
- Høxbroe Michaelsen S, Ornstrup MJ, Poulsen MM, et al.: Long-term follow-up of RET Y791F carriers in Denmark 1994-2017: A National Cohort Study. J Surg Oncol 119 (6): 687-693, 2019.[PUBMED Abstract]
- Siqueira DR, Ceolin L, Ferreira CV, et al.: Role of RET genetic variants in MEN2-associated pheochromocytoma. Eur J Endocrinol 170 (6): 821-8, 2014.[PUBMED Abstract]
- Ceolin L, Siqueira DR, Romitti M, et al.: Molecular basis of medullary thyroid carcinoma: the role of RET polymorphisms. Int J Mol Sci 13 (1): 221-39, 2012.[PUBMED Abstract]
- Robledo M, Gil L, Pollán M, et al.: Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res 63 (8): 1814-7, 2003.[PUBMED Abstract]
- Kaczmarek-Ryś M, Ziemnicka K, Pławski A, et al.: Modifying impact of RET gene haplotypes on medullary thyroid carcinoma clinical course. Endocr Relat Cancer 25 (4): 421-436, 2018.[PUBMED Abstract]
- Margraf RL, Crockett DK, Krautscheid PM, et al.: Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30 (4): 548-56, 2009.[PUBMED Abstract]
- Wells SA, Donis-Keller H: Current perspectives on the diagnosis and management of patients with multiple endocrine neoplasia type 2 syndromes. Endocrinol Metab Clin North Am 23 (1): 215-28, 1994.[PUBMED Abstract]
- Gardet V, Gatta B, Simonnet G, et al.: Lessons from an unpleasant surprise: a biochemical strategy for the diagnosis of pheochromocytoma. J Hypertens 19 (6): 1029-35, 2001.[PUBMED Abstract]
- Pacak K, Ilias I, Adams KT, et al.: Biochemical diagnosis, localization and management of pheochromocytoma: focus on multiple endocrine neoplasia type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. J Intern Med 257 (1): 60-8, 2005.[PUBMED Abstract]
- Raue F, Kraimps JL, Dralle H, et al.: Primary hyperparathyroidism in multiple endocrine neoplasia type 2A. J Intern Med 238 (4): 369-73, 1995.[PUBMED Abstract]
- Milos IN, Frank-Raue K, Wohllk N, et al.: Age-related neoplastic risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germ line RET Cys634Trp (TGC>TGG) mutation. Endocr Relat Cancer 15 (4): 1035-41, 2008.[PUBMED Abstract]
- Marsh DJ, McDowall D, Hyland VJ, et al.: The identification of false positive responses to the pentagastrin stimulation test in RET mutation negative members of MEN 2A families. Clin Endocrinol (Oxf) 44 (2): 213-20, 1996.[PUBMED Abstract]
- Elisei R, Romei C, Renzini G, et al.: The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab 97 (2): 426-35, 2012.[PUBMED Abstract]
- Rich TA, Feng L, Busaidy N, et al.: Prevalence by age and predictors of medullary thyroid cancer in patients with lower risk germline RET proto-oncogene mutations. Thyroid 24 (7): 1096-106, 2014.[PUBMED Abstract]
- Qi XP, Zhao JQ, Du ZF, et al.: Prophylactic thyroidectomy for MEN 2-related medullary thyroid carcinoma based on predictive testing for RET proto-oncogene mutation and basal serum calcitonin in China. Eur J Surg Oncol 39 (9): 1007-12, 2013.[PUBMED Abstract]
- Machens A, Lorenz K, Dralle H: Individualization of lymph node dissection in RET (rearranged during transfection) carriers at risk for medullary thyroid cancer: value of pretherapeutic calcitonin levels. Ann Surg 250 (2): 305-10, 2009.[PUBMED Abstract]
- Castagna MG, Fugazzola L, Maino F, et al.: Reference range of serum calcitonin in pediatric population. J Clin Endocrinol Metab 100 (5): 1780-4, 2015.[PUBMED Abstract]
- Schreinemakers JM, Vriens MR, Valk GD, et al.: Factors predicting outcome of total thyroidectomy in young patients with multiple endocrine neoplasia type 2: a nationwide long-term follow-up study. World J Surg 34 (4): 852-60, 2010.[PUBMED Abstract]
- Skinner MA, Moley JA, Dilley WG, et al.: Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 353 (11): 1105-13, 2005.[PUBMED Abstract]
- Szinnai G, Meier C, Komminoth P, et al.: Review of multiple endocrine neoplasia type 2A in children: therapeutic results of early thyroidectomy and prognostic value of codon analysis. Pediatrics 111 (2): E132-9, 2003.[PUBMED Abstract]
- Brauckhoff M, Machens A, Lorenz K, et al.: Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: a changing perspective. Ann Surg 259 (4): 800-6, 2014.[PUBMED Abstract]
- Niccoli-Sire P, Murat A, Baudin E, et al.: Early or prophylactic thyroidectomy in MEN 2/FMTC gene carriers: results in 71 thyroidectomized patients. The French Calcitonin Tumours Study Group (GETC). Eur J Endocrinol 141 (5): 468-74, 1999.[PUBMED Abstract]
- Wells SA, Skinner MA: Prophylactic thyroidectomy, based on direct genetic testing, in patients at risk for the multiple endocrine neoplasia type 2 syndromes. Exp Clin Endocrinol Diabetes 106 (1): 29-34, 1998.[PUBMED Abstract]
- Franc S, Niccoli-Sire P, Cohen R, et al.: Complete surgical lymph node resection does not prevent authentic recurrences of medullary thyroid carcinoma. Clin Endocrinol (Oxf) 55 (3): 403-9, 2001.[PUBMED Abstract]
- Zenaty D, Aigrain Y, Peuchmaur M, et al.: Medullary thyroid carcinoma identified within the first year of life in children with hereditary multiple endocrine neoplasia type 2A (codon 634) and 2B. Eur J Endocrinol 160 (5): 807-13, 2009.[PUBMED Abstract]
- Hansen HS, Torring H, Godballe C, et al.: Is thyroidectomy necessary in RET mutations carriers of the familial medullary thyroid carcinoma syndrome? Cancer 89 (4): 863-7, 2000.[PUBMED Abstract]
- Moley JF, Skinner M, Gillanders WE, et al.: Management of the Parathyroid Glands During Preventive Thyroidectomy in Patients With Multiple Endocrine Neoplasia Type 2. Ann Surg 262 (4): 641-6, 2015.[PUBMED Abstract]
- Machens A, Schneyer U, Holzhausen HJ, et al.: Prospects of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab 90 (4): 2029-34, 2005.[PUBMED Abstract]
- Razvi S, Hostalek U: Therapeutic challenges in the application of serum thyroid stimulating hormone testing in the management of patients with hypothyroidism on replacement thyroid hormone therapy: a review. Curr Med Res Opin 35 (7): 1215-1220, 2019.[PUBMED Abstract]
- Sawin CT, Geller A, Hershman JM, et al.: The aging thyroid. The use of thyroid hormone in older persons. JAMA 261 (18): 2653-5, 1989.[PUBMED Abstract]
- Baloch Z, Carayon P, Conte-Devolx B, et al.: Laboratory medicine practice guidelines. Laboratory support for the diagnosis and monitoring of thyroid disease. Thyroid 13 (1): 3-126, 2003.[PUBMED Abstract]
- Seib CD, Harari A, Conte FA, et al.: Utility of serum thyroglobulin measurements after prophylactic thyroidectomy in patients with hereditary medullary thyroid cancer. Surgery 156 (2): 394-8, 2014.[PUBMED Abstract]
- Samaan NA, Schultz PN, Hickey RC: Medullary thyroid carcinoma: prognosis of familial versus nonfamilial disease and the role of radiotherapy. Horm Metab Res Suppl 21: 21-5, 1989.[PUBMED Abstract]
- Scherübl H, Raue F, Ziegler R: Combination chemotherapy of advanced medullary and differentiated thyroid cancer. Phase II study. J Cancer Res Clin Oncol 116 (1): 21-3, 1990.[PUBMED Abstract]
- Wells SA, Robinson BG, Gagel RF, et al.: Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 30 (2): 134-41, 2012.[PUBMED Abstract]
- Fox E, Widemann BC, Chuk MK, et al.: Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res 19 (15): 4239-48, 2013.[PUBMED Abstract]
- Kraft IL, Akshintala S, Zhu Y, et al.: Outcomes of Children and Adolescents with Advanced Hereditary Medullary Thyroid Carcinoma Treated with Vandetanib. Clin Cancer Res 24 (4): 753-765, 2018.[PUBMED Abstract]
- Elisei R, Schlumberger MJ, Müller SP, et al.: Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol 31 (29): 3639-46, 2013.[PUBMED Abstract]
- Schlumberger M, Elisei R, Müller S, et al.: Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol 28 (11): 2813-2819, 2017.[PUBMED Abstract]
- Sherman SI, Clary DO, Elisei R, et al.: Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer 122 (24): 3856-3864, 2016.[PUBMED Abstract]
- Carlomagno F, Guida T, Anaganti S, et al.: Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23 (36): 6056-63, 2004.[PUBMED Abstract]
- Subbiah V, Velcheti V, Tuch BB, et al.: Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 29 (8): 1869-1876, 2018.[PUBMED Abstract]
- Drilon A, Hu ZI, Lai GGY, et al.: Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol 15 (3): 151-167, 2018.[PUBMED Abstract]
- Redaelli S, Plaza-Menacho I, Mologni L: Novel targeted therapeutics for MEN2. Endocr Relat Cancer 25 (2): T53-T68, 2018.[PUBMED Abstract]
- Mulligan LM: Progress and potential impact of RET kinase targeting in cancer. Expert Rev Proteomics 13 (7): 631-3, 2016.[PUBMED Abstract]
- Lenders JW, Duh QY, Eisenhofer G, et al.: Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99 (6): 1915-42, 2014.[PUBMED Abstract]
- Castinetti F, Qi XP, Walz MK, et al.: Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol 15 (6): 648-55, 2014.[PUBMED Abstract]
- Lairmore TC, Ball DW, Baylin SB, et al.: Management of pheochromocytomas in patients with multiple endocrine neoplasia type 2 syndromes. Ann Surg 217 (6): 595-601; discussion 601-3, 1993.[PUBMED Abstract]
- Inabnet WB, Caragliano P, Pertsemlidis D: Pheochromocytoma: inherited associations, bilaterality, and cortex preservation. Surgery 128 (6): 1007-11;discussion 1011-2, 2000.[PUBMED Abstract]
- Scholten A, Valk GD, Ulfman D, et al.: Unilateral subtotal adrenalectomy for pheochromocytoma in multiple endocrine neoplasia type 2 patients: a feasible surgical strategy. Ann Surg 254 (6): 1022-7, 2011.[PUBMED Abstract]
- Grubbs EG, Rich TA, Ng C, et al.: Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J Am Coll Surg 216 (2): 280-9, 2013.[PUBMED Abstract]
- Walz MK, Alesina PF, Wenger FA, et al.: Posterior retroperitoneoscopic adrenalectomy--results of 560 procedures in 520 patients. Surgery 140 (6): 943-8; discussion 948-50, 2006.[PUBMED Abstract]
- Walz MK, Alesina PF, Wenger FA, et al.: Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World J Surg 30 (5): 899-908, 2006.[PUBMED Abstract]
- Perrier ND, Kennamer DL, Bao R, et al.: Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg 248 (4): 666-74, 2008.[PUBMED Abstract]
- Behrman SW, Bahr MH, Dickson PV, et al.: The microbiology of secondary and postoperative pancreatic infections: implications for antimicrobial management. Arch Surg 146 (5): 613-9, 2011.[PUBMED Abstract]
- Evans DB, Perrier ND: On "Posterior retroperitoneoscopic adrenalectomy--results of 560 procedures in 520 patients". Surgery 140 (6): 951-2, 2006.[PUBMED Abstract]
- Dickson PV, Jimenez C, Chisholm GB, et al.: Posterior retroperitoneoscopic adrenalectomy: a contemporary American experience. J Am Coll Surg 212 (4): 659-65; discussion 665-7, 2011.[PUBMED Abstract]
- Cabalag MS, Mann GB, Gorelik A, et al.: Posterior retroperitoneoscopic adrenalectomy: outcomes and lessons learned from initial 50 cases. ANZ J Surg 85 (6): 478-82, 2015.[PUBMED Abstract]
- Norton JA, Brennan MF, Wells SA Jr: Surgical Management of Hyperparathyroidism. In: Bilezikian JP, Marcus R, Levine MA: The Parathyroids: Basic and Clinical Concepts. New York: Raven Press, 1994, pp 531-551.[PUBMED Abstract]
- Scholten A, Schreinemakers JM, Pieterman CR, et al.: Evolution of surgical treatment of primary hyperparathyroidism in patients with multiple endocrine neoplasia type 2A. Endocr Pract 17 (1): 7-15, 2011 Jan-Feb.[PUBMED Abstract]
- Machens A, Dralle H: Advances in risk-oriented surgery for multiple endocrine neoplasia type 2. Endocr Relat Cancer 25 (2): T41-T52, 2018.[PUBMED Abstract]
- Khan MI, Waguespack SG, Hu MI: Medical management of postsurgical hypoparathyroidism. Endocr Pract 17 (Suppl 1): 18-25, 2011 Mar-Apr.[PUBMED Abstract]
- Stålberg P, Carling T: Familial parathyroid tumors: diagnosis and management. World J Surg 33 (11): 2234-43, 2009.[PUBMED Abstract]
- Peacock M, Bilezikian JP, Klassen PS, et al.: Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab 90 (1): 135-41, 2005.[PUBMED Abstract]
- Schuffenecker I, Ginet N, Goldgar D, et al.: Prevalence and parental origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Le Groupe d'Etude des Tumeurs a Calcitonine. Am J Hum Genet 60 (1): 233-7, 1997.[PUBMED Abstract]
- Norum RA, Lafreniere RG, O'Neal LW, et al.: Linkage of the multiple endocrine neoplasia type 2B gene (MEN2B) to chromosome 10 markers linked to MEN2A. Genomics 8 (2): 313-7, 1990.[PUBMED Abstract]
- Carlson KM, Bracamontes J, Jackson CE, et al.: Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet 55 (6): 1076-82, 1994.[PUBMED Abstract]
- Kitamura Y, Scavarda N, Wells SA, et al.: Two maternally derived missense mutations in the tyrosine kinase domain of the RET protooncogene in a patient with de novo MEN 2B. Hum Mol Genet 4 (10): 1987-8, 1995.[PUBMED Abstract]
- Rich TA, Liu M, Etzel CJ, et al.: Comparison of attitudes regarding preimplantation genetic diagnosis among patients with hereditary cancer syndromes. Fam Cancer 13 (2): 291-9, 2014.[PUBMED Abstract]
- Freyer G, Ligneau B, Schlumberger M, et al.: Quality of life in patients at risk of medullary thyroid carcinoma and followed by a comprehensive medical network: trends for future evaluations. Ann Oncol 12 (10): 1461-5, 2001.[PUBMED Abstract]
- Freyer G, Dazord A, Schlumberger M, et al.: Psychosocial impact of genetic testing in familial medullary-thyroid carcinoma: a multicentric pilot-evaluation. Ann Oncol 10 (1): 87-95, 1999.[PUBMED Abstract]
- Grosfeld FJ, Lips CJ, Ten Kroode HF, et al.: Psychosocial consequences of DNA analysis for MEN type 2. Oncology (Huntingt) 10 (2): 141-6; discussion 146, 152, 157, 1996.[PUBMED Abstract]
- Johnston LB, Chew SL, Trainer PJ, et al.: Screening children at risk of developing inherited endocrine neoplasia syndromes. Clin Endocrinol (Oxf) 52 (2): 127-36, 2000.[PUBMED Abstract]
- MacDonald DJ, Lessick M: Hereditary cancers in children and ethical and psychosocial implications. J Pediatr Nurs 15 (4): 217-25, 2000.[PUBMED Abstract]
- Grosfeld FJ, Lips CJ, Beemer FA, et al.: Psychological risks of genetically testing children for a hereditary cancer syndrome. Patient Educ Couns 32 (1-2): 63-7, 1997 Sep-Oct.[PUBMED Abstract]
- Giarelli E: Multiple endocrine neoplasia type 2a (MEN2a): a call for psycho-social research. Psychooncology 11 (1): 59-73, 2002 Jan-Feb.[PUBMED Abstract]
- Grosfeld FJ, Lips CJ, Beemer FA, et al.: Distress in MEN 2 family members and partners prior to DNA test disclosure. Multiple endocrine neoplasia type 2. Am J Med Genet 91 (1): 1-7, 2000.[PUBMED Abstract]
- Grosfeld FJ, Beemer FA, Lips CJ, et al.: Parents' responses to disclosure of genetic test results of their children. Am J Med Genet 94 (4): 316-23, 2000.[PUBMED Abstract]
- Giarelli E: Bringing threat to the fore: participating in lifelong surveillance for genetic risk of cancer. Oncol Nurs Forum 30 (6): 945-55, 2003 Nov-Dec.[PUBMED Abstract]
- Schultz PN: Providing information to patients with a rare cancer: using Internet discussion forums to address the needs of patients with medullary thyroid carcinoma. Clin J Oncol Nurs 6 (4): 219-22, 2002 Jul-Aug.[PUBMED Abstract]
- 多発性内分泌腫瘍4型
-
序
多発性内分泌腫瘍4型(MEN4)は、臨床所見が他のMEN症候群と重複する新規のまれな症候群である。現在までに報告されているMEN4の確定症例19例の最も一般的な表現型は、原発性副甲状腺機能亢進症(PHPT)で、下垂体腺腫がこれに続く。MEN4は、腫瘍抑制遺伝子のCDKN1B遺伝子(12p13.1)における生殖細胞病原性多様体が原因で生じる。[ 1 ]この症候群は最初にラット(MENX)で発見され[ 2 ]、その後ヒト(MEN4)でも発見された。この症候群は、多発性内分泌腫瘍1型(MEN1)-likeの表現型を有する。MEN1関連表現型を有する患者におけるCDKN1B多様体の発生率を推定することは困難であるが、およそ1.5~3.7%であると考えられる。[ 3 ][ 4 ][ 5 ]MEN4表現型に至る病原性多様体は、常染色体優性形式で伝達される。
臨床診断
MEN4と報告された症例の約80%が、副甲状腺腫瘍によるPHPTに罹患する。PHPTはMEN4の方がMEN1よりもり高い年齢で発症し(それぞれの平均年齢、~56歳 vs ~25歳)、女性が優勢である。[ 6 ]外科的切除後にPHPTの再発は報告されておらず、このことはMEN4におけるPHPTがMEN1におけるPHPTよりも全般的に軽度の疾患であることを示している可能性がある。MEN4における下垂体病変はこの疾患の2番目に一般的な発現であり、報告された症例の約37%が罹患する。MEN4における下垂体腺腫は多様であり、非機能性、ソマトトロピノーマ、プロラクチノーマ、またはコルチコトロピノーマタイプが含まれる。これらの病変に対する診断時年齢も大幅に異なっており、30~79歳に及ぶ。MEN4であることが報告された最も年齢の低い患者は、30歳で先端巨大症を発症した。[ 2 ]膵神経内分泌腫瘍(NET)はまれで、少数の症例しか報告されていない。これには、膵十二指腸または消化管NETが含まれており、非機能性のものもあればホルモン活性を有し、ガストリン、インスリン、副腎皮質刺激ホルモン、血管活性腸管ポリペプチドなどいくつかの物質を分泌するものもある。副腎腫瘍はMEN1では頻度の高い所見であるが、MEN4では非機能性の両側性副腎結節の症例が1例のみ報告されている。[ 5 ]MEN1で一般的に報告される脂肪腫、血管線維腫、コラゲノーマなどの皮膚症状は、MEN4では報告されていない。遺伝子型-表現型の相関は分かっていない。
遺伝的特徴、遺伝的形質、および遺伝子検査
CDKN1B多様体は、細胞周期進行を制御する腫瘍抑制遺伝子と推定されるp27Kip1(一般的にはp27またはKIP1と呼ばれる)をコードする。この遺伝子の変化はp27蛋白発現の大幅な低下につながり、制御不能な細胞周期進行を誘発する。p27の1つのアレルの欠失は多くのヒトのがんで頻繁に起こるイベントであるが、ヒトのがんで残りのアレルが変異するか、ヘテロ接合性の消失により欠失するのはまれである。[ 7 ]CDKN1Bにおける体細胞変異または生殖細胞病原性多様体は、散発性PHPT、小腸NET、リンパ腫、および乳がんの患者でも同定されている。こうした知見は、他の新生物に対するCDKN1Bの腫瘍感受性遺伝子としての新たな役割を実証している。[ 8 ][ 9 ][ 10 ]
現在までのところ、CDKN1B生殖細胞多様体を有する症例は19例のみが医学文献で報告されている。[ 8 ]フレームシフト、ナンセンス、またはミスセンス多様体の病原性生殖細胞多様体13例が記述されている。[ 11 ][ 12 ]
MEN1-likeの特徴を有し、MEN1遺伝子検査の結果が陰性の発端者または個人には、遺伝カウンセリングおよびMEN4の検査が提案される。MEN4診断は、CDKN1B多様体に対する遺伝子検査を用いてのみ確定される。実地臨床では、無症候性または症候性のPHPTを有し、さらに若年(典型的に30歳未満)で多腺性疾患、副甲状腺がん、または異型腺腫を有する患者、あるいは家族歴を有するか、症候性疾患の証拠が認められるのにMEN1またはRETが陰性の患者は、認可を受けた検査室を利用したCDKN1B遺伝子検査の候補である。[ 8 ]疾患が証明された患者については、MEN1の特徴の有無にかかわらず第一度近親者にもスクリーニングが提案される。CDKN1B生殖細胞多様体が同定されると、MEN4に対する定期的な臨床的生化学スクリーニングを促すべきである。
サーベイランス
CDKN1B病原性多様体のキャリアにはサーベイランスを実施すべきであるが、ガイドラインはまだ確立されていない。[ 8 ][ 13 ]現在のところ、サーベイランスは主に臨床的に実施され、成長ホルモン過剰の証拠に集中しており、インスリン様成長因子1について年1回の生化学的評価およびPHPTに対する年1回の血液検査が行われる。[ 13 ]既知のキャリアに対するサーベイランスは青年期に開始される。画像検査の役割は確立されていない。
転帰
MEN1病原性多様体陽性症例293例およびMEN1病原性多様体陰性症例30例(全員にMEN1表現型が認められた)を対象にした研究では、病原性多様体陰性コホートでは中年期以降に疾患が発現し、余命が良好であったことが示された。[ 14 ]この知見をMEN4に適用する際の制限の1つは、これら30人のMEN1陰性患者のうち、CDKN1B陽性であったのは1人のみであったことである。
参考文献- Marinoni I, Pellegata NS: p27kip1: a new multiple endocrine neoplasia gene? Neuroendocrinology 93 (1): 19-28, 2011.[PUBMED Abstract]
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al.: Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A 103 (42): 15558-63, 2006.[PUBMED Abstract]
- Georgitsi M, Raitila A, Karhu A, et al.: Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab 92 (8): 3321-5, 2007.[PUBMED Abstract]
- Agarwal SK, Mateo CM, Marx SJ: Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 94 (5): 1826-34, 2009.[PUBMED Abstract]
- Molatore S, Marinoni I, Lee M, et al.: A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat 31 (11): E1825-35, 2010.[PUBMED Abstract]
- Lee M, Pellegata NS: Multiple endocrine neoplasia type 4. Front Horm Res 41: 63-78, 2013.[PUBMED Abstract]
- Philipp-Staheli J, Payne SR, Kemp CJ: p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res 264 (1): 148-68, 2001.[PUBMED Abstract]
- Alrezk R, Hannah-Shmouni F, Stratakis CA: MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer 24 (10): T195-T208, 2017.[PUBMED Abstract]
- Malanga D, De Gisi S, Riccardi M, et al.: Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol 166 (3): 551-60, 2012.[PUBMED Abstract]
- Occhi G, Regazzo D, Trivellin G, et al.: A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet 9 (3): e1003350, 2013.[PUBMED Abstract]
- Georgitsi M: MEN-4 and other multiple endocrine neoplasias due to cyclin-dependent kinase inhibitors (p27(Kip1) and p18(INK4C)) mutations. Best Pract Res Clin Endocrinol Metab 24 (3): 425-37, 2010.[PUBMED Abstract]
- Lee M, Pellegata NS: Multiple endocrine neoplasia syndromes associated with mutation of p27. J Endocrinol Invest 36 (9): 781-7, 2013.[PUBMED Abstract]
- Wasserman JD, Tomlinson GE, Druker H, et al.: Multiple Endocrine Neoplasia and Hyperparathyroid-Jaw Tumor Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res 23 (13): e123-e132, 2017.[PUBMED Abstract]
- de Laat JM, van der Luijt RB, Pieterman CR, et al.: MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med 14 (1): 182, 2016.[PUBMED Abstract]
- 家族性褐色細胞腫および傍神経節腫症候群
-
序
傍神経節腫(PGL)および褐色細胞腫(PHEO)は、クロム親和性細胞から発生するまれな腫瘍で、カテコールアミンと神経ペプチドを合成、蓄積、分泌する能力がある。個々には二次性高血圧を呈することがある。2004年に世界保健機関は、副腎腫瘍をPHEOとして特徴を明らかにした。[ 1 ]傍神経節腫の用語は、非副腎(つまり副腎外)腫瘍にのみ用いられ、副交感神経に沿う傍神経節または交感神経幹からさまざまな部位に発生することがある。PGLは、頭頸部、腹部、または骨盤にみられることがある。交感神経鎖から発生するもののみが分泌性である。頭蓋底または頭頸部にみられるPGLは、典型的にグロムス細胞、頸動脈小体の近く、迷走神経沿い、または頸静脈窩で発生し、通常は副交感神経系の傍神経節由来であるため、カテコールアミンを分泌することはまれである。[ 2 ][ 3 ]最も認識しやすい腫瘍は、頸動脈小体で発見される。頸部より下の部位に発生するPGLの多くは、上縦隔または膀胱に位置する。[ 3 ]これらの腫瘍は無症状の場合があるため、一般集団における発生率は不定であるが、30,000~100,000人に1人の割合である。[ 3 ]1件の剖検研究では、2,000人に1人という非常に高い発生率が示されたが、これは潜伏腫瘍の頻度の高さを示唆している。[ 4 ]PGLは性比率が等しく、あらゆる年齢で発生する可能性があるが、40~50歳の発生率が最も高い。[ 5 ][ 6 ]
臨床記述
PGLおよびPHEOは、遺伝性症候群の顕性化として、つまり遺伝性のいくつかのPGL/PHEO症候群の1つにおける単独腫瘍として散発的に発生することがある。
PGLおよびPHEOは典型的に増殖が緩徐な腫瘍であり、臨床的に注目されるようになるまで長年にわたって変化しないものもある。反対に、少数ではあるが、この種の腫瘍が悪性でより侵攻性の高い臨床経過をたどる場合もある。PGLおよびPHEOの悪性腫瘍は、非クロム親和性組織の原発腫瘍から遠隔部位に転移がみられることにより定義される。転移が多く見られる部位は、骨、肝臓、肺である。[ 1 ][ 7 ]
良性腫瘍と悪性腫瘍を区別する上で信頼性の高い分子的、免疫組織化学的、または遺伝的な予測因子は存在しないが[ 8 ]、いくつかの研究で、多変量解析により、SDHBキャリア[ 9 ][ 10 ]、より若い患者[ 10 ]、副腎外腫瘍の患者[ 10 ]、および腫瘍がより大きい個人[ 11 ]で悪性腫瘍の発生率が高いことが示されている。一部の専門家は、周辺組織への局所浸潤を悪性の別のマーカーとみなしている。[ 12 ][ 13 ]他の専門家は、局所的に浸潤した腫瘍は、遠隔転移した腫瘍より潜行性の経過をたどる傾向があるため、この分類に同意していない。[ 14 ]そのため、PGLにおける悪性の割合を推定することは困難である;悪性の割合は5~20%と報告されている。[ 15 ][ 16 ][ 17 ][ 18 ]
PGLおよびPHEOの臨床診断
PGLは、腫瘍の発生部位および腫瘍が分泌性かどうかにより、さまざまな症状を引き起こすことがある。頭頸部のPGLでは、まれにカテコールアミンが上昇することがある。分泌性のPGLおよびPHEOでは、高血圧、頭痛、頻脈、発汗、および紅潮が現れることがある。一般的に非分泌性の腫瘍は無痛であるため、病変の増殖が周辺組織に達し腫瘤効果が発現するまで注意が向けられない。頭頸部PGLの患者は、頸部側方の腫瘤の肥大、嗄声、ホルネル症候群、拍動性耳鳴、めまい、顔面麻痺、霧視を呈することがある。[ 19 ]
カテコールアミン過剰が臨床的に明らかな患者に対して生化学検査を一般的に実施して、腫瘍の分泌性について評価する。[ 20 ]この評価は、尿および/または血漿のメタネフリン分画(ノルメタネフリンおよびメタネフリン)の測定による実施が最も良好で、カテコールアミン(ノルメタネフリン、ドパミン、およびエピネフリン)を直接測定するよりも、高い感度および特異度が得られる。[ 21 ][ 22 ][ 23 ]血漿メタネフリン濃度を測定する患者では、静脈内カテーテルを挿入し、患者を15~20分間仰臥位にさせた後に血液を採取する。[ 24 ]さらに、採血の8~12時間前から、患者には食品もしくはカフェイン含有飲料、喫煙、または活発な運動への参加を控えさせる必要がある。[ 24 ]
PGLの画像検査は診断の中心である;初期評価には頸部および胸部のコンピュータ断層撮影(CT)が含まれる。磁気共鳴画像法(MRI)も頭頸部で有用である。PGLでは一般的に均質な像が認められ、造影剤の静脈内投与後に強い造影が得られる。隣接する血管および骨格構造と腫瘍を区別するために、MRIを使用することもある。T2強調画像で、2cm以上の腫瘍は、典型的な「ごま塩(salt and pepper)」状の像、つまり信号の欠落が散見されるまばらな領域と血管分布の増大による信号強度の高い領域とが混在した像を呈する傾向が高い。[ 25 ]
解剖学的画像検査と併用した核医学画像検査、特にソマトスタチン受容体シンチグラフィ(SRS)は、病変の位置と拡がりを特定するために有用となりうる(多病巣性 vs 遠隔転移巣)。[ 26 ]良性腫瘍は、ヨウ素123-メタヨードベンジルグアニジン(123I-MIBG)画像検査よりもSRSに対する感度が高いことが報告されている。感度は、頭頸部領域で腹部のPGLまたはPHEOよりも高い(それぞれ91% vs 40%および42%)。[ 27 ]SRSは転移腫瘍の発見においてMIBGよりも優れていることが報告されている(それぞれ95% vs 23%)。[ 27 ]しかしながら、123I-MIBGはPHEOに対する感度が非常に高く[ 27 ]、ポジトロン放射断層撮影-コンピュータ断層撮影(PET-CT)はPGLに対する特異度が非常に高い。PGLおよび/またはPHEOに対するフッ素18-ジヒドロキシフェニルアラニン(18F-DOPA)、18F-フルオロドパミン、またはPET-CTによる機能的画像検査法は、頭頸部腫瘍の位置特定に特に有用であろう。データによると、腫瘍部位の特定に用いられるPETトレーサーの選択は、さまざまな腫瘍の代謝活動に基づき、患者の遺伝状態に注意して実施する必要がある。[ 9 ]SDHxおよびVHLに病原性多様体を有する患者では、18F-フルデオキシグルコース活性が高い傾向にあり、これは低酸素症に対応した遺伝子の活性化に関連することが示唆されている。[ 9 ][ 28 ]一部のSDHB腫瘍は18F-DOPAを濃縮する働きがわずかであるため、SDHx病原性多様体の患者はそうしたスキャンで偽陰性の結果が出ることがある。ガリウム68-DOTATATE PET/CTは、転移病変を有する患者で病変の拡がりを判定できる可能性のある画像診断法として有望なことを示している。[ 29 ]VHL病原性多様体に伴う腫瘍でも、同様にMIBGスキャンで見逃されることがある。[ 9 ]
PHEOの画像検査は、通常副腎の専用CTからなる。多発性内分泌腫瘍2型(MEN2)であるか、MEN2のリスクが高い個人における生化学的スクリーニングでPHEOが示唆される場合は、MRIまたはCTなどの位置確認のための検査を実施できる。[ 30 ]診断の確定は、ヨウ素131-MIBGシンチグラフィまたはPET画像検査を用いて行える。[ 30 ][ 31 ][ 32 ][ 33 ]
遺伝的特徴、遺伝的形質、および遺伝子検査
散発性PHEOまたはPGLとみられるかなりの割合の人が生殖細胞病原性多様体のキャリアである。散発性PHEOとみられる患者の最大33%および散発性PGLとみられる患者の最大40%が、実際に既知のPGL/PHEO感受性遺伝子の1つに識別可能な生殖細胞病原性多様体を有する。[ 16 ][ 34 ][ 35 ][ 36 ][ 37 ][ 38 ][ 39 ]ある研究では、腫瘍が1ヵ所のみで家族歴が陰性の患者では、遺伝性病原性多様体がみられる確率は11.6%であったが[ 16 ]、このような患者の41%に病原性多様体を検出したグループもある。[ 38 ][ 40 ]PGL/PHEOで米国国立がん研究所に紹介された21歳未満の患者55人を対象にした1件のレトロスペクティブ・レビューでは、80%の患者に生殖細胞病原性多様体が認められた。[ 41 ](小児のPGL/PHEOに関する詳しい情報については、小児の褐色細胞腫と傍神経節腫の治療に関するPDQ要約を参照のこと。)例えば、SDHB病原性多様体キャリアの中でさえ、浸透度が低く、疾患発症の遅れが認められ、本疾患の遺伝性をさらに覆い隠す可能性がある。[ 42 ]そのため、PHEOまたはPGL患者では、散発性腫瘍であるとみられる患者であっても、これらの疾患に関連する病原性多様体の頻度が高いため、すべて遺伝子検査を検討してもよい。[ 43 ]
PGLおよびPHEOは、フォン・ヒッペル-リンダウ病(VHL)、MEN2、神経線維腫症1型、カーニー-ストラタキス症候群、家族性傍神経節腫(FPGL)症候群など、十分に明らかにされたいくつかの腫瘍感受性症候群の1つとみなすことができる。FPGLは、次の4遺伝子のいずれかに生じた病原性多様体によって引き起こされることが最も多い:SDHA、SDHB、SDHC、およびSDHD(まとめてSDHxと呼ばれる)。SDHxの蛋白は、コハク酸デヒドロゲナーゼ(SDH)複合体の一部を構成しており、ミトコンドリアの内膜に位置し、細胞エネルギーの代謝に重要な役割を果たしている。[ 44 ]SDHBにおける病原性多様体が最も多く、次がSDHDで、SDHCおよびSDHAにおける病原性多様体はまれである。つい最近、FPGL/PHEOでSDHAF2(SDH5とも呼ばれる)、TMEM127、およびMAX遺伝子における病原性多様体が報告されたが[ 45 ][ 46 ][ 47 ][ 48 ]、これらの多様体はまれである。腫瘍形成の機序は依然として特定困難である。ある研究によると、SDHx関連腫瘍は、神経内分泌組織分化に関わる重要な遺伝子の発現減少に関連している高メチル化因子の表現型を示すことが示唆される。[ 49 ]
FPGLの遺伝パターンは、関与している遺伝子によって左右される。ほとんどの家系が従来の常染色体優性遺伝を示すが、SDHAF2およびSDHDに病原性多様体を認める家系では、ほぼ例外なく、この表現型が父系から伝達されたことを示している。言い換えると、この病原性多様体は母方または父方から受け継がれうるが、腫瘍が発生するのは、その病原性多様体が父から遺伝した場合のみである。[ 50 ][ 51 ]SDHDまたはSDHAF2の病原性多様体が原因ではないFPGLの症例では、罹患者の第一度近親者(FDR)が病原性多様体を保有する確率は50%であり、PGLを発症するリスクが高い。浸透度の低い病原性多様体が認められる家系では、家族歴が陰性とみなされる可能性があることから、その家系に病原性多様体が確認された場合は、罹患していないすべてのFDRに対して遺伝子検査を勧めることが重要である。
遺伝性のPHEOおよびPGL症候群に対する遺伝子検査は、ほとんどが公表されたアルゴリズムに基づいており[ 43 ]、その場合の検査は、腫瘍の種類および部位、診断時年齢、家族歴、悪性腫瘍の存在などの因子に基づいて、段階的に実施する。[ 16 ][ 52 ][ 53 ]このアプローチにより、費用対効果の高い臨床的特徴に基づいた標的検査が可能となっている。しかしながら、ここ数年で、次世代の遺伝子配列決定(NGS)技術により遺伝子検査の費用が劇的に低下しており、2~3の遺伝子を検査する従来の費用で、現在では10~30の遺伝子の病原性多様体の検査ができるようになっている。これらの検査は、非定型症状または重複した臨床的特徴を認める個人および家族により適切な可能性がある。多重遺伝子検査パネルに関連する費用がこのまま低下していけば、PGLおよびPHEOに対する現行の検査アルゴリズムはすぐに時代遅れになる可能性がある。2013年のシリーズで、PGLおよびPHEOの85サンプルについて、NGSパネル検査を用いて、PGLで既知の10の感受性遺伝子が解析された;NGSアッセイおよび解析により、98.7%の感度が示された。[ 54 ]多重遺伝子パネルを介したスクリーニングにより、検出率が中程度に増加している。PHEO患者87人を対象にした小規模シリーズにおいて、25.3%の個人(87人中22人)が10のPGL/PHEO関連遺伝子を含んだスクリーニングパネルで生殖細胞病原性多様体を有することが明らかにされた;11.7%がVHLに、6.8%がRETに、2.3%がSDHDに、2.3%がMAXに、1.1%がSDHBに、および1.1%がTMEM127に生殖細胞病原性多様体を有した。[ 55 ]見たところ散発性の腫瘍が74.7%の患者(87人中65人)で認められた。
遺伝子型-表現型の相関
FPGL/PHEOにおける腫瘍の種類および部位、発症時の年齢、および生涯の浸透度は、遺伝子多様体によってさまざまである。これらの関係は、遺伝子検査およびスクリーニングを決断する指針として役立つ可能性があるが、この疾患では高度の変動性がみられることを考えると、注意しなければならない。FPGL/PHEO症候群はゲノム刷り込みが起こるまれな遺伝性疾患の1つである。例えば、SDHD病原性多様体は通常、母親から遺伝している場合は通常活性化されず、FPGL/PHEO症候群のリスクは増加しない。しかしながら、この遺伝子が父親から遺伝している場合、病原性多様体のスイッチが入り、リスクが増加する。
SDHDの病原性多様体は、主に副交感神経系PGLのリスク増加と関連している。これらは多巣性で、頭頸部に発生することが多い一方で、SDHBキャリアの腫瘍は腹部に発生することが多い。[ 56 ][ 57 ]複数のシリーズにより、SDHDキャリアでは、頭頸部腫瘍のリスクが71%であることが示されたが、これに対して、SDHBキャリアにおけるリスクは27~29%であった。[ 18 ][ 56 ]あるシリーズによると、SDHDキャリアでは、部位や種類にかかわらずPGLを発症する生涯リスクが50歳で77%と高いことが推定され[ 56 ]、別のシリーズによると、70歳で90%と推定された。[ 57 ]文献で報告された1,700例を超える症例のレビューでは、同様な推定値が得られ、生涯の浸透度は86%であることが示唆される。[ 58 ]SDHDキャリアにおける悪性の割合は5%を下回る。[ 58 ]
SDHB遺伝子の病原性多様体は症候性PGLと関連しているが、PHEOおよび副交感神経系PGLでも報告されている。SDHB関連のPGLは、頭頸部よりも腹部および縦隔に発生することが多い。1,700症例のレビューでは、生涯浸透度が77%であることが示唆された。[ 58 ]しかしながら、浸透度を検討した初期の研究では、無症状の病原性多様体キャリアの組み入れが制限され、若い年齢で罹患したことを強く示す個人のサンプリングのために、多くの研究が確認バイアスを受けていた。家族ベースおよび集団ベースの研究では、50歳までに9~35%のより低い浸透度推定値が明らかになった。[ 42 ][ 59 ][ 60 ][ 61 ][ 62 ]他の研究では、生涯浸透度が42~50%であると推定されている。[ 62 ][ 63 ]SDHDキャリアにおける浸透度は男性よりも女性の方が低い可能性があるという証拠がある。[ 63 ]悪性の割合は、SDHBの方が他のSDH遺伝子よりも高く、ほとんどのシリーズで最大3分の1の患者の腫瘍が悪性である。[ 56 ][ 57 ]SDHBの病原性多様体でも、消化管間質腫瘍(GIST)、腎細胞がん、甲状腺乳頭がんなど、他のいくつかの腫瘍および悪性疾患との関連が指摘されている。[ 56 ][ 57 ]
SDHCの病原性多様体はまれで、PGL全体の0.5%を占めると推定される。[ 58 ]あるシリーズで、40歳までに複数のPGLまたは単独のPGLと診断された患者153人のうち、SDHC病原性多様体が認められたのは3人(2%)であった。[ 35 ]別のシリーズで、頭頸部PGL登録から得た発端者121人における病原性多様体の発生率は4%(121人中5人)であったことが示された。[ 64 ]SDHC病原性多様体は、頭頸部PGLの原因となることが最も多いが、少数ながら腹部PGLの患者でも認められている。[ 16 ][ 65 ]SDHB、SDHC、およびSDHDの病原性多様体は、PGLとGISTの2徴を特徴とするカーニー-ストラタキス症候群を引き起こすこともある。[ 66 ]
SDHA、SDHAF2、MAX、およびTMEM127の病原性多様体も報告されている;総合すると、症例全体に占める割合は2~3%を下回る。SDHAの両アレル性病原性多様体は、古くから常染色体劣性疾患である遺伝性若年性脳症/リー症候群を引き起こすことが知られていたが[ 67 ]、片アレル性病原性多様体がPGL発症リスク増加に関係することは最近まで知られていなかった。1件のシリーズで、オランダのPGL患者393人のコホートにおけるSDHA病原性多様体の発生率は7.6%であったことが示された。[ 68 ]腫瘍は最も一般的に頭頸部に発生し、続いて副腎および腹部(副腎外)に発生する。[ 69 ][ 70 ]オランダの同じシリーズ[ 68 ]において、非発端者の病原性多様体キャリアに対して推定される浸透度は、70歳までに10%であった。当初、SDHAF2の病原性多様体は、頭頸部PGLにのみ認められていた。[ 48 ]MAX遺伝子は、無関係な3症例のエクソーム配列解析により、PHEOの感受性遺伝子として2011年に初めて報告された。[ 45 ]3つの異なった生殖細胞病原性多様体が同定され、同グループによる59症例の追跡シリーズでは、さらに5つの多様体が同定された。MAX蛋白は、神経堤細胞腫瘍の発生および進行に重要な役割を果たしている。[ 71 ]TMEM127遺伝子は、染色体2q11.2に位置しており、多数の細胞プロセスを調節しているmTORの発現抑制因子であることが知られている膜貫通蛋白をコードしている。TMEM127病原性多様体を有する患者23人のレビューでは、96%(23人中22人)がPHEOであり、9%(23人中2人)がPGLであったことが示された。[ 58 ]
European-American-Asian Pheochromocytoma-Paraganglioma Registry Study Groupから追加された患者58人を対象とした研究では、まれな素因遺伝子のキャリア数が以前の報告数の倍以上であり、その内訳はSDHA(n = 29)、SDHAF2(n = 1)、MAX(n = 8)、TMEM127(n = 20)であった。[ 72 ]この研究では、SDHA病原性多様体キャリアの12%、およびTMEM127キャリアの10%に悪性疾患が認められ、これらの率は以前の推定を有意に上回っていた。副腎外腫瘍はコホートで多く認められ(48%)、特に、SDHAキャリアに多く(79%)、そのうちのかなりの割合(44%)に頭頸部腫瘍がみられた。しかしGISTは、このコホートのSDHAキャリアに一切認められず、以前に調査されたコホートで頻繁に報告されたこととは対照的である。SDHA関連腫瘍は8歳前後の患者に発生した。MAX病原性多様体を伴う腫瘍は、ほぼすべてが副腎にみられ、多くの場合に両側性であった。総じて40歳時点でのPGL/PHEO発生の浸透度は、MAX病原性多様体キャリアで73%、TMEM127キャリアで41%、SDHAキャリアで39%と推定された。浸透度は陽性の近親者についても算出され、これらの個人ではSDHAキャリアの発端患者に比べて有意に低かった(13%)が、MAXまたはTMEM127の発端者および非発端者に対しては有意な差がなかった。このような比較的少数の研究は、前述のように、選択バイアスや診断バイアスが生じやすいことに十分留意する必要がある。例えば、このコホートの家族の22%しかカスケードスクリーニングを受けていないことは、浸透度の計算に影響を及ぼす。加えて、転移病変の発生率が高いことには三次医療機関の診断バイアスが含まれている可能性があり、GIST腫瘍がみられないことは、登録データがPGL/PHEOに限定されているために関連腫瘍の全体が捉えられていないことが原因で生じている可能性がある。[ 73 ]
サーベイランス
SDH遺伝子の1つに生殖細胞病原性多様体が確認されている患者は、PGL、PHEO、腎腫瘍、GISTを発症するリスクが有意に高い。PHEOおよびPGLは典型的に緩徐な増殖パターンを示すが、野放しに増殖させると腫瘤による影響が生じ、最終的に神経学的障害を来すことがある。さらに、これらの腫瘍の大半は良性であるが、一部は悪性転換を来すことがある。このため、早期発見および切除により患者に対するリスクを最小化できることから、規定期間での腫瘍の増殖性についての定期的スクリーニングがきわめて重要である。[ 10 ]理想的プロトコルを明確にするために行われた研究は少ないが、感度が高く、放射線曝露量がわずかであることから、全身MRIがスクリーニングについての妥当な方法として提案されている。[ 43 ][ 74 ]1件の研究で、SDHx病原性多様体のキャリア37人が10歳からの年1回の生化学検査および年1回または2年ごとの全身MRIを受けた。[ 75 ]このスクリーニングプロトコルで5人の患者に6つの腫瘍が確認された。MRIの感度は87.5%、特異度は94.7%であった。生化学検査の感度は37.5%と有意に低く、特異度は94.9%とMRIと同様であった。[ 75 ]157人の患者を対象としたレトロスペクティブ研究がSDH病原性多様体のキャリアにおける頭頸部傍神経節腫の発見について高速造影血管MRIプロトコルを評価した。[ 76 ]このプロトコルは感度および特異度がそれぞれ88.7%および93.7%と高かった。別のグループは、患者157人のコホートに関する自らの経験を解析し、悪性腫瘍のリスクが最大のものを同定するために、腫瘍特性への連続的クエリーのアルゴリズムおよびそれによるより厳格なサーベイランスを提案した。副腎外部位、家族歴陽性、遺伝子検査陽性、または4cmを超える腫瘍の大きさを含む以下の特徴のいずれか1つの存在は、悪性腫瘍を同定する100%の感度および42.5%の特異度と関係していた。著者らはさらに、これらの患者で副腎腫瘍が小さい(4cm以下)場合、このようなコホートでは、平均追跡期間7.3年後に悪性腫瘍が発生しなかったことから、長期のサーベイランスを段階的に減らせる可能性があると提案した。[ 10 ]これらの知見について、別の施設でプロスペクティブな検証ができれば、有益であると考えられる。
SDH病原性多様体キャリアのサーベイランスのための至適画像検査プロトコルは依然として明らかではないが、年1回の生化学検査および臨床的サーベイランスが考慮可能である。生化学検査は、血漿遊離メタネフリン/カテコールアミンまたは分画カテコールアミンの24時間尿排泄(利用可であればドパミン代謝物のメトキシチラミンを含む)の測定により実施可能である。臨床的サーベイランスに身体診察と血圧測定を含めてもよい。臨床的サーベイランスおよび生化学検査は5~10歳に開始するか、家系の中で最も早い診断時年齢より10歳早く開始する。[ 77 ][ 78 ]
証拠レベル:4
介入
術前管理
内科的管理は、PGL/PHEOの外科的切除への橋渡しである。カテコールアミン分泌過多の証拠がない患者では、術前の薬物療法は必須ではないが、ホルモン検査の結果にかかわらず、その使用を支持する人もいる。[ 24 ]薬物療法の目的は、手術の少なくとも10~14日前から高血圧をコントロールすることである。[ 79 ]管理の目的は、術中の高血圧クリーゼ、心不整脈、肺水腫、および心虚血を避けるため、術前に高血圧を呈していない患者でも、カテコールアミン誘発性合併症を予防することである。カテコールアミン過剰を適切に防止できなかった場合は、周術期に高血圧クリーゼおよび致死性不整脈により死亡するリスクが劇的に上昇するとともに、腫瘍切除後に高血圧クリーゼを発症することがある。[ 80 ][ 81 ]
さまざまなレジメンを比較するランダム化比較試験がない状況では、広く推奨できるアプローチはない。血圧をコントロールし、血液量を増やすために、α-アドレナリン受容体遮断薬のフェノキシベンザミン(Dibenzyline)が最も頻繁に使用される。[ 24 ]プラゾシン、テラゾシン、またはドキサゾシンといった他のα遮断薬も使用されており、成功を収めている;これらの薬物は、より特異的なα-1アドレナリン作用競合的拮抗薬であり、フェノキシベンザミンより消失半減期が短い。[ 82 ][ 83 ]フェノキシベンザミンのα受容体への非競合的結合は、その長寿命性と併せて、薬物の持続性作用をもたらす可能性があり、術後低血圧を経験する患者もいる。[ 24 ][ 84 ]ある研究によると、ドキサゾシン徐放製剤による治療を受けた患者では、フェノキシベンザミンが投与された患者よりも、周術期の血行動態変化が安定しており、術前の血圧コントロールまでの時間が短いことが明らかになった。[ 84 ]
α遮断薬を開始すると、これらの患者では典型的に血液量が低下するため、血液量の増強がしばしば必要になる。[ 85 ][ 86 ]α遮断薬による血管拡張作用に加え、高ナトリウム食の摂取および多量の水分摂取、または術前の生理食塩水点滴により、血液量増強が達成できる場合がある。適切な遮断の臨床所見は、鼻づまりまたはめまいの症状である。
ニカルジピンまたはニフェジピンのようなカルシウムチャネル遮断薬も、術前の高血圧コントロールに採用されている。[ 87 ]難治性高血圧に対しては、αおよびβ遮断薬と併用して、カルシウムチャネル遮断薬を使用してもよいし、α遮断薬による副作用に耐えられない患者では、第二選択治療薬として単独で使用してもよい。[ 24 ]
病原性多様体が同定されている場合は、外科治療の計画およびアプローチが変わる可能性があるため、術前画像検査の検討が必要である。[ 41 ](画像検査法に関する詳しい情報については、本要約のPGLおよびPHEOの臨床診断のセクションを参照のこと。)
手術
PGLおよびPHEOには、外科的切除が選択される治療法である。切開切除および腹腔鏡アプローチはいずれも安全であるが、施行可能な場合、腹腔鏡下切除が好ましい。[ 77 ][ 88 ]腫瘍が大きい(6~7cmを超える)場合、腹腔鏡検査の限定された視野内では技術的に困難となるリスクが高いことから、切開切除が一般に推奨される。露出およびアプローチの手段は、腫瘍の解剖学的位置を基にする。大動脈および傍大動脈領域への直接アクセスは、後方アプローチにより達成できる。直接的で安全かつ有効である。[ 89 ]完全切除には、腫瘍全体を十分に露出させることが重要である。ロボットの支援は、解剖領域の三次元拡大図が得られるため、選択された症例で有用な可能性がある。[ 90 ]背面からの後腹膜鏡下副腎摘出術の効力および安全性は確立されているが、進行中の研究で家族性症候群における本アプローチの妥当性が検討されている(NCT02618694を参照のこと)。
PGLは、大動脈分岐の上方、下腸間膜動脈の起点(Zuckerkandl器官)の下方、または膀胱円蓋部の近くの大動脈周辺後腹膜交感神経鎖に局在することが多い。[ 91 ][ 92 ]悪性PGLは、周囲に分布している血管に癒着している高密度の線維性被膜を有し、完全切除が困難になる可能性がある。[ 92 ]所属リンパ節は、悪性腫瘍が浸潤していることがあり、それが術前に疑われたり、術中に認められたりした場合は、所属リンパ節郭清を施行してもよい。
遺伝子検査は、同時性または異時性の両側性病変が遺伝性PHEOで非常に多いことから、再発または対側疾患のリスクに関する情報を得て、切除範囲(例、皮質を温存するかどうか)の指針とするため、初回手術前に実施するのが最善である。生殖細胞病原性多様体の手術前の情報は、皮質を残す副腎摘出術に関連する変数に大きな影響を及ぼす。皮質の温存は、病原性多様体を有することが明らかになった患者では、対側腫瘍を発症するリスクが高いため重要となる。皮質の温存は、対側の副腎摘出術による将来の副腎皮質機能不全の可能性を低下させる。悪性疾患のリスクが高いSDHBキャリアでは、この検討を慎重に比較する必要がある。108人の患者コホートを対象にした1件の研究において、生殖細胞病原性多様体を有する患者の33%には遺伝性症候群の家族歴が認められず、SDHB生殖細胞病原性多様体を有する患者の36%には家族歴が認められず、発症時にPGL/PHEOの既往もみられなかった。[ 93 ]50年近くに及ぶ1件のレトロスペクティブ・シリーズで、片側性PHEOが認められ、片側全副腎摘出術を受けた患者49人中15人(30%)が初回診断後中央値8.2年(範囲、1~20年)で対側副腎にPHEOを発症した。[ 94 ]対側副腎にPHEOを発症した15人の患者のうち、8人がMEN2Aで、2人がMEN2B、2人がVHL、1人が家族性PHEOであった。対側腫瘍を発症するリスクは時間の経過とともに増大し、中央値で6年後に25%の患者が腫瘍を発症し、中央値で32年後には43%が腫瘍を発症した。皮質を残す手術は、副腎皮質機能不全のリスクと生涯にわたるステロイド補充の必要性を最小限に抑えることができるため、魅力的な選択肢である。大規模な患者シリーズで、皮質を残す手術では、皮質温存後の再発率が3~7%であったのに対し、完全切除後の再発率(副腎床における再発)は2~3%であった。[ 94 ][ 95 ]いずれの研究でもステロイド依存性の頻度は、皮質を残さない手技を受けた患者より皮質を残す手技を受けた患者の方が低かった(86%に対して57%)。皮質を残す手技の後に副腎皮質機能不全を発症したのは患者39人中1人(3%)であった;全副腎摘出術後に副腎皮質機能不全を発症したのは患者25人中5人(20%)であった。[ 94 ]これらの研究の著者らは、最初は片側性疾患とみられる患者を含め、遺伝性PHEO患者に対する有効な選択肢として皮質を残す手術を推奨している。
証拠レベル:5
参考文献- Inherited tumour syndromes. In: Lloyd RV, Osamura RY, Klöppel G, et al.: WHO Classification of Tumours of Endocrine Organs. 4th ed. Lyon, France: International Agency for Research on Cancer, 2017, pp. 262–66.[PUBMED Abstract]
- Offergeld C, Brase C, Yaremchuk S, et al.: Head and neck paragangliomas: clinical and molecular genetic classification. Clinics (Sao Paulo) 67 (Suppl 1): 19-28, 2012.[PUBMED Abstract]
- Raygada M, Pasini B, Stratakis CA: Hereditary paragangliomas. Adv Otorhinolaryngol 70: 99-106, 2011.[PUBMED Abstract]
- McNeil AR, Blok BH, Koelmeyer TD, et al.: Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med 30 (6): 648-52, 2000.[PUBMED Abstract]
- O'Riordain DS, Young WF, Grant CS, et al.: Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg 20 (7): 916-21; discussion 922, 1996.[PUBMED Abstract]
- Erickson D, Kudva YC, Ebersold MJ, et al.: Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 86 (11): 5210-6, 2001.[PUBMED Abstract]
- Eisenhofer G, Lenders JW, Timmers H, et al.: Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 57 (3): 411-20, 2011.[PUBMED Abstract]
- Jovanovic R, Kostadinova-Kunovska S, Bogoeva B, et al.: Histological features, Ki-67 and Bcl-2 immunohistochemical expression and their correlation with the aggressiveness of pheochromocytomas. Prilozi 33 (2): 23-40, 2012.[PUBMED Abstract]
- Taïeb D, Neumann H, Rubello D, et al.: Modern nuclear imaging for paragangliomas: beyond SPECT. J Nucl Med 53 (2): 264-74, 2012.[PUBMED Abstract]
- Dhir M, Li W, Hogg ME, et al.: Clinical Predictors of Malignancy in Patients with Pheochromocytoma and Paraganglioma. Ann Surg Oncol 24 (12): 3624-3630, 2017.[PUBMED Abstract]
- Eisenhofer G, Lenders JW, Siegert G, et al.: Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 48 (11): 1739-49, 2012.[PUBMED Abstract]
- Eisenhofer G, Bornstein SR, Brouwers FM, et al.: Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Cancer 11 (3): 423-36, 2004.[PUBMED Abstract]
- Zarnegar R, Kebebew E, Duh QY, et al.: Malignant pheochromocytoma. Surg Oncol Clin N Am 15 (3): 555-71, 2006.[PUBMED Abstract]
- Medeiros LJ, Wolf BC, Balogh K, et al.: Adrenal pheochromocytoma: a clinicopathologic review of 60 cases. Hum Pathol 16 (6): 580-9, 1985.[PUBMED Abstract]
- Walz MK, Alesina PF, Wenger FA, et al.: Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World J Surg 30 (5): 899-908, 2006.[PUBMED Abstract]
- Mannelli M, Castellano M, Schiavi F, et al.: Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 94 (5): 1541-7, 2009.[PUBMED Abstract]
- Lee JH, Barich F, Karnell LH, et al.: National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer 94 (3): 730-7, 2002.[PUBMED Abstract]
- Niemeijer ND, Rijken JA, Eijkelenkamp K, et al.: The phenotype of SDHB germline mutation carriers: a nationwide study. Eur J Endocrinol 177 (2): 115-125, 2017.[PUBMED Abstract]
- DeLellis RA, Lloyd RV, Heitz PU, et al., eds.: Pathology and Genetics of Tumours of Endocrine Organs. Lyon, France: IARC Press, 2004. World Health Organization classification of tumours, vol. 8.[PUBMED Abstract]
- Grossman A, Pacak K, Sawka A, et al.: Biochemical diagnosis and localization of pheochromocytoma: can we reach a consensus? Ann N Y Acad Sci 1073: 332-47, 2006.[PUBMED Abstract]
- Lenders JW, Pacak K, Walther MM, et al.: Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287 (11): 1427-34, 2002.[PUBMED Abstract]
- Eisenhofer G, Lenders JW, Linehan WM, et al.: Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med 340 (24): 1872-9, 1999.[PUBMED Abstract]
- Sawka AM, Jaeschke R, Singh RJ, et al.: A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 88 (2): 553-8, 2003.[PUBMED Abstract]
- Chen H, Sippel RS, O'Dorisio MS, et al.: The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 39 (6): 775-83, 2010.[PUBMED Abstract]
- Olsen WL, Dillon WP, Kelly WM, et al.: MR imaging of paragangliomas. AJR Am J Roentgenol 148 (1): 201-4, 1987.[PUBMED Abstract]
- Gimenez-Roqueplo AP, Dahia PL, Robledo M: An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 44 (5): 328-33, 2012.[PUBMED Abstract]
- Michałowska I, Ćwikła JB, Pęczkowska M, et al.: Usefulness of Somatostatin Receptor Scintigraphy (Tc-[HYNIC, Tyr3]-Octreotide) and 123I-Metaiodobenzylguanidine Scintigraphy in Patients with SDHx Gene-Related Pheochromocytomas and Paragangliomas Detected by Computed Tomography. Neuroendocrinology 101 (4): 321-30, 2015.[PUBMED Abstract]
- Span PN, Rao JU, Oude Ophuis SB, et al.: Overexpression of the natural antisense hypoxia-inducible factor-1alpha transcript is associated with malignant pheochromocytoma/paraganglioma. Endocr Relat Cancer 18 (3): 323-31, 2011.[PUBMED Abstract]
- Janssen I, Blanchet EM, Adams K, et al.: Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res 21 (17): 3888-95, 2015.[PUBMED Abstract]
- Pacak K, Eisenhofer G, Ahlman H, et al.: Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 3 (2): 92-102, 2007.[PUBMED Abstract]
- Lips CJ, Landsvater RM, Höppener JW, et al.: Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Engl J Med 331 (13): 828-35, 1994.[PUBMED Abstract]
- van der Harst E, de Herder WW, Bruining HA, et al.: [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. J Clin Endocrinol Metab 86 (2): 685-93, 2001.[PUBMED Abstract]
- Pacak K, Linehan WM, Eisenhofer G, et al.: Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 134 (4): 315-29, 2001.[PUBMED Abstract]
- Jafri M, Whitworth J, Rattenberry E, et al.: Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin Endocrinol (Oxf) 78 (6): 898-906, 2013.[PUBMED Abstract]
- Pęczkowska M, Kowalska A, Sygut J, et al.: Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol (Oxf) 79 (6): 817-23, 2013.[PUBMED Abstract]
- Karasek D, Frysak Z, Pacak K: Genetic testing for pheochromocytoma. Curr Hypertens Rep 12 (6): 456-64, 2010.[PUBMED Abstract]
- Neumann HP, Bausch B, McWhinney SR, et al.: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346 (19): 1459-66, 2002.[PUBMED Abstract]
- Fishbein L, Merrill S, Fraker DL, et al.: Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 20 (5): 1444-50, 2013.[PUBMED Abstract]
- Gómez AM, Soares DC, Costa AAB, et al.: Pheochromocytoma and paraganglioma: implications of germline mutation investigation for treatment, screening, and surveillance. Arch Endocrinol Metab 63 (4): 369-375, 2019.[PUBMED Abstract]
- Bacca A, Sellari Franceschini S, Carrara D, et al.: Sporadic or familial head neck paragangliomas enrolled in a single center: clinical presentation and genotype/phenotype correlations. Head Neck 35 (1): 23-7, 2013.[PUBMED Abstract]
- Babic B, Patel D, Aufforth R, et al.: Pediatric patients with pheochromocytoma and paraganglioma should have routine preoperative genetic testing for common susceptibility genes in addition to imaging to detect extra-adrenal and metastatic tumors. Surgery 161 (1): 220-227, 2017.[PUBMED Abstract]
- Rijken JA, Niemeijer ND, Corssmit EP, et al.: Low penetrance of paraganglioma and pheochromocytoma in an extended kindred with a germline SDHB exon 3 deletion. Clin Genet 89 (1): 128-32, 2016.[PUBMED Abstract]
- Lenders JW, Duh QY, Eisenhofer G, et al.: Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99 (6): 1915-42, 2014.[PUBMED Abstract]
- Hussain I, Husain Q, Baredes S, et al.: Molecular genetics of paragangliomas of the skull base and head and neck region: implications for medical and surgical management. J Neurosurg 120 (2): 321-30, 2014.[PUBMED Abstract]
- Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, et al.: Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43 (7): 663-7, 2011.[PUBMED Abstract]
- Neumann HP, Sullivan M, Winter A, et al.: Germline mutations of the TMEM127 gene in patients with paraganglioma of head and neck and extraadrenal abdominal sites. J Clin Endocrinol Metab 96 (8): E1279-82, 2011.[PUBMED Abstract]
- Burnichon N, Lepoutre-Lussey C, Laffaire J, et al.: A novel TMEM127 mutation in a patient with familial bilateral pheochromocytoma. Eur J Endocrinol 164 (1): 141-5, 2011.[PUBMED Abstract]
- Bayley JP, Kunst HP, Cascon A, et al.: SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol 11 (4): 366-72, 2010.[PUBMED Abstract]
- Letouzé E, Martinelli C, Loriot C, et al.: SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23 (6): 739-52, 2013.[PUBMED Abstract]
- van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, et al.: Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet 2 (8675): 1291-4, 1989.[PUBMED Abstract]
- Baysal BE: Mitochondrial complex II and genomic imprinting in inheritance of paraganglioma tumors. Biochim Biophys Acta 1827 (5): 573-7, 2013.[PUBMED Abstract]
- Amar L, Bertherat J, Baudin E, et al.: Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23 (34): 8812-8, 2005.[PUBMED Abstract]
- Neumann HP, Erlic Z, Boedeker CC, et al.: Clinical predictors for germline mutations in head and neck paraganglioma patients: cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res 69 (8): 3650-6, 2009.[PUBMED Abstract]
- Rattenberry E, Vialard L, Yeung A, et al.: A comprehensive next generation sequencing-based genetic testing strategy to improve diagnosis of inherited pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 98 (7): E1248-56, 2013.[PUBMED Abstract]
- Sbardella E, Cranston T, Isidori AM, et al.: Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine 59 (1): 175-182, 2018.[PUBMED Abstract]
- Neumann HP, Pawlu C, Peczkowska M, et al.: Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292 (8): 943-51, 2004.[PUBMED Abstract]
- Ricketts CJ, Forman JR, Rattenberry E, et al.: Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 31 (1): 41-51, 2010.[PUBMED Abstract]
- Welander J, Söderkvist P, Gimm O: Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 18 (6): R253-76, 2011.[PUBMED Abstract]
- Schiavi F, Milne RL, Anda E, et al.: Are we overestimating the penetrance of mutations in SDHB? Hum Mutat 31 (6): 761-2, 2010.[PUBMED Abstract]
- Solis DC, Burnichon N, Timmers HJ, et al.: Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin Genet 75 (4): 354-63, 2009.[PUBMED Abstract]
- Hes FJ, Weiss MM, Woortman SA, et al.: Low penetrance of a SDHB mutation in a large Dutch paraganglioma family. BMC Med Genet 11: 92, 2010.[PUBMED Abstract]
- Rijken JA, Niemeijer ND, Jonker MA, et al.: The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin Genet 93 (1): 60-66, 2018.[PUBMED Abstract]
- Jochmanova I, Wolf KI, King KS, et al.: SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. J Cancer Res Clin Oncol 143 (8): 1421-1435, 2017.[PUBMED Abstract]
- Schiavi F, Boedeker CC, Bausch B, et al.: Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 294 (16): 2057-63, 2005.[PUBMED Abstract]
- Peczkowska M, Cascon A, Prejbisz A, et al.: Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab 4 (2): 111-5, 2008.[PUBMED Abstract]
- Pasini B, McWhinney SR, Bei T, et al.: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 16 (1): 79-88, 2008.[PUBMED Abstract]
- Horváth R, Abicht A, Holinski-Feder E, et al.: Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry 77 (1): 74-6, 2006.[PUBMED Abstract]
- van der Tuin K, Mensenkamp AR, Tops CMJ, et al.: Clinical Aspects of SDHA-Related Pheochromocytoma and Paraganglioma: A Nationwide Study. J Clin Endocrinol Metab 103 (2): 438-445, 2018.[PUBMED Abstract]
- Burnichon N, Brière JJ, Libé R, et al.: SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 19 (15): 3011-20, 2010.[PUBMED Abstract]
- Korpershoek E, Favier J, Gaal J, et al.: SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab 96 (9): E1472-6, 2011.[PUBMED Abstract]
- Grandori C, Cowley SM, James LP, et al.: The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 16: 653-99, 2000.[PUBMED Abstract]
- Bausch B, Schiavi F, Ni Y, et al.: Clinical Characterization of the Pheochromocytoma and Paraganglioma Susceptibility Genes SDHA, TMEM127, MAX, and SDHAF2 for Gene-Informed Prevention. JAMA Oncol 3 (9): 1204-1212, 2017.[PUBMED Abstract]
- Fishbein L, Nathanson KL: Pheochromocytoma and Paraganglioma Susceptibility Genes: Estimating the Associated Risk of Disease. JAMA Oncol 3 (9): 1212-1213, 2017.[PUBMED Abstract]
- Schiffman JD: No child left behind in SDHB testing for paragangliomas and pheochromocytomas. J Clin Oncol 29 (31): 4070-2, 2011.[PUBMED Abstract]
- Jasperson KW, Kohlmann W, Gammon A, et al.: Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Fam Cancer 13 (2): 257-65, 2014.[PUBMED Abstract]
- Gravel G, Niccoli P, Rohmer V, et al.: The value of a rapid contrast-enhanced angio-MRI protocol in the detection of head and neck paragangliomas in SDHx mutations carriers: a retrospective study on behalf of the PGL.EVA investigators. Eur Radiol 26 (6): 1696-704, 2016.[PUBMED Abstract]
- Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, et al.: Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 16 (2): 391-400, 2009.[PUBMED Abstract]
- Lefebvre M, Foulkes WD: Pheochromocytoma and paraganglioma syndromes: genetics and management update. Curr Oncol 21 (1): e8-e17, 2014.[PUBMED Abstract]
- Pacak K: Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 92 (11): 4069-79, 2007.[PUBMED Abstract]
- GRAHAM JB: Pheochromocytoma and hypertension; an analysis of 207 cases. Int Abstr Surg 92 (2): 105-21, 1951.[PUBMED Abstract]
- Goldstein RE, O'Neill JA, Holcomb GW, et al.: Clinical experience over 48 years with pheochromocytoma. Ann Surg 229 (6): 755-64; discussion 764-6, 1999.[PUBMED Abstract]
- Miura Y, Yoshinaga K: Doxazosin: a newly developed, selective alpha 1-inhibitor in the management of patients with pheochromocytoma. Am Heart J 116 (6 Pt 2): 1785-9, 1988.[PUBMED Abstract]
- Prys-Roberts C, Farndon JR: Efficacy and safety of doxazosin for perioperative management of patients with pheochromocytoma. World J Surg 26 (8): 1037-42, 2002.[PUBMED Abstract]
- Zhu Y, He HC, Su TW, et al.: Selective α1-adrenoceptor antagonist (controlled release tablets) in preoperative management of pheochromocytoma. Endocrine 38 (2): 254-9, 2010.[PUBMED Abstract]
- Ross EJ, Prichard BN, Kaufman L, et al.: Preoperative and operative management of patients with phaeochromocytoma. Br Med J 1 (5534): 191-8, 1967.[PUBMED Abstract]
- Hack HA: The perioperative management of children with phaeochromocytoma. Paediatr Anaesth 10 (5): 463-76, 2000.[PUBMED Abstract]
- Proye C, Thevenin D, Cecat P, et al.: Exclusive use of calcium channel blockers in preoperative and intraoperative control of pheochromocytomas: hemodynamics and free catecholamine assays in ten consecutive patients. Surgery 106 (6): 1149-54, 1989.[PUBMED Abstract]
- Vargas HI, Kavoussi LR, Bartlett DL, et al.: Laparoscopic adrenalectomy: a new standard of care. Urology 49 (5): 673-8, 1997.[PUBMED Abstract]
- Perrier ND, Kennamer DL, Bao R, et al.: Posterior retroperitoneoscopic adrenalectomy: preferred technique for removal of benign tumors and isolated metastases. Ann Surg 248 (4): 666-74, 2008.[PUBMED Abstract]
- Dickson PV, Jimenez C, Chisholm GB, et al.: Posterior retroperitoneoscopic adrenalectomy: a contemporary American experience. J Am Coll Surg 212 (4): 659-65; discussion 665-7, 2011.[PUBMED Abstract]
- Ober WB: Emil Zuckerkandl and his delightful little organ. Pathol Annu 18 Pt 1: 103-19, 1983.[PUBMED Abstract]
- Mundschenk J, Lehnert H: Malignant pheochromocytoma. Exp Clin Endocrinol Diabetes 106 (5): 373-6, 1998.[PUBMED Abstract]
- Nockel P, El Lakis M, Gaitanidis A, et al.: Preoperative genetic testing in pheochromocytomas and paragangliomas influences the surgical approach and the extent of adrenal surgery. Surgery 163 (1): 191-196, 2018.[PUBMED Abstract]
- Grubbs EG, Rich TA, Ng C, et al.: Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J Am Coll Surg 216 (2): 280-9, 2013.[PUBMED Abstract]
- Castinetti F, Qi XP, Walz MK, et al.: Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol 15 (6): 648-55, 2014.[PUBMED Abstract]
- カーニー-ストラタキス症候群
-
臨床記述
カーニー-ストラタキス症候群(CSS;カーニー-ストラタキスの二徴としても知られる)は2002年に初めて報告された。この症候群は名前は似ているが、カーニー複合およびカーニーの三徴とは全く異なる(表6を参照のこと)。CSSは、不完全浸透を示すコハク酸デヒドロゲナーゼ(SDH)サブユニットB、C、またはD(SDHx)遺伝子における常染色体優性生殖細胞病原性多様体を特徴とする。罹患者は、多病巣性の局所で侵攻性を示す消化管間質腫瘍(GIST)および多発性の頸部、胸腔内、および腹腔内傍神経節腫(PGL)を比較的若年で発症する。[ 1 ][ 2 ][ 3 ]CSS関連GISTおよびPGLは、散発性に発生するより一般的なこれらの疾患とは異なる表現型を呈する;その結果、CSS患者における画像検査、治療、およびサーベイランスにおける特有の特徴を理解することが重要である。
表6.カーニー-ストラタキス症候群、カーニーの三徴、およびカーニー複合の比較 症候群 遺伝パターン 発症時平均年齢(歳) 罹患する性別 関連病変 病原性多様体 腫瘍の挙動 AD = 常染色体優性;GIST = 消化管間質腫瘍;F = 女性;M = 男性。 カーニー-ストラタキス症候群[ 1 ][ 3 ][ 4 ] AD 23 M、F 傍神経節腫、胃類上皮細胞型GIST SDHxの生殖細胞病原性多様体がよく認められる;KITまたはPDGFRAの病原性多様体は認められない GIST転移が認められるが、遷延性の経過;傍神経節腫は侵攻性 カーニーの三徴[ 4 ][ 5 ][ 6 ] なし <30 95%超がF 肺軟骨腫、傍神経節腫、胃類上皮細胞型GIST KITまたはPDGFRAの病原性多様体は認められない;まれに、SDHx病原性多様体(1シリーズで9.5%)[ 7 ] GIST転移が認められるが、遷延性の経過 カーニー複合[ 8 ][ 9 ] AD 20 M、F 黒子、粘液腫、シュワン腫、甲状腺濾胞状腺腫またはがん、原発性色素性かつ結節性の副腎皮質疾患、下垂体腺腫 PRKAR1A生殖細胞病原性多様体 該当せず 遺伝的特徴、遺伝的形質、および遺伝子検査
CSS関連GISTの腫瘍発生は、大多数のGISTでみられるようなKITまたはPDGFRA遺伝子における機能獲得型変異よりも、むしろコハク酸デヒドロゲナーゼ欠乏が関与しているようである。[ 10 ]SDH欠乏はまた、小児型GISTの特徴的な所見である;CSS関連GISTは、これらの腫瘍に類似した、若年での発症(年齢中央値、19歳)、胃への特異性、多病巣性、イマチニブに対する抵抗性といった臨床所見を呈する。[ 3 ][ 11 ][ 12 ][ 13 ]さらに、SDH欠乏GISTは所属リンパ節、腹膜腔、および肝臓に頻繁に転移するため、腫瘍径および細胞分裂の割合では転移の可能性または生存を正確に予測できない;しかしながら、長期の生存が一般的である。[ 6 ][ 14 ]
CSSに関与する遺伝子の遺伝子検査に関する詳しい情報については、本要約の家族性褐色細胞腫およびPGL症候群のセクションの遺伝的特徴、遺伝的形質、および遺伝子検査のサブセクションを参照のこと。
サーベイランス
CSSの自然史はほとんど解明されていないが、専門家は、進行中のサーベイランスに次のものを含めるように推奨している:貧血およびカテコールアミン過剰の症状に焦点を当てた年1回の現病歴の聴取を行う患者の綿密な追跡、身体診察、PGLの再発を発見するための血漿メタネフリン濃度およびクロモグラニンAによる生化学分析、および横断的画像検査。PGLの多くはカテコールアミンを分泌しないが、PGLにおいてクロモグラニンAが高くなることが明らかにされており、腫瘍再発に対する有用なマーカーである可能性がある。スクリーニングに適した画像検査法は現時点で不明であるが、フッ素18-フルデオキシグルコース・ポジトロン放射断層撮影-コンピュータ断層撮影(18F-FDG PET-CT)は副腎外のPGLおよびGISTの同定において感度が高い。CTによる電離放射線曝露のリスクがあるため、年1回のサーベイランスにはMRIの使用を提唱する者もいる。[ 15 ][ 16 ]
証拠レベル:4
介入
多発性原発GISTおよびPGLはCSSでは一般的であるため、手術の計画前に腫瘍の範囲を正確に確認するための術前の画像検査が、何よりも重要である。ほとんどの患者が初診時にCTまたは磁気共鳴画像法(MRI)による画像検査を既に受けている。どちらの画像検査法もPGLの同定に対する感度がきわめて優れているが、これらの腫瘍はびまん性の性質を有するため、機能的画像検査の追加が推奨される。18F-FDG PET-CTは、SDHx関連PGLの同定においてヨウ素123-メタヨードベンジルグアニジンよりも優れており、GISTでは代謝活性が高いため、これらの同定における感度がきわめて優れている。[ 15 ][ 17 ]したがって、CSSの患者を含めてSDHx病原性多様体を有する患者では、すべてのGISTおよびPGLを最適に発見し、病期分類するために18F-FDG PET-CTが望ましい機能的画像検査法である。[ 16 ]原発性PGLの同定には18F-フルオロ-L-ジヒドロキシフェニルアラニン(18F-FDOPA)PET-CTが優れている一方、転移の同定には18F-FDG PET-CTが優れていることを示唆する証拠がある。
CSSの患者を対象にしたプロスペクティブな治療研究は実施されていない;したがって、推奨は、限られた臨床経験、単一のケースシリーズ、臨床像が似た遺伝的に類似した腫瘍からの外挿に基づいている。CSS関連GISTおよびPGLに対する治療の中心は、腫瘍の外科的完全切除である。手術の時期は腫瘍の発生に相関する。外科的切除は腹腔鏡下手術または開腹手術のいずれでも達成できる。PGLについては、血管再建はまれである。PGLは一般に傍大動脈領域に認められるが、大血管の再建が必要となることはまれである。GIST腫瘍は楔状切除および一次的閉鎖と再吻合術により切除可能である。完全切除が達成された患者で生存が最も長くなるため、切除断端陰性を確実に達成することが重要である。[ 18 ]同時性病変が認められるまれな状況では、患者が耐えられるようであれば合併切除が適切である。より一般的には、腫瘍は異時性に発生する(GISTが最初に発生);各腫瘍の診断時に個別に切除される。
複数に分離したGISTが頻繁に認められるため、術前の徹底的な内視鏡検査および胃の完全な術中精査が必須である。多病巣性の頻度が高く、腫瘍再発の可能性が高いからといって、予防的な胃全摘術はこれに伴う合併症発生率が実質的に高くなるため、正当化されない。さらに、胃全摘術は一般的に、現在の疾患負担によって全摘術より少ない切除が受け入れられない場合にのみ実施される。このために、肉眼的切除断端陰性での胃楔状切除が手術の目標である。[ 19 ]切除時に疑わしいリンパ節があれば、一般的にすべて検体が採取される。局所進行CSS関連GISTはいくぶん潜行性の経過を示すことを示唆する証拠がある[ 20 ];したがって、術前の画像検査または腹部探索に基づいてリンパ節転移の懸念があれば原発腫瘍の切除を躊躇する必要はない。局所進行成人型GISTにおいて切除可能性を改善するまたは切除の負担を軽減するために、術前イマチニブの役割が広く記述されている一方、局所進行SDH欠乏GISTに効果がある可能性は低い。[ 21 ]これらの腫瘍に対して、ソラフェニブ、スニチニブ、ダサチニブ、ニロチニブなどの第二選択標的薬物が、補助療法の設定において有益な可能性があることを示唆する証拠がある。[ 22 ][ 23 ]現時点で、術前補助療法の設定におけるこれらの薬物の使用を支持するデータは存在しない。
CSS関連PGLの治療に関して、患者は一般的に、周術期の心疾患罹病率および死亡率を最低限に抑えるため術前のα遮断薬から開始される。PGLは典型的に、膀胱および大動脈分岐部から上縦隔および頭頸部に及ぶ一連の大動脈周囲領域に発生する。GISTの治療と同様に、手術の目標は既知の病変をすべて切除することである。この目標を達成するために術前の画像検査および術中の探索は不可欠である。多発性腫瘍が一般的である;病変が両側副腎に認められる場合、外科医は、患者を生涯にわたって致死性のアジソン病クリーゼのリスクを伴うステロイド依存状態に陥らせてしまう可能性に直面する。このような状況では、皮質を残す副腎摘出術の実施に熟練した外科医に助言を求めてもよい。
証拠レベル:5
参考文献- Carney JA, Stratakis CA: Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 108 (2): 132-9, 2002.[PUBMED Abstract]
- McWhinney SR, Pasini B, Stratakis CA, et al.: Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med 357 (10): 1054-6, 2007.[PUBMED Abstract]
- Pasini B, McWhinney SR, Bei T, et al.: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 16 (1): 79-88, 2008.[PUBMED Abstract]
- Gaal J, Stratakis CA, Carney JA, et al.: SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol 24 (1): 147-51, 2011.[PUBMED Abstract]
- Agaimy A, Pelz AF, Corless CL, et al.: Epithelioid gastric stromal tumours of the antrum in young females with the Carney triad: a report of three new cases with mutational analysis and comparative genomic hybridization. Oncol Rep 18 (1): 9-15, 2007.[PUBMED Abstract]
- Zhang L, Smyrk TC, Young WF, et al.: Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol 34 (1): 53-64, 2010.[PUBMED Abstract]
- Boikos SA, Xekouki P, Fumagalli E, et al.: Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur J Hum Genet 24 (4): 569-73, 2016.[PUBMED Abstract]
- Boikos SA, Stratakis CA: Carney complex: pathology and molecular genetics. Neuroendocrinology 83 (3-4): 189-99, 2006.[PUBMED Abstract]
- Correa R, Salpea P, Stratakis CA: Carney complex: an update. Eur J Endocrinol 173 (4): M85-97, 2015.[PUBMED Abstract]
- Hensen EF, Bayley JP: Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam Cancer 10 (2): 355-63, 2011.[PUBMED Abstract]
- Agaram NP, Laquaglia MP, Ustun B, et al.: Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res 14 (10): 3204-15, 2008.[PUBMED Abstract]
- Miettinen M, Wang ZF, Sarlomo-Rikala M, et al.: Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol 35 (11): 1712-21, 2011.[PUBMED Abstract]
- Sawhney SA, Chapman AD, Carney JA, et al.: Incomplete Carney triad--a review of two cases. QJM 102 (9): 649-53, 2009.[PUBMED Abstract]
- Rege TA, Wagner AJ, Corless CL, et al.: "Pediatric-type" gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior. Am J Surg Pathol 35 (4): 495-504, 2011.[PUBMED Abstract]
- Ayala-Ramirez M, Callender GG, Kupferman ME, et al.: Paraganglioma syndrome type 1 in a patient with Carney-Stratakis syndrome. Nat Rev Endocrinol 6 (2): 110-5, 2010.[PUBMED Abstract]
- Timmers HJ, Kozupa A, Chen CC, et al.: Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol 25 (16): 2262-9, 2007.[PUBMED Abstract]
- Timmers HJ, Chen CC, Carrasquillo JA, et al.: Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 94 (12): 4757-67, 2009.[PUBMED Abstract]
- Abadin SS, Ayala-Ramirez M, Jimenez C, et al.: Impact of surgical resection for subdiaphragmatic paragangliomas. World J Surg 38 (3): 733-41, 2014.[PUBMED Abstract]
- Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)--update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.[PUBMED Abstract]
- Maki RG, Blay JY, Demetri GD, et al.: Key Issues in the Clinical Management of Gastrointestinal Stromal Tumors: An Expert Discussion. Oncologist 20 (7): 823-30, 2015.[PUBMED Abstract]
- Ganjoo KN, Villalobos VM, Kamaya A, et al.: A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann Oncol 25 (1): 236-40, 2014.[PUBMED Abstract]
- Gill AJ, Chou A, Vilain R, et al.: Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol 34 (5): 636-44, 2010.[PUBMED Abstract]
- Janeway KA, Albritton KH, Van Den Abbeele AD, et al.: Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 52 (7): 767-71, 2009.[PUBMED Abstract]
- 家族性非髄様甲状腺がん
-
臨床記述
さまざまな組織学的亜型が存在する乳頭がんおよび濾胞がんは、甲状腺の濾胞細胞から発生し、分化型甲状腺がんまたは甲状腺非髄様がん(NMTC)と総称される。甲状腺乳頭がん(PTC)は最も多い甲状腺がんの病型であり、全症例の85%近くを占め、世界中で急速に増加している。[ 1 ][ 2 ]
放射線曝露、特に小児期の曝露は、甲状腺がん発生の原因因子として広く研究されてきたが、この因子はごく少数の症例しか説明しない。[ 3 ][ 4 ]甲状腺がんの発生に関する最も強力な危険因子の1つは、家族性甲状腺非髄様がん(FNMTC)と呼ばれる疾患の家族歴である。FNMTCの正確な発生率を確定することは、各研究間で遺伝性病態の認定に用いられている基準が異なるために困難である。しかし、罹患した近親者の数とそれらとの関係(すなわち、第一度近親者、第二度近親者など)、遺伝パターン、共存する甲状腺の病態の有無などの基準は、広く定義する必要がある。
遺伝性疾患と散発性疾患の区別をさらに混同させる因子は、手術や剖検の10~15%に偶発的な微小がんがみられるという有病率の高さである。[ 5 ]大部分の家系内では継承される遺伝子が特定されていないため、背景にあるこの疾患の高い有病率により、他の家系員における甲状腺悪性腫瘍のリスク評価が困難になっている。こうした不確実さは、特に遺伝性疾患の境界例で、例えば甲状腺がんを有する第二度近親者がいる場合などに、問題となることがある。
FNMTCは、より大きな症候群の一部として発生し他の臓器の腫瘍を伴う場合や、単独の病態として発生する場合がある。表7は、NMTCに関連する各種の遺伝性症候群を示している。
遺伝的特徴、遺伝的形質、および遺伝子検査
多発性内分泌腫瘍2型に関連する家族性甲状腺髄様がん(FMTC)の遺伝学は、十分に確立されている。遺伝的因子も明らかにNMTCに寄与しており、あらゆるがん部位のうち最も遺伝率が高い部位の1つに数えられ、患者の近親者の相対リスクは5~10倍であり、特に(女性の)同胞では顕著である。[ 6 ][ 7 ][ 8 ][ 9 ]乳頭がんが主で、濾胞がんの亜型も含まれるFNMTCは、NMTC全症例の5~10%を占めると考えられている。[ 6 ][ 10 ][ 11 ]注目したいのは、家系内の罹患者が2人しかいなければ、疾患が実際には散発性である可能性が40~60%であるが、家系内に3人以上罹患者がいる場合は、96%の確率でこの疾患に遺伝性の要素が関与していることである。[ 12 ]少数の例外としてNMTCを伴う数種類のまれな遺伝的症候群も存在するが、FNMTCの大半は無症候性であり、基礎にある遺伝的素因ははっきりしない。それでも、FNMTC家系(pedigree)は不完全浸透性と発現のばらつきを伴う常染色体優性の明確なメンデル型遺伝パターンを示すため、家族性がんという用語は多少の誤解を招きかねない。[ 6 ][ 13 ][ 14 ][ 15 ][ 16 ]しかし、FMTCとは異なり、FNMTCは多遺伝子性疾患であり、単一の座が大多数の症例または特定が容易な表現型に関与することはなく、浸透度の低い複数のアレルと環境的因子によって変化することが多い。[ 17 ]
症候性FNMTCの除外
無症候性FNMTCの臨床的遺伝子検査が存在しないため、リスクのある家系の識別は、臨床医が甲状腺のがんや疾患を呈する患者に対し、徹底した臨床検査と詳細な個人歴および家族歴の聴取を入念に行うことに頼らざるを得ない。FNMTCを示唆する病歴の特徴には、複数世代にわたる発症、早期発症型両側性/多病巣性の甲状腺腫瘍(特に男性)とより侵攻性の臨床経過、良性の甲状腺病変を伴っていることなどがある。[ 18 ]FNMTCは最終的に除外診断、つまり先にコーデン症候群や家族性大腸腺腫症といったNMTCに関連する他の家族性がん素因症候群を除外する必要があるため、精密な検査が非常に重要である。こうしたFNMTCに対する鑑別診断を表7に要約している。ただし、NMTCとマッキューン・オルブライト症候群、ポイツ・ジェガース症候群、毛細血管拡張性運動失調症、多発性内分泌腫瘍1型症候群との関連は、まだはっきりと確立されていない。
表7. 甲状腺非髄様がんに関連する遺伝性症候群a 症候群 遺伝子 遺伝形式 甲状腺がんの発生率(%) 甲状腺がんの種類 甲状腺外の臨床的特徴 FAP = 家族性大腸腺腫症;FTC = 甲状腺濾胞がん;MNG = 多発結節性甲状腺腫;PTC = 甲状腺乳頭がん。 a出典:Nose,[ 19 ] Sturgeon et al.,[ 20 ] and Vriens et al.[ 18 ] FAP/ガードナー症候群 APC 常染色体優性 2 PTC(篩状型) 消化管腺腫性ポリープ;ガードナー症候群では、デスモイド腫瘍、過剰歯、頭蓋の線維性異形成、骨腫、類表皮嚢胞、網膜上皮の肥厚などもみられる。 コーデン症候群(PTEN過誤腫症候群) PTEN(まれにSDHx、KLLN、AKT1、PIK3CA) 常染色体優性 10–35 FTC、PTC 乳房、子宮内膜、甲状腺、腎臓、消化管、脳、皮膚の悪性腫瘍および過誤腫。 カーニー複合 PRKAR1α 常染色体優性 11–15 FTC、PTC 軟部組織の粘液腫、皮膚および粘膜の色素沈着(青色母斑)、神経鞘腫、副腎、下垂体、精巣の腫瘍。 ウェルナー症候群 WRN 常染色体劣性 18 FTC、未分化PTC 早老(成人性プロジェリア)、強皮症様の皮膚変化、白内障、皮下のカルシウム沈着、筋萎縮症、糖尿病。 DICER1症候群 DICER1 常染色体優性 不明 PTC(およびMNG) 家族性胸膜肺芽腫症候群;嚢胞性腎腫;卵巣セルトリ・ライディッヒ細胞腫。 マッキューン・オルブライト症候群 GNAS モザイク体細胞変異 不明 FTC 多骨性線維性骨異形成症、カフェオレ斑、下垂体、副腎、性腺組織の内分泌機能亢進。 ポイツ・ジェガース症候群 STK11(LKB1) 常染色体優性 不明 主にPTC 小腸の過誤腫、粘膜皮膚の色素沈着過剰、精巣セルトリ細胞腫。 毛細血管拡張性運動失調症 ATM 常染色体劣性 不明 主にPTC 小脳性運動失調および眼振、眼皮膚毛細血管拡張症、免疫不全、リンパ網内系のがん。 多発性内分泌腫瘍1型(MEN1) MEN1 常染色体優性 不明 主にPTC 副甲状腺腫、胃腸膵管系内分泌腫瘍、下垂体前葉腫瘍。 無症候性FNMTCに関連する遺伝子と遺伝的多様体の特定
FNMTCに関連する遺伝学的多様性の全貌を明らかにするために各種の手法が用いられており、そのうちの主要な1つであるゲノムワイド連鎖解析では、ゲノム全体に均等に分布するマイクロサテライトマーカーと、複数の家系員が罹患した情報量の多い大規模家系(pedigree) を利用する。FNMTCに関連している遺伝子座は15以上判明しており、それらは表8に要約されている。斜体で示されている遺伝子座は、これまでに特定された感受性遺伝子の位置を表しており、その他の座に存在する原因遺伝子は未だ不明である。最初の4座はマイクロサテライト連鎖解析により特定された。他の座は、高密度に存在する一塩基多型(SNP)アレイやマイクロRNAアレイによって特定されることが増えており、より新しい結果は次世代塩基配列決定法によって明らかにされている。(連鎖解析および次世代塩基配列決定法に関する詳しい情報については、がん遺伝学の概要のPDQ要約を参照のこと。)これらの研究のほとんどは、FNMTCに一致する家系(pedigree) の家族群を対象として行われたが、2つの遺伝子座はより大規模な集団レベルでのSNPアレイ解析で特定された。複数の研究で注目すべきこととして、散発性のNMTCに関連する体細胞性の変化を最もよく起こす遺伝子、すなわちBRAF、RET、RET/PTC、MET、MEK1、MEK2、RAS、NTRKが、FNMTCに関与するものから除外されている点がある。[ 21 ]
表8. 無症候性の家族性非髄様甲状腺がん(Familial Nonmedullary Thyroid Cancer) 感受性遺伝子座 遺伝子座 位置 腫瘍の種類 サンプルサイズ 研究の種類 元のコホート国 年 参照文献 MNG1 14q31 PTCを伴うMNG 1 家系(kindred) マイクロサテライト連鎖解析 カナダ 1997 [ 22 ] 18 MNG 2 PTC TCO 19p13.2 好酸性細胞型PTC 1家系(kindred) マイクロサテライト連鎖解析 フランス 1998 [ 23 ][ 24 ][ 25 ][ 26 ] 20家族 6 MNG 3 PTC 49 NMTC fPTC/PRN 1q21 PRNを伴うPTC 1家系(kindred) マイクロサテライト連鎖解析 米国 2000 [ 27 ] 5 PTC 2 PRN NMTC1 2q21 PTC(濾胞型) 1家系(kindred)、80家系(pedigree) マイクロサテライト連鎖解析 タスマニア 2001 [ 25 ][ 26 ][ 28 ] 19家族 49 NMTC FTEN 8p23.1-p22 PTC(古典型) 1家系(kindred) 10K SNPアレイ ポルトガル 2008 [ 29 ] 11良性例 5 NMTC 不明 8q24 黒色腫を伴うPTC 26家族 50K SNPアレイ 米国 2009 [ 30 ] FOXE1 9q22.33 PTC/FTC 60家族 300K SNPアレイ アイスランド/スペイン/米国 2009 [ 31 ] 197 PTC/FTC NKX2-1/TITF-1 14q13.3 PTCおよびMNG 60家族 300K SNPアレイ アイスランド/米国/スペイン 2009 [ 31 ] 197 PTC/FTC 不明 6q22 PTC/FTC(古典型) 38家族 50K SNPアレイ 米国/イタリア 2009 [ 32 ] 49 PTC miR-886-3p 5q31.2 PTC 21 PTC 3K miRNAアレイ 米国 2011 [ 33 ] 7 FNMTC 10の正常な甲状腺組織 miR-20a 13q31.3 PTC 21 PTC 3K miRNAアレイ 米国 2011 [ 33 ] 7 FNMTC 10の正常な甲状腺組織 テロメア-テロメラーゼ複合体(TERT、TRF1、TFR2、RAP1、TIN2、TPP1、POT1) 5p15.3(TERT)など。 PTC 47 PTC 2008 [ 34 ] SRGAP1 12q14 PTC 38家族 250K SNPアレイ 米国/ポーランド 2013 [ 35 ] HAPB2 10q25-26 PTC、濾胞状腺腫 1家系(kindred) WES 米国 2015 [ 36 ] 7 PTC RTFC(c14orf93) 14q11.2 PTC 15家族 WES 中国 2017 [ 37 ] 連鎖解析により特定された感受性遺伝子座
MNG1、TCO、fPTC/PRN、NMTC1は、複数の罹患者を含む家族で特定されたFNMTC感受性遺伝子座として提案され、表8に要約されている。これらの遺伝子座に対する連鎖については、相反する証拠が存在する。MNG1では、複数の多発結節性甲状腺腫(MNG)がみられるカナダの1家系(kindred)でのみ強力な証拠が得られており、追加の124家族で行われた連鎖解析ではMNG1とFNMTCとの関連は明らかにならなかった。したがって、この遺伝子座はMNG単独に対して重要であるが、FNMTCに対しては重要でない可能性がある。[ 22 ][ 23 ][ 27 ][ 38 ][ 39 ][ 40 ]TCOはFNMTC症例の少数を説明するが、好酸性細胞型腫瘍を伴うFNMTCに特異的であり、これは確認されたFNMTC症例の大多数には該当しないまれな形態である。[ 23 ][ 24 ][ 25 ][ 26 ]fPTC/PRNも、PTCに乳頭状腎新生物が伴う、まれなFNMTCの亜型であるが、報告された元の家族以外にこの表現型が認められる家族は特定されていない。[ 23 ][ 27 ][ 40 ]NMTC1は、PTCの別のまれな亜型である濾胞型の素因となるようである。古典的PTCと好酸性腫瘍もこの遺伝子座に関連があるが、程度はより低い。[ 25 ][ 25 ][ 28 ]2001年に各国の22のFNMTC家族を対象として実施された包括的な変異および連鎖解析によると、1家族のみにおいて、前述のものを含む既知の感受性遺伝子座(この例ではTCO)に対する有意な連鎖が認められた。[ 23 ]この累積された証拠から、これらのFNMTC遺伝子座はごく一部の家族で疾患を説明することが示唆され、これはFNMTCが遺伝的異質性および遺伝子座異質性を示すという見方に一致する。
ゲノムワイドSNPアレイにより特定された感受性遺伝子座
表8には、徐々に密度を高くしたSNPアレイにより特定された、5つのFNMTC遺伝子座も掲載されている。最初のSNPアレイを用いたFNMTC研究は、2008年にポルトガルの家族を対象として、マイクロサテライト解析と併せて実施された。[ 29 ]この家族にはPTCの家系員が5人(4例は古典例で1例は濾胞型)と良性甲状腺疾患の家系員が11人いた。この研究では、感受性遺伝子座が8p23.1-p22に特定され、FTEN(familial thyroid epithelial neoplasia)と命名された。8q24は26のFNMTC家族(PTCを有する)のSNPアレイを使用した連鎖解析から特定された最初の遺伝子座である。1家族において、3世代にわたるPTCと黒色腫(およびMNG)が認められたが、黒色腫は他の25家族では報告されなかった。8q24領域における遺伝子の塩基配列決定では候補となる病原性多様体は特定されなかったが、遺伝子発現解析により、PTCで下方制御されるノンコーディングRNA遺伝子であるAK023948(PTSCC1)が関与している可能性が指摘された。[ 30 ]
2009年に、アイスランドでPTCまたはFTCの197例に対する集団レベルの研究が実施され、アイスランド人の対照37,196人の遺伝子型との比較が行われた。[ 31 ]遺伝子FOXE1とNKX2-1の近傍にそれぞれ存在する9q22.33と14q13.3の2つの遺伝子座には、高い統計的有意性が認められた。特に2つのSNPであるrs944289(NKX2-1近傍)とrs965513(FOXE1近傍)は、PTCおよびFTCのリスク増大に関連していた。これらの結果は米国(検査対象726人)およびスペイン(検査対象1,433人)の2つの大規模コホートでも確認され、またさらに他のコホートでも、特にFOXE1において、注目される別のSNPがいくつか見つかっている。[ 31 ][ 41 ][ 42 ]FOXE1は、甲状腺の形成、分化、機能に重要な役割を担う甲状腺転写因子を産出するため、現在でもFNMTCに関して注目される遺伝子である。[ 43 ]NKX2.1/TITF-1遺伝子も、甲状腺転写因子をコードする。生殖細胞多様体A339Vは、MNGまたはPTC/MNGを有するFNMTCの2家族で報告されている。[ 44 ]しかし、この関連は後続する研究の他の家族では確認できていない。[ 45 ]最後に、米国とイタリアの大規模コホート(28のFNMTC家族の個人110人、患者49人)について、50K SNPアレイを用いた研究が実施された。これらの家族の大多数で、古典的PTCが認められた。プール解析により、既知の1q21遺伝子座(PRN)に加え、6q22に新しい遺伝子座が見つかった。[ 32 ]
マイクロRNA(miRNA)感受性遺伝子座
miRNAは遺伝子発現を調節する微小なノンコーディングRNAである。全ゲノムmiRNAマイクロアレイを用いて、散発性NMTCの21例と家族性NMTCの7例、および正常な甲状腺組織標本10例の評価が行われた。[ 33 ]定量的逆転写-ポリメラーゼ連鎖反応(RT-PCR)により、miR-20a(13q31.3)およびmiR-886-3p(5q31.2)という2つのmiRNAが散発性と家族性のNMTCで別様に発現していたことが確かめられた。いずれのNMTCも、正常な甲状腺組織と比較して4倍、下方制御されていた。miR-886-3pを用いた細胞株トランスフェクション研究では、これが細胞増殖および細胞遊走に重要な役割を果たし、DNA複製および焦点接着斑の経路に関与する遺伝子を調節することが確認された。[ 33 ]さらに、pre-miR-146a(rs2910164)の多型がmiRNA発現に影響していることが明らかになり、PTC患者608人の腫瘍においてかなりの割合で特定されたことからは、この多型がPTCの遺伝的素因に寄与しており、体細胞性変化を介して腫瘍形成に何らかの役割を果たしている可能性が示唆される。[ 46 ]遺伝子調節機構の役割と、それが遺伝子発現およびFNMTC腫瘍形成に及ぼす影響について、さらなる研究が必要である。
テロメア-テロメラーゼ複合体
テロメアは、タンデムリピートで構成され、染色体安定性の維持に重要な非コード化染色体末端である。テロメア長はテロメラーゼ複合体によって維持され、この複合体には、テロメラーゼ逆転写酵素(TERT)とともにTRF1、TFR2、RAP1、TIN2、TPP1、POT1という別の6種類の蛋白が含まれる。[ 47 ]短くなったテロメア長は、がんの発生に寄与しうる染色体不安定性に関連する。また、テロメア-テロメラーゼ複合体は、FNMTCの素因となりうる別の遺伝的機序として、注目される研究対象である。2008年に、FNMTC患者のコホートに対する研究で、定量PCRおよび蛍光in situハイブリダイゼーションを用いて、相対テロメア長の評価が行われた。[ 34 ]その結果、家族性PTC患者のテロメア長は、罹患していない家族および散発性PTC患者に比べて、有意に短いことが明らかになった。さらにこのグループでは、FNMTCがんにおいてテロメアが比較的脆弱で、断片化する率が高いことも判明した。[ 48 ]FNMTCにおけるテロメア長に関する2番目の研究でも、患者13人のテロメア長は非罹患家族31人より短かった。[ 49 ]しかしこの研究では、相対テロメア長は、テロメア複合体遺伝子であるhTERT、TRF1、TFR2、RAP1、TIN2、TPP1、POT1のそれぞれのコピー数や発現の変化に関連していなかった。別の複数の研究では、FNMTCと散発性PTCの症例の間にテロメア長の有意差は一切みられなかった。[ 50 ]FNMTCの素因になるテロメア-テロメラーゼ複合体の役割や機序は、解明されていない。
近年特定された他のFNMTC感受性遺伝子と多様体
SRGAP1は、2013年に米国とポーランドのPTCを有する38のFNMTC家族のゲノムワイド連鎖解析によって特定された遺伝子である。[ 35 ]4つの生殖細胞ミスセンス多様体が特定されたが、Q149HおよびA275Tという2つの多様体は、2つの家族において分離されたが800例の散発例では分離されなかったため、特に注目に値する。SRGAP1はニューロン内で低分子量G蛋白CDC42を制御するほか、細胞遊走性に影響を及ぼす。[ 51 ]機能分析により、SRGAP1のQ149HおよびR617Cの両多様体により、CDC42を不活性化する能力が損なわれる機能喪失型の変化が生じ、腫瘍形成につながる可能性があることが示された。[ 35 ]さらなる研究により、他のFNMTCコホートでこの遺伝子の関連性を検証する必要がある。
HAPB2は、2015年にPTCと濾胞状腺腫を有するFNMTC家系(kindred)の7人について、非罹患の配偶者を対照として行われた全エクソーム配列解析(WES)により特定された。[ 36 ]すべての症例で、特異的な生殖細胞多様体のG534Eがヘテロ接合状態で認められた。この研究グループは、次世代塩基配列決定法により、Cancer Genome AtlasのNMTC例の4.7%にもこの多様体を見出した。この多様体は、正常な甲状腺組織または散発性PTCより、罹患した家族の甲状腺新生物で蛋白発現が多いことに関連していた。G534Eの機能検査では、コロニー形成と細胞遊走の増加が明らかになり、腫瘍抑制機能の喪失が示唆された。注目すべきは、著者らが1000 Genomes Project(phase III)およびHapMap3を利用して、一般集団での頻度である1%を基準とし、この家系(kindred)で特定された多様体を選別したことである。しかし、後のコメント論文で、Exome Aggregation Consortium(ExAC)(全集団で2.22%、フィンランド人以外のヨーロッパ人で3.29%)やNational Heart, Lung, and Blood Institute(NHLBI)Grand Opportunity Exome Sequencing Projectデータベース(全体で5.5%、ヨーロッパ系のアメリカ人で3.88%)などの公共のデータベースに基づいて、より高いG534E多様体の頻度が報告されていることが指摘された。[ 52 ][ 53 ][ 54 ]ほかにも、FNMTCにおけるHAPB2 G534Eの頻度を確認するために多くの研究が実施され、多様な結果が得られている。この多様体は、中国の12のPTCを伴うFNMTC家族では(散発性PTC患者217人でも)特定されず、[ 54 ]また中東の11のFNMTC家族でも特定されなかったが、[ 55 ]米国の研究により、PTCがみられる複数の独立したFNMTC家系(kindred)で分離されることが示された。[ 56 ]他の数件の研究では、G534E多様体が、たとえコホート全体で特定されても、あるいは家族内で疾患により分離されなくても、対照と散発例において家族性PTC例と同程度か、より頻繁に認められることが示された。[ 55 ][ 57 ][ 58 ][ 59 ]したがって、HAPB2 G534E多様体の頻度は複数の祖先と集団の間で異なっており、ヨーロッパ系の祖先をもつ集団では低~中程度の頻度であるが、アジア系および中東系の集団では低頻度か発生しないようである。この多様体の役割とFNMTCとの関連を確定するには、より大規模な妥当性研究が必要である。
最後に、中国のFNMTC家族のWESにより、RTFC(c14orf93)が特定された。[ 37 ]FNMTCに関連する候補遺伝子として3つの遺伝子(RTFC、PYGL、BMP4)が特定されたが、RTFC遺伝子だけが、飢餓状態で甲状腺がん細胞の生存を促し、細胞の遊走とコロニー形成能を促進する発がん性を有することが示された。特にRTFCのV205M(c.613G>C)多様体は、FNMTC患者において特定されたが、非罹患者からなる対照では特定されなかったため、重要である。さらにRTFCの発がん性変異が2つ(R115QおよびG209D)、散発性NMTC患者で特定された。総合的にみて、ExACの東アジア人集団にみられるこれらの多様体の頻度は全集団での頻度より高いことから、東アジアのFNMTC家族においては、この遺伝子が最も関連性の高いものである可能性がある。この遺伝子とこれらの多様体について、より大規模な妥当性研究を実施する必要がある。
要約すると、FNMTC家族で複数の感受性遺伝子座が特定されているが、無症候性FNMTCの大多数を説明する単一の遺伝子座はなく、臨床遺伝子検査の十分な実施理由になる強力な関連性を有する遺伝子も特定されていない。WESなどの新しい配列決定技術を用いることで、今後、新しい遺伝子の発見と評価が可能になるであろう。感受性遺伝子の特定により、スクリーニングと早期診断が可能になり、ひいては患者と家族の転帰の改善につながると考えられる。
サーベイランス
分化型甲状腺がんは、遺伝性か散発性かにかかわらず、疾患の臨床病理学的特徴によっては再発率が高い可能性がある。疾患再発は、最初の診断から40年経過後でも発生しうる。[ 60 ]そのため、再発疾患に対するサーベイランスは、こうした腫瘍を有する患者の長期管理において重要な役割を果たす。最適なフォローアップ戦略は、初期腫瘍の特徴と患者の治療に対する反応との両方に応じて異なる。[ 61 ]幸いなことに、ほとんどの患者では疾患の再発リスクは低く、それに応じてサーベイランスの強度は比較的低い。こうした症例における術後の評価は、頸部超音波検査と血清サイログロブリンの測定が中心である。[ 61 ]
サイログロブリンは、良性と悪性の両方の甲状腺濾胞細胞によって産生される蛋白で、分化型甲状腺がん患者では腫瘍マーカーとして使用される。サイログロブリン測定は甲状腺全摘術後の感度が最も高いため、サイログロブリンの検出-特に血清濃度の増加傾向-はしばしば疾患再発または進行の初期指標である。[ 62 ]しかしながら、この腫瘍マーカーの使用についてはいくつかの注意点を認識しておくことが重要である。測定ごとに血清甲状腺刺激ホルモン(TSH)とサイログロブリン抗体の値を同時に評価することが必須である。サイログロブリンはTSH値の増加とともに上昇する;したがって、サイログロブリン値の上昇は疾患の進行または単純にTSH値の増加を示している可能性がある。さらに、サイログロブリン抗体の存在はサイログロブリンの正確な測定を妨げる可能性があり、ほとんどの症例で腫瘍マーカーの見せかけの低下を招く。[ 63 ]このような症例では、疾患状態の代替のマーカーとして抗体価を使用できる。[ 61 ]腫瘍マーカーとしてのサイログロブリン使用に関する最後の注意点は、値の傾向を正確に評価するため、各測定時に検査は同じ検査室で実施しなければならないということである;それぞれのアッセイが、同じ血清サンプルで異なるサイログロブリン値を示す可能性がある。[ 61 ]持続性または再発疾患を評価するための血清サイログロブリンの測定は、治療完了から3~6ヵ月経過時に実施され、その後は持続性または再発疾患の懸念に応じて、定期的に監視される。[ 61 ]一部の患者、特に甲状腺濾胞がん患者または疾患の再発または残存が臨床的に強く疑われる患者では、刺激サイログロブリン検査(甲状腺ホルモンを中止しTSH最低値が30mIU/Lに達した後またはrecombinant TSH注入後)が有用なことがある。
患者が放射性ヨードを受けているか、手術のみを受けているかにかかわらず、前頸部のすべての区画の注意深い超音波検査は、ほとんどの病変がこの領域に存在しているため疾患の再発または残存があるかどうかを判断するために重要なツールである。最初の超音波検査は一般的に、手術から6~12ヵ月後に実施される。[ 61 ]疾患の残存が懸念される場合、超音波検査はより早期に実施されるが、音波検査者は術後腫脹による偽陽性所見の可能性を認識しておくことが重要である。その後の頸部超音波評価の時期と必要性については、患者の再発リスクと血清サイログロブリンの状態によって異なる。[ 61 ]
血清サイログロブリン検査と併用する超音波検査は、リンパ節病変の同定について非常に感度が高く、歴史的にサーベイランスの中心であった放射性ヨウ素による診断的全身スキャンよりもはるかに優れている。[ 62 ]
介入
甲状腺結節が検出された後、追加の精密検査として、甲状腺の完全な超音波検査と頸部全体の超音波検査による頸部中央および側方リンパ節の評価などを行う。結節の大きさ、画像所見、患者が有する関連のある危険因子から疑われる結節の細胞診検査には、穿刺吸引法(FNA)が適応である。[ 64 ][ 65 ]現在のほとんどのガイドラインは、10mm以上のすべての結節についてFNA生検を推奨している。最大径が10mm未満の結節でも、X線画像検査で悪性を疑わせる所見が得られた場合は、細胞診検査が必要な場合がある。悪性腫瘍が疑われる超音波所見には、低エコー、複雑な結節または充実性結節、血管分布、不規則な境界、石灰化などがある。術前の包括的な頸部超音波検査は、手術前に疑わしい結節のFNA生検を施行する機会になるだけでなく、外科医が適切な手術の計画を立て、外科手技と付随するリスクに関して患者とのカウンセリングを行う契機にもなる。[ 66 ]ポジトロン放射断層撮影スキャンは甲状腺結節の評価には推奨されないが、別の理由で行われたスキャンで、取り込まれた造影剤の集積が甲状腺に認められることがある。サーベイランス期間中の偶発的なフッ素18-フルオロデオキシグルコースの結合活性の増強と結節サイズの増大(容積の50%以上)も、結節のFNA生検の適応となることがある。[ 61 ]
細胞診検査と確定できない甲状腺結節
Bethesda Thyroid Cytology Classificationは甲状腺生検の細胞学的解釈を標準化している。病理学的な結果は、以下の6つのカテゴリーのいずれかに分類される。[ 67 ]
生検で証明された悪性結節(または悪性の疑いのある結節)を有する患者は、後述のように外科的切除が必要である。AUS/FLUSに分類される結節は、構造的または細胞学的異型性の程度から良性の診断が除外されるが、悪性と診断するには異型性の程度が不十分であるため、不確定カテゴリーに含まれる。[ 67 ]こうした病変では一般に、繰り返しFNAが施行され、結節の臨床所見が変化した場合や後の生検で再びAUS/FLUS分類の結果が出た場合に、外科的切除が行われる。
甲状腺がんの外科治療
FNMTCの診断を受けた患者は、散発例に比べて疾患の侵攻性が高くなる場合がある。[ 68 ]家系に罹患者が3人いる場合は、より若い年齢での腫瘍の特性が侵攻性である可能性が高い;これから、より積極的な外科的管理が必要になる場合があることが示唆される。[ 69 ]多中心性病変、リンパ節転移、局所浸潤、侵攻性疾患の再発の頻度が高まるリスクがあるため、ほとんどの専門家が甲状腺全摘術を支持する。[ 68 ][ 70 ]ほとんどの外科医は、FNMTC患者において、リンパ節転移がX線像や臨床所見または術中に疑われるか生検で証明された場合に、甲状腺全摘術と区域に基づくリンパ節群の治療的郭清を妥当とすることに同意するであろう。しかし、初回の甲状腺摘出術施行時に頸部中央区域の腫大していないリンパ節に対する適切な治療については議論がある。例えば、一部の研究グループは、FNMTCが既知の患者全員にルーチンで予防的中央区域リンパ節郭清(PCND)を施行し、局所再発のリスクを低下させる方法を提唱しているが、生存利益を支持する具体的でプロスペクティブなランダム化されたデータは存在しない。[ 71 ][ 72 ]2件のレトロスペクティブ研究(遺伝性甲状腺がんに限定したものではない)はPCNDに伴う疾患再発率の低下を報告しているが、[ 73 ][ 74 ]2件のメタアナリシスでは、PCNDによる再発率の臨床的に有意な低下は認められなかった。[ 75 ][ 76 ]American Thyroid Association(ATA)の最新の推奨事項では、T3/T4乳頭がん(家族性もそうでないものも)患者で、臨床的に側方リンパ節転移が認められるか、情報を用いて放射性ヨード療法などのさらなる治療が計画される場合は、レベルVIの予防的または両側リンパ節郭清が推奨される。またATAは、この推奨事項は利用可能な外科的専門知識に照らし、PCNDが周術期の罹病率を上昇させうることを認識した上で解釈されるべきであるとしている。[ 61 ]現在、ルーチンではなく選択的なPCNDは、決定過程の指針として最も合理的な選択肢とみられる。[ 77 ]
甲状腺全摘術の後、患者は生涯にわたる甲状腺ホルモン補充療法を受ける必要がある。[ 61 ]レボチロキシン補充療法の用量は約1.6μg/kg/日であり、その後適切なレベルのTSH抑制が得られるまで漸増する。[ 78 ][ 79 ]TSH抑制の程度も患者の病態、再発リスク、侵襲的なTSH抑制による心血管系および骨合併症の個人的リスク[ 61 ]、および腫瘍の臨床病理学的な特徴に基づいて個別化される。患者は一般的に、臨床検査評価と超音波検で構成される再発に対するサーベイランスレジメンを受ける。乳頭がんおよび濾胞がんでは、チロキシンとTSHが甲状腺抑制のレベルを示し、サイログロブリンとサイログロブリン抗体の値は、起こりうる再発や転移の重要なマーカーとなる。
参考文献- Aschebrook-Kilfoy B, Schechter RB, Shih YC, et al.: The clinical and economic burden of a sustained increase in thyroid cancer incidence. Cancer Epidemiol Biomarkers Prev 22 (7): 1252-9, 2013.[PUBMED Abstract]
- Aschebrook-Kilfoy B, Ward MH, Sabra MM, et al.: Thyroid cancer incidence patterns in the United States by histologic type, 1992-2006. Thyroid 21 (2): 125-34, 2011.[PUBMED Abstract]
- Bhatti P, Veiga LH, Ronckers CM, et al.: Risk of second primary thyroid cancer after radiotherapy for a childhood cancer in a large cohort study: an update from the childhood cancer survivor study. Radiat Res 174 (6): 741-52, 2010.[PUBMED Abstract]
- Mazonakis M, Tzedakis A, Damilakis J, et al.: Thyroid dose from common head and neck CT examinations in children: is there an excess risk for thyroid cancer induction? Eur Radiol 17 (5): 1352-7, 2007.[PUBMED Abstract]
- Pazaitou-Panayiotou K, Capezzone M, Pacini F: Clinical features and therapeutic implication of papillary thyroid microcarcinoma. Thyroid 17 (11): 1085-92, 2007.[PUBMED Abstract]
- Stoffer SS, Van Dyke DL, Bach JV, et al.: Familial papillary carcinoma of the thyroid. Am J Med Genet 25 (4): 775-82, 1986.[PUBMED Abstract]
- Goldgar DE, Easton DF, Cannon-Albright LA, et al.: Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst 86 (21): 1600-8, 1994.[PUBMED Abstract]
- Frich L, Glattre E, Akslen LA: Familial occurrence of nonmedullary thyroid cancer: a population-based study of 5673 first-degree relatives of thyroid cancer patients from Norway. Cancer Epidemiol Biomarkers Prev 10 (2): 113-7, 2001.[PUBMED Abstract]
- Hemminki K, Eng C, Chen B: Familial risks for nonmedullary thyroid cancer. J Clin Endocrinol Metab 90 (10): 5747-53, 2005.[PUBMED Abstract]
- Loh KC: Familial nonmedullary thyroid carcinoma: a meta-review of case series. Thyroid 7 (1): 107-13, 1997.[PUBMED Abstract]
- Lupoli G, Vitale G, Caraglia M, et al.: Familial papillary thyroid microcarcinoma: a new clinical entity. Lancet 353 (9153): 637-9, 1999.[PUBMED Abstract]
- Charkes ND: On the prevalence of familial nonmedullary thyroid cancer in multiply affected kindreds. Thyroid 16 (2): 181-6, 2006.[PUBMED Abstract]
- Lote K, Andersen K, Nordal E, et al.: Familial occurrence of papillary thyroid carcinoma. Cancer 46 (5): 1291-7, 1980.[PUBMED Abstract]
- Houlston RS, Stratton MR: Genetics of non-medullary thyroid cancer. QJM 88 (10): 685-93, 1995.[PUBMED Abstract]
- Burgess JR, Duffield A, Wilkinson SJ, et al.: Two families with an autosomal dominant inheritance pattern for papillary carcinoma of the thyroid. J Clin Endocrinol Metab 82 (2): 345-8, 1997.[PUBMED Abstract]
- Malchoff CD, Malchoff DM: Familial nonmedullary thyroid carcinoma. Cancer Control 13 (2): 106-10, 2006.[PUBMED Abstract]
- Khan A, Smellie J, Nutting C, et al.: Familial nonmedullary thyroid cancer: a review of the genetics. Thyroid 20 (7): 795-801, 2010.[PUBMED Abstract]
- Vriens MR, Suh I, Moses W, et al.: Clinical features and genetic predisposition to hereditary nonmedullary thyroid cancer. Thyroid 19 (12): 1343-9, 2009.[PUBMED Abstract]
- Nosé V: Familial thyroid cancer: a review. Mod Pathol 24 (Suppl 2): S19-33, 2011.[PUBMED Abstract]
- Sturgeon C, Clark OH: Familial nonmedullary thyroid cancer. Thyroid 15 (6): 588-93, 2005.[PUBMED Abstract]
- Hou P, Xing M: Absence of germline mutations in genes within the MAP kinase pathway in familial non-medullary thyroid cancer. Cell Cycle 5 (17): 2036-9, 2006.[PUBMED Abstract]
- Bignell GR, Canzian F, Shayeghi M, et al.: Familial nontoxic multinodular thyroid goiter locus maps to chromosome 14q but does not account for familial nonmedullary thyroid cancer. Am J Hum Genet 61 (5): 1123-30, 1997.[PUBMED Abstract]
- Bevan S, Pal T, Greenberg CR, et al.: A comprehensive analysis of MNG1, TCO1, fPTC, PTEN, TSHR, and TRKA in familial nonmedullary thyroid cancer: confirmation of linkage to TCO1. J Clin Endocrinol Metab 86 (8): 3701-4, 2001.[PUBMED Abstract]
- Canzian F, Amati P, Harach HR, et al.: A gene predisposing to familial thyroid tumors with cell oxyphilia maps to chromosome 19p13.2. Am J Hum Genet 63 (6): 1743-8, 1998.[PUBMED Abstract]
- McKay JD, Thompson D, Lesueur F, et al.: Evidence for interaction between the TCO and NMTC1 loci in familial non-medullary thyroid cancer. J Med Genet 41 (6): 407-12, 2004.[PUBMED Abstract]
- Prazeres HJ, Rodrigues F, Soares P, et al.: Loss of heterozygosity at 19p13.2 and 2q21 in tumours from familial clusters of non-medullary thyroid carcinoma. Fam Cancer 7 (2): 141-9, 2008.[PUBMED Abstract]
- Malchoff CD, Sarfarazi M, Tendler B, et al.: Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab 85 (5): 1758-64, 2000.[PUBMED Abstract]
- McKay JD, Lesueur F, Jonard L, et al.: Localization of a susceptibility gene for familial nonmedullary thyroid carcinoma to chromosome 2q21. Am J Hum Genet 69 (2): 440-6, 2001.[PUBMED Abstract]
- Cavaco BM, Batista PF, Sobrinho LG, et al.: Mapping a new familial thyroid epithelial neoplasia susceptibility locus to chromosome 8p23.1-p22 by high-density single-nucleotide polymorphism genome-wide linkage analysis. J Clin Endocrinol Metab 93 (11): 4426-30, 2008.[PUBMED Abstract]
- He H, Nagy R, Liyanarachchi S, et al.: A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Res 69 (2): 625-31, 2009.[PUBMED Abstract]
- Gudmundsson J, Sulem P, Gudbjartsson DF, et al.: Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet 41 (4): 460-4, 2009.[PUBMED Abstract]
- Suh I, Filetti S, Vriens MR, et al.: Distinct loci on chromosome 1q21 and 6q22 predispose to familial nonmedullary thyroid cancer: a SNP array-based linkage analysis of 38 families. Surgery 146 (6): 1073-80, 2009.[PUBMED Abstract]
- Xiong Y, Zhang L, Holloway AK, et al.: MiR-886-3p regulates cell proliferation and migration, and is dysregulated in familial non-medullary thyroid cancer. PLoS One 6 (10): e24717, 2011.[PUBMED Abstract]
- Capezzone M, Cantara S, Marchisotta S, et al.: Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J Clin Endocrinol Metab 93 (10): 3950-7, 2008.[PUBMED Abstract]
- He H, Bronisz A, Liyanarachchi S, et al.: SRGAP1 is a candidate gene for papillary thyroid carcinoma susceptibility. J Clin Endocrinol Metab 98 (5): E973-80, 2013.[PUBMED Abstract]
- Gara SK, Jia L, Merino MJ, et al.: Germline HABP2 Mutation Causing Familial Nonmedullary Thyroid Cancer. N Engl J Med 373 (5): 448-55, 2015.[PUBMED Abstract]
- Liu C, Yu Y, Yin G, et al.: C14orf93 (RTFC) is identified as a novel susceptibility gene for familial nonmedullary thyroid cancer. Biochem Biophys Res Commun 482 (4): 590-596, 2017.[PUBMED Abstract]
- McKay JD, Williamson J, Lesueur F, et al.: At least three genes account for familial papillary thyroid carcinoma: TCO and MNG1 excluded as susceptibility loci from a large Tasmanian family. Eur J Endocrinol 141 (2): 122-5, 1999.[PUBMED Abstract]
- Lesueur F, Stark M, Tocco T, et al.: Genetic heterogeneity in familial nonmedullary thyroid carcinoma: exclusion of linkage to RET, MNG1, and TCO in 56 families. NMTC Consortium. J Clin Endocrinol Metab 84 (6): 2157-62, 1999.[PUBMED Abstract]
- Cavaco BM, Batista PF, Martins C, et al.: Familial non-medullary thyroid carcinoma (FNMTC): analysis of fPTC/PRN, NMTC1, MNG1 and TCO susceptibility loci and identification of somatic BRAF and RAS mutations. Endocr Relat Cancer 15 (1): 207-15, 2008.[PUBMED Abstract]
- Bullock M, Duncan EL, O'Neill C, et al.: Association of FOXE1 polyalanine repeat region with papillary thyroid cancer. J Clin Endocrinol Metab 97 (9): E1814-9, 2012.[PUBMED Abstract]
- Landa I, Ruiz-Llorente S, Montero-Conde C, et al.: The variant rs1867277 in FOXE1 gene confers thyroid cancer susceptibility through the recruitment of USF1/USF2 transcription factors. PLoS Genet 5 (9): e1000637, 2009.[PUBMED Abstract]
- Pereira JS, da Silva JG, Tomaz RA, et al.: Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine 49 (1): 204-14, 2015.[PUBMED Abstract]
- Ngan ES, Lang BH, Liu T, et al.: A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst 101 (3): 162-75, 2009.[PUBMED Abstract]
- Cantara S, Capuano S, Formichi C, et al.: Lack of germline A339V mutation in thyroid transcription factor-1 (TITF-1/NKX2.1) gene in familial papillary thyroid cancer. Thyroid Res 3 (1): 4, 2010.[PUBMED Abstract]
- Jazdzewski K, Murray EL, Franssila K, et al.: Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci U S A 105 (20): 7269-74, 2008.[PUBMED Abstract]
- Fu D, Collins K: Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol Cell 28 (5): 773-85, 2007.[PUBMED Abstract]
- Cantara S, Pisu M, Frau DV, et al.: Telomere abnormalities and chromosome fragility in patients affected by familial papillary thyroid cancer. J Clin Endocrinol Metab 97 (7): E1327-31, 2012.[PUBMED Abstract]
- He M, Bian B, Gesuwan K, et al.: Telomere length is shorter in affected members of families with familial nonmedullary thyroid cancer. Thyroid 23 (3): 301-7, 2013.[PUBMED Abstract]
- Jendrzejewski J, Tomsic J, Lozanski G, et al.: Telomere length and telomerase reverse transcriptase gene copy number in patients with papillary thyroid carcinoma. J Clin Endocrinol Metab 96 (11): E1876-80, 2011.[PUBMED Abstract]
- Wong K, Ren XR, Huang YZ, et al.: Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell 107 (2): 209-21, 2001.[PUBMED Abstract]
- Tomsic J, He H, de la Chapelle A: HABP2 Mutation and Nonmedullary Thyroid Cancer. N Engl J Med 373 (21): 2086, 2015.[PUBMED Abstract]
- Sponziello M, Durante C, Filetti S: HABP2 Mutation and Nonmedullary Thyroid Cancer. N Engl J Med 373 (21): 2085-6, 2015.[PUBMED Abstract]
- Zhou EY, Lin Z, Yang Y: HABP2 Mutation and Nonmedullary Thyroid Cancer. N Engl J Med 373 (21): 2084-5, 2015.[PUBMED Abstract]
- Alzahrani AS, Murugan AK, Qasem E, et al.: HABP2 Gene Mutations Do Not Cause Familial or Sporadic Non-Medullary Thyroid Cancer in a Highly Inbred Middle Eastern Population. Thyroid 26 (5): 667-71, 2016.[PUBMED Abstract]
- Zhang T, Xing M: HABP2 G534E Mutation in Familial Nonmedullary Thyroid Cancer. J Natl Cancer Inst 108 (6): djv415, 2016.[PUBMED Abstract]
- Tomsic J, Fultz R, Liyanarachchi S, et al.: HABP2 G534E Variant in Papillary Thyroid Carcinoma. PLoS One 11 (1): e0146315, 2016.[PUBMED Abstract]
- Sahasrabudhe R, Stultz J, Williamson J, et al.: The HABP2 G534E variant is an unlikely cause of familial non-medullary thyroid cancer. J Clin Endocrinol Metab : jc20153928, 2015.[PUBMED Abstract]
- Weeks AL, Wilson SG, Ward L, et al.: HABP2 germline variants are uncommon in familial nonmedullary thyroid cancer. BMC Med Genet 17 (1): 60, 2016.[PUBMED Abstract]
- Mazzaferri EL, Jhiang SM: Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med 97 (5): 418-28, 1994.[PUBMED Abstract]
- Haugen BR, Alexander EK, Bible KC, et al.: 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 26 (1): 1-133, 2016.[PUBMED Abstract]
- Pacini F, Molinaro E, Castagna MG, et al.: Recombinant human thyrotropin-stimulated serum thyroglobulin combined with neck ultrasonography has the highest sensitivity in monitoring differentiated thyroid carcinoma. J Clin Endocrinol Metab 88 (8): 3668-73, 2003.[PUBMED Abstract]
- Spencer CA, Takeuchi M, Kazarosyan M, et al.: Serum thyroglobulin autoantibodies: prevalence, influence on serum thyroglobulin measurement, and prognostic significance in patients with differentiated thyroid carcinoma. J Clin Endocrinol Metab 83 (4): 1121-7, 1998.[PUBMED Abstract]
- Pellegriti G, Frasca F, Regalbuto C, et al.: Worldwide increasing incidence of thyroid cancer: update on epidemiology and risk factors. J Cancer Epidemiol 2013: 965212, 2013.[PUBMED Abstract]
- Kwak JY: Indications for fine needle aspiration in thyroid nodules. Endocrinol Metab (Seoul) 28 (2): 81-5, 2013.[PUBMED Abstract]
- Marshall CL, Lee JE, Xing Y, et al.: Routine pre-operative ultrasonography for papillary thyroid cancer: effects on cervical recurrence. Surgery 146 (6): 1063-72, 2009.[PUBMED Abstract]
- Cibas ES, Ali SZ: The Bethesda System for Reporting Thyroid Cytopathology. Thyroid 19 (11): 1159-65, 2009.[PUBMED Abstract]
- Mazeh H, Benavidez J, Poehls JL, et al.: In patients with thyroid cancer of follicular cell origin, a family history of nonmedullary thyroid cancer in one first-degree relative is associated with more aggressive disease. Thyroid 22 (1): 3-8, 2012.[PUBMED Abstract]
- El Lakis M, Giannakou A, Nockel PJ, et al.: Do patients with familial nonmedullary thyroid cancer present with more aggressive disease? Implications for initial surgical treatment. Surgery 165 (1): 50-57, 2019.[PUBMED Abstract]
- Mazeh H, Sippel RS: Familial nonmedullary thyroid carcinoma. Thyroid 23 (9): 1049-56, 2013.[PUBMED Abstract]
- Mazzaferri EL, Doherty GM, Steward DL: The pros and cons of prophylactic central compartment lymph node dissection for papillary thyroid carcinoma. Thyroid 19 (7): 683-9, 2009.[PUBMED Abstract]
- McLeod DS, Sawka AM, Cooper DS: Controversies in primary treatment of low-risk papillary thyroid cancer. Lancet 381 (9871): 1046-57, 2013.[PUBMED Abstract]
- Moo TA, McGill J, Allendorf J, et al.: Impact of prophylactic central neck lymph node dissection on early recurrence in papillary thyroid carcinoma. World J Surg 34 (6): 1187-91, 2010.[PUBMED Abstract]
- Perrino M, Vannucchi G, Vicentini L, et al.: Outcome predictors and impact of central node dissection and radiometabolic treatments in papillary thyroid cancers < or =2 cm. Endocr Relat Cancer 16 (1): 201-10, 2009.[PUBMED Abstract]
- Shan CX, Zhang W, Jiang DZ, et al.: Routine central neck dissection in differentiated thyroid carcinoma: a systematic review and meta-analysis. Laryngoscope 122 (4): 797-804, 2012.[PUBMED Abstract]
- Zetoune T, Keutgen X, Buitrago D, et al.: Prophylactic central neck dissection and local recurrence in papillary thyroid cancer: a meta-analysis. Ann Surg Oncol 17 (12): 3287-93, 2010.[PUBMED Abstract]
- Moreno MA, Edeiken-Monroe BS, Siegel ER, et al.: In papillary thyroid cancer, preoperative central neck ultrasound detects only macroscopic surgical disease, but negative findings predict excellent long-term regional control and survival. Thyroid 22 (4): 347-55, 2012.[PUBMED Abstract]
- Jin J, Allemang MT, McHenry CR: Levothyroxine replacement dosage determination after thyroidectomy. Am J Surg 205 (3): 360-3; discussion 363-4, 2013.[PUBMED Abstract]
- Mistry D, Atkin S, Atkinson H, et al.: Predicting thyroxine requirements following total thyroidectomy. Clin Endocrinol (Oxf) 74 (3): 384-7, 2011.[PUBMED Abstract]
- 本要約の変更点(05/29/2020)
-
PDQがん情報要約は定期的に見直され、新情報が利用可能になり次第更新される。本セクションでは、上記の日付における本要約最新変更点を記述する。
要旨
本文で以下の記述が改訂された;リスク軽減のための甲状腺摘出術の実施時期は、特定のRET多様体に伴うリスクが指針となるが、本手技の至適な実施時期の決定にカルシトニンの基礎値を用いることができる。
序
本文に以下の記述が追加された;現在家族性甲状腺髄様がん(FMTC)は、MEN2Aの亜型とみなされる(引用、参考文献1としてWells et al.)。
参考文献2としてWorld Health Organization(WHO)が追加された。
参考文献7としてMazeh et al.が追加された。
多発性内分泌腫瘍1型(MEN1)
参考文献27としてVentura al.が追加された。
本文に以下の記述が追加された;膵十二指腸神経内分泌腫瘍の外科的な摘出術は、再発率が膵体尾部切除術と比較して高く、内分泌機能不全の発生率がWhipple法と比較して低いという関連が認められている(引用、参考文献71としてRatnayake et al.)。さらに、参考文献73としてNell et al.が追加された。
MEN2
本文に以下の記述が追加された;MEN2で転移性褐色細胞腫(PHEO)が報告されている。
本文で以下の記述が改訂された;米国において、MTCは毎年診断される甲状腺がん新規症例の1~2%を占める(引用、参考文献15としてHowlader et al.)。さらに、本文で以下の記述が改訂された;米国で診断される甲状腺がん症例の約75%が散発性である。
本文で以下の記述が改訂された;毎年診断される甲状腺がん新規症例のうち、1~2%がMTCである。
参考文献24としてRandle et al.が追加された。
本文で以下の記述が改訂された;診断時の病期が早期であることに加え、MTCの生存率の向上と関連しているその他の因子には、腫瘍が小さいこと、診断時の年齢が低いこと、生化学的または遺伝学的スクリーニングによる診断であることなどがある。
ヒルシュスプルング病(HSCR)を伴うMEN2Aのサブセクションは広範囲にわたって改訂された。
本文で以下の記述が改訂された;MEN2A症例の最大50%は、FMTC亜型であり、PHEOまたは副甲状腺腺腫/過形成が認められず、生殖細胞RET病原性多様体およびMTCのみが認められる家族または一人の個人として定義される。この定義は、単独の診断として以前のFMTCの分類に置き換えられる。さらに、本文で以下の記述が改訂された;現在FMTCは、MEN2Aの亜型とみなされ、MEN2A症候群の他の症状が発現しないか、発現が遅れる。現在の管理ガイドラインでは、純粋なFMTCと考えられる患者も、PHEOおよび副甲状腺機能亢進症についてスクリーニングするよう推奨している。
参考文献72としてCastinetti et al.が追加された。
家族性PHEOおよび傍神経節腫(PGL)症候群
参考文献1としてWHOが追加された。
家族性非髄様甲状腺がん(Familial Nonmedullary Thyroid Cancer[FNMTC])
本文に以下の記述が追加された;家系にFNMTC罹患者が3人いる場合は、より若い年齢での腫瘍の特性が侵攻性である可能性が高い;これから、より積極的な外科的管理が必要になる場合があることが示唆される(引用、参考文献69としてEl Lakis et al.)。
本要約はPDQ Cancer Genetics Editorial Boardが作成と内容の更新を行っており、編集に関してはNCIから独立している。本要約は独自の文献レビューを反映しており、NCIまたはNIHの方針声明を示すものではない。PDQ要約の更新におけるPDQ編集委員会の役割および要約の方針に関する詳しい情報については、本PDQ要約についておよびPDQ® - NCI's Comprehensive Cancer Databaseを参照のこと。
- 本PDQ要約について
-
本要約の目的
医療専門家向けの本PDQがん情報要約では、内分泌および神経内分泌腫瘍の遺伝学について、包括的な、専門家の査読を経た、そして証拠に基づいた情報を提供する。本要約は、がん患者を治療する臨床家に情報を与え支援するための情報資源として作成されている。これは医療における意思決定のための公式なガイドラインまたは推奨事項を提供しているわけではない。
査読者および更新情報
本要約は編集作業において米国国立がん研究所(NCI)とは独立したPDQ Cancer Genetics Editorial Boardにより定期的に見直され、随時更新される。本要約は独自の文献レビューを反映しており、NCIまたは米国国立衛生研究所(NIH)の方針声明を示すものではない。
委員会のメンバーは毎月、最近発表された記事を見直し、記事に対して以下を行うべきか決定する:
要約の変更は、発表された記事の証拠の強さを委員会のメンバーが評価し、記事を本要約にどのように組み入れるべきかを決定するコンセンサス過程を経て行われる。
本要約の内容に関するコメントまたは質問は、NCIウェブサイトのEmail UsからCancer.govまで送信のこと。要約に関する質問またはコメントについて委員会のメンバー個人に連絡することを禁じる。委員会のメンバーは個別の問い合わせには対応しない。
証拠レベル
本要約で引用される文献の中には証拠レベルの指定が記載されているものがある。これらの指定は、特定の介入やアプローチの使用を支持する証拠の強さを読者が査定する際、助けとなるよう意図されている。PDQ Cancer Genetics Editorial Boardは、証拠レベルの指定を展開する際に公式順位分類を使用している。
本要約の使用許可
PDQは登録商標である。PDQ文書の内容は本文として自由に使用できるが、完全な形で記し定期的に更新しなければ、NCI PDQがん情報要約とすることはできない。しかし、著者は“NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: 【本要約からの抜粋を含める】.”のような一文を記述してもよい。
本PDQ要約の好ましい引用は以下の通りである:
PDQ® Cancer Genetics Editorial Board.PDQ Genetics of Endocrine and Neuroendocrine Neoplasias.Bethesda, MD: National Cancer Institute.Updated <MM/DD/YYYY>.Available at: https://www.cancer.gov/types/thyroid/hp/medullary-thyroid-genetics-pdq.Accessed <MM/DD/YYYY>.[PMID: 26389271]
本要約内の画像は、PDQ要約内での使用に限って著者、イラストレーター、および/または出版社の許可を得て使用されている。PDQ情報以外での画像の使用許可は、所有者から得る必要があり、米国国立がん研究所(National Cancer Institute)が付与できるものではない。本要約内のイラストの使用に関する情報は、多くの他のがん関連画像とともにVisuals Online(2,000以上の科学画像を収蔵)で入手できる。
免責条項
これらの要約内の情報は、保険払い戻しの決定基準として使用されるべきものではない。保険の適用範囲に関する詳しい情報については、Cancer.govのManaging Cancer Careページで入手できる。
お問い合わせ
Cancer.govウェブサイトについての問い合わせまたはヘルプの利用に関する詳しい情報は、Contact Us for Helpページに掲載されている。質問はウェブサイトのEmail UsからもCancer.govに送信可能である。